Global Transcriptomic Analysis of Zebrafish Glucagon Receptor Mutant Reveals Its Regulated Metabolic Network


Abstract
1. Introduction
2. Results
2.1. The Transcriptome of Wildtype and Gcgr−/− Larvae
2.2. Gene Ontology (GO) Enrichment Analysis of Differentially Expressed Genes
2.3. Kyoto Encyclopedia of Genes and Genomes (KEGG) Analysis Reveals that GCGR Deficiency Alters Multiple Metabolic Pathways
2.4. GCGR Regulates Lipid Metabolism
2.5. GCGR Regulates Carbohydrate Metabolism
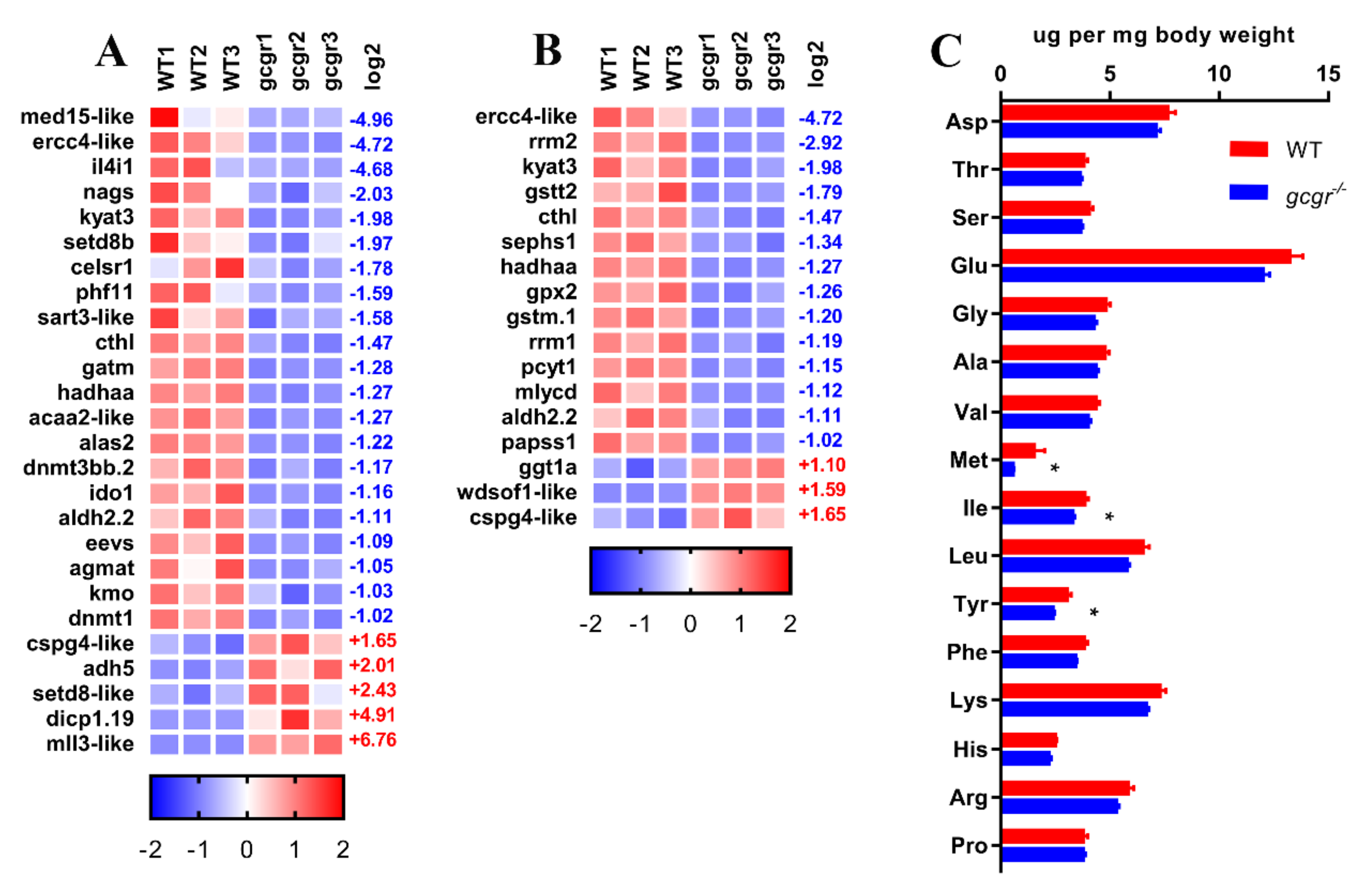
2.6. GCGR Regulates Amino Acid Metabolism
2.7. Verification of Transcriptome Data by qRT-PCR
3. Discussion
4. Materials and Methods
4.1. Zebrafish Lines and Maintenance
4.2. RNA Extraction, cDNA Library Preparation, Sequencing
4.3. Bioinformatics Analysis of RNA Sequence Data
4.4. Analysis of Differentially Expressed Genes (DEGs)
4.5. Quantitative RT-PCR
4.6. Oil Red Staining
4.7. 2-NBDG Uptake Test
4.8. Glucose Exposure and α-Cells Counting
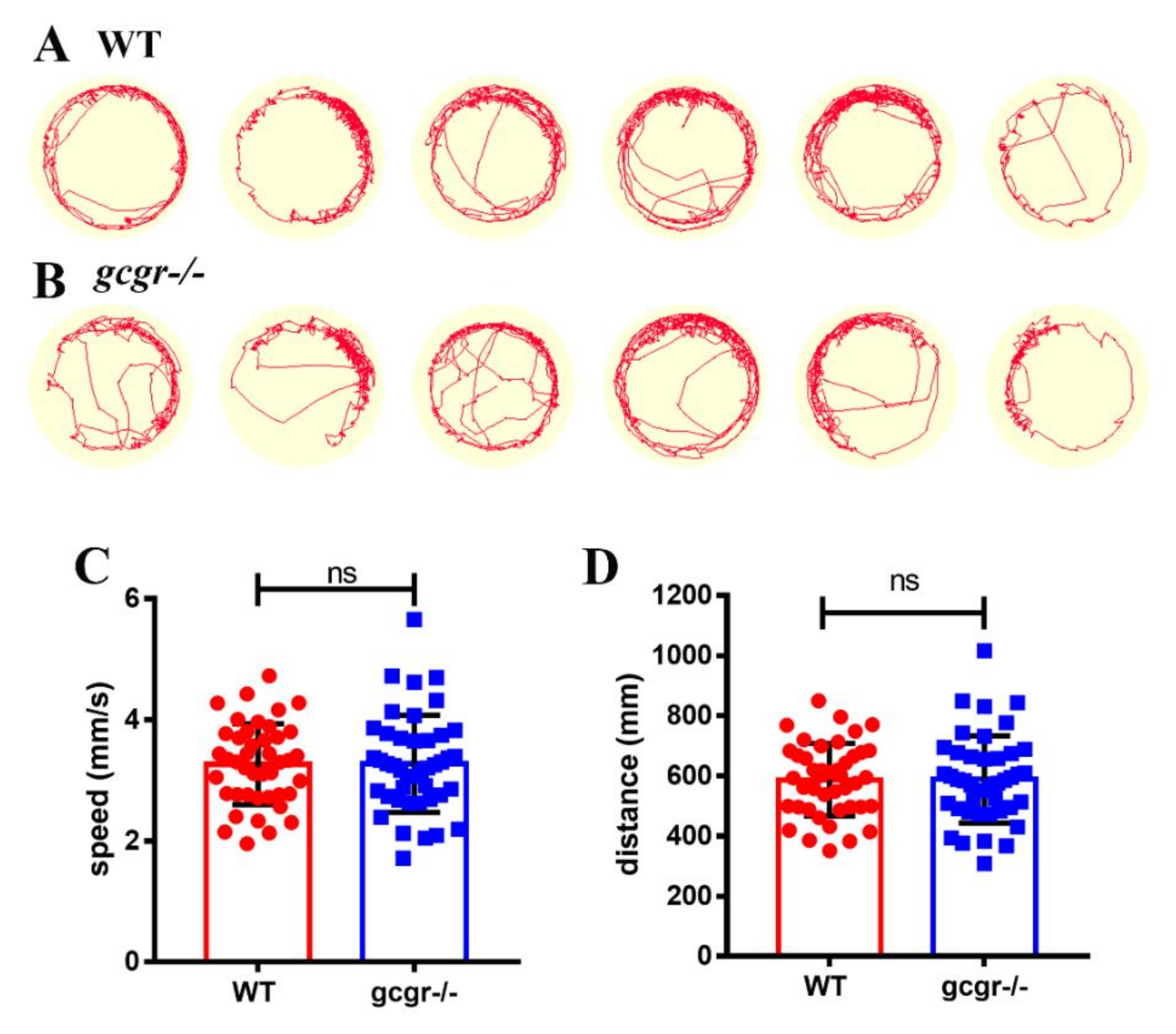
4.9. Behavior Test
4.10. Amino Acid Measurement
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Campbell, J.E.; Drucker, D.J. Islet alpha cells and glucagon—Critical regulators of energy homeostasis. Nat. Rev. Endocrinol. 2015, 11, 329–338. [Google Scholar] [CrossRef]
- Zhang, H.; Qiao, A.; Yang, D.; Yang, L.; Dai, A.; de Graaf, C.; Reedtz-Runge, S.; Dharmarajan, V.; Zhang, H.; Han, G.W.; et al. Structure of the full-length glucagon class B G-protein-coupled receptor. Nature 2017, 546, 259–264. [Google Scholar] [CrossRef]
- Siu, F.Y.; He, M.; de Graaf, C.; Han, G.W.; Yang, D.; Zhang, Z.; Zhou, C.; Xu, Q.; Wacker, D.; Joseph, J.S.; et al. Structure of the human glucagon class B G-protein-coupled receptor. Nature 2013, 499, 444–449. [Google Scholar] [CrossRef] [PubMed]
- Lok, S.; Kuijper, J.L.; Jelinek, L.J.; Kramer, J.M.; Whitmore, T.E.; Sprecher, C.A.; Mathewes, S.; Grant, F.J.; Biggs, S.H.; Rosenberg, G.B.; et al. The human glucagon receptor encoding gene: Structure, cDNA sequence and chromosomal localization. Gene 1994, 140, 203–209. [Google Scholar] [CrossRef]
- Charron, M.J.; Vuguin, P.M. Lack of glucagon receptor signaling and its implications beyond glucose homeostasis. J. Endocrinol. 2015, 224, R123–R130. [Google Scholar] [CrossRef] [PubMed]
- Gelling, R.W.; Du, X.Q.; Dichmann, D.S.; Romer, J.; Huang, H.; Cui, L.; Obici, S.; Tang, B.; Holst, J.J.; Fledelius, C.; et al. Lower blood glucose, hyperglucagonemia, and pancreatic alpha cell hyperplasia in glucagon receptor knockout mice. Proc. Natl. Acad. Sci. USA 2003, 100, 1438–1443. [Google Scholar] [CrossRef]
- Conarello, S.L.; Jiang, G.; Mu, J.; Li, Z.; Woods, J.; Zycband, E.; Ronan, J.; Liu, F.; Roy, R.S.; Zhu, L.; et al. Glucagon receptor knockout mice are resistant to diet-induced obesity and streptozotocin-mediated beta cell loss and hyperglycaemia. Diabetologia 2007, 50, 142–150. [Google Scholar] [CrossRef]
- Lee, Y.; Wang, M.Y.; Du, X.Q.; Charron, M.J.; Unger, R.H. Glucagon receptor knockout prevents insulin-deficient type 1 diabetes in mice. Diabetes 2011, 60, 391–397. [Google Scholar] [CrossRef]
- Wang, M.Y.; Yan, H.; Shi, Z.; Evans, M.R.; Yu, X.; Lee, Y.; Chen, S.; Williams, A.; Philippe, J.; Roth, M.G.; et al. Glucagon receptor antibody completely suppresses type 1 diabetes phenotype without insulin by disrupting a novel diabetogenic pathway. Proc. Natl. Acad. Sci. USA 2015, 112, 2503–2508. [Google Scholar] [CrossRef]
- Mu, J.; Qureshi, S.A.; Brady, E.J.; Muise, E.S.; Candelore, M.R.; Jiang, G.; Li, Z.; Wu, M.S.; Yang, X.; Dallas-Yang, Q.; et al. Anti-diabetic efficacy and impact on amino acid metabolism of GRA1, a novel small-molecule glucagon receptor antagonist. PLoS ONE 2012, 7, e49572. [Google Scholar] [CrossRef]
- Okamoto, H.; Kim, J.; Aglione, J.; Lee, J.; Cavino, K.; Na, E.; Rafique, A.; Kim, J.H.; Harp, J.; Valenzuela, D.M.; et al. Glucagon Receptor Blockade With a Human Antibody Normalizes Blood Glucose in Diabetic Mice and Monkeys. Endocrinology 2015, 156, 2781–2794. [Google Scholar] [CrossRef] [PubMed]
- Dallas-Yang, Q.; Shen, X.; Strowski, M.; Brady, E.; Saperstein, R.; Gibson, R.E.; Szalkowski, D.; Qureshi, S.A.; Candelore, M.R.; Fenyk-Melody, J.E.; et al. Hepatic glucagon receptor binding and glucose-lowering in vivo by peptidyl and non-peptidyl glucagon receptor antagonists. Eur. J. Pharmacol. 2004, 501, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Scheen, A.J.; Paquot, N.; Lefebvre, P.J. Investigational glucagon receptor antagonists in Phase I and II clinical trials for diabetes. Exp. Opin. Investig. Drugs 2017, 26, 1373–1389. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, H.; Winzell, M.S.; Brand, C.L.; Fosgerau, K.; Gelling, R.W.; Nishimura, E.; Ahren, B. Glucagon receptor knockout mice display increased insulin sensitivity and impaired beta-cell function. Diabetes 2006, 55, 3463–3469. [Google Scholar] [CrossRef] [PubMed]
- Ouhilal, S.; Vuguin, P.; Cui, L.; Du, X.Q.; Gelling, R.W.; Reznik, S.E.; Russell, R.; Parlow, A.F.; Karpovsky, C.; Santoro, N.; et al. Hypoglycemia, hyperglucagonemia, and fetoplacental defects in glucagon receptor knockout mice: A role for glucagon action in pregnancy maintenance. Am. J. Physiol. Endocrinol. Metab. 2012, 302, E522–E531. [Google Scholar] [CrossRef] [PubMed]
- Vuguin, P.M.; Kedees, M.H.; Cui, L.; Guz, Y.; Gelling, R.W.; Nejathaim, M.; Charron, M.J.; Teitelman, G. Ablation of the glucagon receptor gene increases fetal lethality and produces alterations in islet development and maturation. Endocrinology 2006, 147, 3995–4006. [Google Scholar] [CrossRef] [PubMed]
- Guan, H.P.; Yang, X.; Lu, K.; Wang, S.P.; Castro-Perez, J.M.; Previs, S.; Wright, M.; Shah, V.; Herath, K.; Xie, D.; et al. Glucagon receptor antagonism induces increased cholesterol absorption. J. Lipid Res. 2015, 56, 2183–2195. [Google Scholar] [CrossRef]
- Kim, T.; Nason, S.; Holleman, C.; Pepin, M.; Wilson, L.; Berryhill, T.F.; Wende, A.R.; Steele, C.; Young, M.E.; Barnes, S.; et al. Glucagon Receptor Signaling Regulates Energy Metabolism via Hepatic Farnesoid X Receptor and Fibroblast Growth Factor 21. Diabetes 2018, 67, 1773–1782. [Google Scholar] [CrossRef]
- Townsend, L.K.; Medak, K.D.; Knuth, C.M.; Peppler, W.T.; Charron, M.J.; Wright, D.C. Loss of glucagon signaling alters white adipose tissue browning. FASEB J. 2019, 33, 4824–4835. [Google Scholar] [CrossRef]
- Longuet, C.; Sinclair, E.M.; Maida, A.; Baggio, L.L.; Maziarz, M.; Charron, M.J.; Drucker, D.J. The glucagon receptor is required for the adaptive metabolic response to fasting. Cell Metab. 2008, 8, 359–371. [Google Scholar] [CrossRef]
- Gild, M.L.; Tsang, V.; Samra, J.; Clifton-Bligh, R.J.; Tacon, L.; Gill, A.J. Hypercalcemia in Glucagon Cell Hyperplasia and Neoplasia (Mahvash Syndrome): A New Association. J. Clin. Endocrinol. Metab. 2018, 103, 3119–3123. [Google Scholar] [CrossRef] [PubMed]
- Larger, E.; Wewer Albrechtsen, N.J.; Hansen, L.H.; Gelling, R.W.; Capeau, J.; Deacon, C.F.; Madsen, O.D.; Yakushiji, F.; De Meyts, P.; Holst, J.J.; et al. Pancreatic alpha-cell hyperplasia and hyperglucagonemia due to a glucagon receptor splice mutation. Endocrinol. Diabetes Metab. Case Rep. 2016, 2016. [Google Scholar] [CrossRef]
- Zhou, C.; Dhall, D.; Nissen, N.N.; Chen, C.R.; Yu, R. Homozygous P86S mutation of the human glucagon receptor is associated with hyperglucagonemia, alpha cell hyperplasia, and islet cell tumor. Pancreas 2009, 38, 941–946. [Google Scholar] [CrossRef] [PubMed]
- Yu, R. Mahvash Disease: 10 Years After Discovery. Pancreas 2018, 47, 511–515. [Google Scholar] [CrossRef]
- Li, M.; Dean, E.D.; Zhao, L.; Nicholson, W.E.; Powers, A.C.; Chen, W. Glucagon receptor inactivation leads to alpha-cell hyperplasia in zebrafish. J. Endocrinol. 2015, 227, 93–103. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Berglund, E.D.; Kang, L.; Lee-Young, R.S.; Hasenour, C.M.; Lustig, D.G.; Lynes, S.E.; Donahue, E.P.; Swift, L.L.; Charron, M.J.; Wasserman, D.H. Glucagon and lipid interactions in the regulation of hepatic AMPK signaling and expression of PPARalpha and FGF21 transcripts in vivo. Am. J. Physiol. Endocrinol. Metab. 2010, 299, E607–E614. [Google Scholar] [CrossRef]
- Yang, N.; Sun, R.; Liao, X.; Aa, J.; Wang, G. UDP-glucuronosyltransferases (UGTs) and their related metabolic cross-talk with internal homeostasis: A systematic review of UGT isoforms for precision medicine. Pharmacol. Res. 2017, 121, 169–183. [Google Scholar] [CrossRef]
- Ali, S.; Lamont, B.J.; Charron, M.J.; Drucker, D.J. Dual elimination of the glucagon and GLP-1 receptors in mice reveals plasticity in the incretin axis. J. Clin. Investig. 2011, 121, 1917–1929. [Google Scholar] [CrossRef]
- Gu, W.; Winters, K.A.; Motani, A.S.; Komorowski, R.; Zhang, Y.; Liu, Q.; Wu, X.; Rulifson, I.C.; Sivits, G., Jr.; Graham, M.; et al. Glucagon receptor antagonist-mediated improvements in glycemic control are dependent on functional pancreatic GLP-1 receptor. Am. J. Physiol. Endocrinol. Metab. 2010, 299, E624–E632. [Google Scholar] [CrossRef]
- Lee, J.; Jung, D.W.; Kim, W.H.; Um, J.I.; Yim, S.H.; Oh, W.K.; Williams, D.R. Development of a highly visual, simple, and rapid test for the discovery of novel insulin mimetics in living vertebrates. ACS Chem. Biol. 2013, 8, 1803–1814. [Google Scholar] [CrossRef]
- Townsend, D.M.; Tew, K.D. The role of glutathione-S-transferase in anti-cancer drug resistance. Oncogene 2003, 22, 7369–7375. [Google Scholar] [CrossRef] [PubMed]
- Hanigan, M.H. Gamma-glutamyl transpeptidase: Redox regulation and drug resistance. Adv. Cancer Res. 2014, 122, 103–141. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, C.; Seino, Y.; Miyahira, H.; Yamamoto, M.; Fukami, A.; Ozaki, N.; Takagishi, Y.; Sato, J.; Fukuwatari, T.; Shibata, K.; et al. Remodeling of hepatic metabolism and hyperaminoacidemia in mice deficient in proglucagon-derived peptides. Diabetes 2012, 61, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Svoboda, M.; Tastenoy, M.; Vertongen, P.; Robberecht, P. Relative quantitative analysis of glucagon receptor mRNA in rat tissues. Mol. Cell. Endocrinol. 1994, 105, 131–137. [Google Scholar] [CrossRef]
- Guzman, C.B.; Zhang, X.M.; Liu, R.; Regev, A.; Shankar, S.; Garhyan, P.; Pillai, S.G.; Kazda, C.; Chalasani, N.; Hardy, T.A. Treatment with LY2409021, a glucagon receptor antagonist, increases liver fat in patients with type 2 diabetes. Diabetes Obes. Metab. 2017, 19, 1521–1528. [Google Scholar] [CrossRef]
- Abu-Hamdah, R.; Rabiee, A.; Meneilly, G.S.; Shannon, R.P.; Andersen, D.K.; Elahi, D. Clinical review: The extrapancreatic effects of glucagon-like peptide-1 and related peptides. J. Clin. Endocrinol. Metab. 2009, 94, 1843–1852. [Google Scholar] [CrossRef]
- Neumann, U.H.; Ho, J.S.S.; Mojibian, M.; Covey, S.D.; Charron, M.J.; Kieffer, T.J. Glucagon receptor gene deletion in insulin knockout mice modestly reduces blood glucose and ketones but does not promote survival. Mol. Metab. 2016, 5, 731–736. [Google Scholar] [CrossRef]
- Damond, N.; Thorel, F.; Moyers, J.S.; Charron, M.J.; Vuguin, P.M.; Powers, A.C.; Herrera, P.L. Blockade of glucagon signaling prevents or reverses diabetes onset only if residual beta-cells persist. Elife 2016, 5, e13828. [Google Scholar] [CrossRef]
- Sharma, A.X.; Quittner-Strom, E.B.; Lee, Y.; Johnson, J.A.; Martin, S.A.; Yu, X.; Li, J.; Lu, J.; Cai, Z.; Chen, S.; et al. Glucagon Receptor Antagonism Improves Glucose Metabolism and Cardiac Function by Promoting AMP-Mediated Protein Kinase in Diabetic Mice. Cell Rep. 2018, 22, 1760–1773. [Google Scholar] [CrossRef]
- Kim, J.; Okamoto, H.; Huang, Z.; Anguiano, G.; Chen, S.; Liu, Q.; Cavino, K.; Xin, Y.; Na, E.; Hamid, R.; et al. Amino Acid Transporter Slc38a5 Controls Glucagon Receptor Inhibition-Induced Pancreatic alpha Cell Hyperplasia in Mice. Cell Metab. 2017, 25, 1348–1361. [Google Scholar] [CrossRef]
- Dean, E.D.; Li, M.; Prasad, N.; Wisniewski, S.N.; Von Deylen, A.; Spaeth, J.; Maddison, L.; Botros, A.; Sedgeman, L.R.; Bozadjieva, N.; et al. Interrupted Glucagon Signaling Reveals Hepatic alpha Cell Axis and Role for L-Glutamine in alpha Cell Proliferation. Cell Metab. 2017, 25, 1362–1373. [Google Scholar] [CrossRef]
- Yang, J.; MacDougall, M.L.; McDowell, M.T.; Xi, L.; Wei, R.; Zavadoski, W.J.; Molloy, M.P.; Baker, J.D.; Kuhn, M.; Cabrera, O.; et al. Polyomic profiling reveals significant hepatic metabolic alterations in glucagon-receptor (GCGR) knockout mice: Implications on anti-glucagon therapies for diabetes. BMC Genom. 2011, 12, 281. [Google Scholar] [CrossRef]
- Galsgaard, K.D.; Winther-Sorensen, M.; Orskov, C.; Kissow, H.; Poulsen, S.S.; Vilstrup, H.; Prehn, C.; Adamski, J.; Jepsen, S.L.; Hartmann, B.; et al. Disruption of glucagon receptor signaling causes hyperaminoacidemia exposing a possible liver-alpha-cell axis. Am. J. Physiol. Endocrinol. Metab. 2018, 314, E93–E103. [Google Scholar] [CrossRef]
- Kimmel, C.B.; Ballard, W.W.; Kimmel, S.R.; Ullmann, B.; Schilling, T.F. Stages of embryonic development of the zebrafish. Dev. Dyn. 1995, 203, 253–310. [Google Scholar] [CrossRef]
- Zecchin, E.; Filippi, A.; Biemar, F.; Tiso, N.; Pauls, S.; Ellertsdottir, E.; Gnugge, L.; Bortolussi, M.; Driever, W.; Argenton, F. Distinct delta and jagged genes control sequential segregation of pancreatic cell types from precursor pools in zebrafish. Dev. Biol. 2007, 301, 192–204. [Google Scholar] [CrossRef]
- Huang, J.; Liang, X.; Xuan, Y.; Geng, C.; Li, Y.; Lu, H.; Qu, S.; Mei, X.; Chen, H.; Yu, T.; et al. A reference human genome dataset of the BGISEQ-500 sequencer. GigaScience 2017, 6, 1–9. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, Y.; Shi, C.; Huang, Z.; Zhang, Y.; Li, S.; Li, Y.; Ye, J.; Yu, C.; Li, Z.; et al. SOAPnuke: A MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. GigaScience 2018, 7, 1–6. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Tarazona, S.; Furio-Tari, P.; Turra, D.; Pietro, A.D.; Nueda, M.J.; Ferrer, A.; Conesa, A. Data quality aware analysis of differential expression in RNA-seq with NOISeq R/Bioc package. Nucl. Acids Res. 2015, 43, e140. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Name | WT1 | WT2 | WT3 | gcgr1 | gcgr2 | gcgr3 |
|---|---|---|---|---|---|---|
| Total raw reads (M) | 21.94 | 21.94 | 21.94 | 21.94 | 21.94 | 21.94 |
| Total clean reads (M) | 21.89 | 21.9 | 21.92 | 21.93 | 21.89 | 21.89 |
| Clean reads ratio (%) | 99.76 | 99.82 | 99.9 | 99.92 | 99.75 | 99.74 |
| Genome total mapping (%) | 90.38 | 89.55 | 90.25 | 90.54 | 90.47 | 90.55 |
| Genome uniquely mapping (%) | 64.66 | 63.98 | 64.69 | 65.12 | 65.53 | 65.34 |
| Genes total mapping (%) | 79.41 | 79.83 | 79.28 | 80.03 | 79.62 | 79.89 |
| Genes uniquely mapping (%) | 69 | 69.02 | 69.03 | 69.21 | 69.13 | 69.21 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, Q.; Hu, M.; Jia, J.; Bai, X.; Liu, C.; Wu, Z.; Chen, W.; Li, M. Global Transcriptomic Analysis of Zebrafish Glucagon Receptor Mutant Reveals Its Regulated Metabolic Network. Int. J. Mol. Sci. 2020, 21, 724. https://doi.org/10.3390/ijms21030724
Kang Q, Hu M, Jia J, Bai X, Liu C, Wu Z, Chen W, Li M. Global Transcriptomic Analysis of Zebrafish Glucagon Receptor Mutant Reveals Its Regulated Metabolic Network. International Journal of Molecular Sciences. 2020; 21(3):724. https://doi.org/10.3390/ijms21030724
Chicago/Turabian StyleKang, Qi, Mengyi Hu, Jianxin Jia, Xuanxuan Bai, Chengdong Liu, Zhiqiang Wu, Wenbiao Chen, and Mingyu Li. 2020. "Global Transcriptomic Analysis of Zebrafish Glucagon Receptor Mutant Reveals Its Regulated Metabolic Network" International Journal of Molecular Sciences 21, no. 3: 724. https://doi.org/10.3390/ijms21030724
APA StyleKang, Q., Hu, M., Jia, J., Bai, X., Liu, C., Wu, Z., Chen, W., & Li, M. (2020). Global Transcriptomic Analysis of Zebrafish Glucagon Receptor Mutant Reveals Its Regulated Metabolic Network. International Journal of Molecular Sciences, 21(3), 724. https://doi.org/10.3390/ijms21030724