Distinct Roles of mTOR Targets S6K1 and S6K2 in Breast Cancer


Department of Microbiology, Immunology and Genetics, University of North Texas Health Science Center, Fort Worth, TX 76107, USA
*
Author to whom correspondence should be addressed.
†
Current address: Department of Molecular Biology and Cell Biology, University of California, Berkeley, CA 94720, USA.
Int. J. Mol. Sci. 2020, 21(4), 1199; https://doi.org/10.3390/ijms21041199
Submission received: 25 January 2020
/
Revised: 6 February 2020
/
Accepted: 7 February 2020
/
Published: 11 February 2020
(This article belongs to the Special Issue mTOR in Human Diseases 2.0)
Abstract
:The mechanistic target of rapamycin (mTOR) is a master regulator of protein translation, metabolism, cell growth and proliferation. It forms two complexes, mTOR complex 1 (mTORC1) and 2 (mTORC2). mTORC1 is frequently deregulated in many cancers, including breast cancer, and is an important target for cancer therapy. The immunosuppressant drug rapamycin and its analogs that inhibit mTOR are currently being evaluated for their potential as anti-cancer agents, albeit with limited efficacy. mTORC1 mediates its function via its downstream targets 40S ribosomal S6 kinases (S6K) and eukaryotic translation initiation factor 4E (eIF4E)-binding protein 1 (4E-BP1). There are two homologs of S6K: S6K1 and S6K2. Most of the earlier studies focused on S6K1 rather than S6K2. Because of their high degree of structural homology, it was generally believed that they behave similarly. Recent studies suggest that while they may share some functions, they may also exhibit distinct or even opposite functions. Both homologs have been implicated in breast cancer, although how they contribute to breast cancer may differ. The purpose of this review article is to compare and contrast the expression, structure, regulation and function of these two S6K homologs in breast cancer.
1. Introduction
Breast cancer is the second leading cause of cancer-related death among women in the United States. Multiple factors contribute to the poor survival and the severity of breast cancer. The mechanistic target of rapamycin (mTOR), also known as the mammalian target of rapamycin, is an important target for breast cancer therapy since it is frequently deregulated in breast cancers and plays a critical role in tumorigenesis [1,2]. mTOR forms two distinct complexes with either raptor (mTORC1) or rictor (mTORC2). mTORC1 acts downstream of the Akt signaling pathway, which is deregulated in approximately 60% of breast cancers and plays critical roles in breast cancer development, progression and resistance to chemotherapeutic drugs [2,3,4,5,6,7,8,9,10,11]. mTORC1 mediates its function via its downstream targets 40S ribosomal S6 kinase (S6K) and 4E-binding protein 1 (4E-BP1) [12].
Studies addressing the highly conserved inducible phosphorylation of ribosomal protein S6 in somatic cells led to the discovery of S6K1 (p70S6Kα) or p70S6 kinase [13,14]. It was originally identified as the serine/threonine kinase that mediated the mitogen-inducible phosphorylation of ribosomal protein S6 (rpS6). However, the observation that rpS6 phosphorylation was not affected in S6K1 knockout mice led to the identification of a close homolog of S6K1, S6K2 (p70S6Kβ), which was later shown to be the major kinase mediating rpS6 phosphorylation [15,16,17,18,19]. S6K1 and S6K2 are encoded by RPS6KB1 on chromosome 17 and RPS6KB2 on chromosome 11, respectively (Table 1). Both genes code for two isoforms each with the use of alternative translation start sites: p70 S6K (S6KαII) and p85 S6K (S6KαI) in the case of S6K1, and p54 S6K (S6KβII) and p56 S6K (S6KβI) for S6K2 [16,20]. The N-terminal extensions of the longer forms of both S6K1 and S6K2 harbor a functional nuclear localization signal (NLS), making them constitutively nuclear. However, the shorter isoforms represent the predominant forms for both homologs and will be referred to as S6K1 and S6K2 henceforth.
Most of the earlier studies focused on p70 S6K1 or S6K1 as the downstream target of mTORC1. It was believed that due to structural similarities, S6K1 and S6K2 share redundant functions. Recent studies, however, challenge this notion [21,22,23,24,25]. Both homologs have been implicated in breast cancer, although they may play distinct roles [26,27]. In this review article, we briefly describe differences in structural aspects, regulation and cellular functions of these two homologs prior to discussing their distinct roles in breast cancer.
2. Structure of S6Ks
Both S6K1 and S6K2 exhibit a modular structure consisting of an N-terminal regulatory region, the kinase domain, followed by the kinase extension domain and a C-terminal regulatory region harboring the autoinhibitory/pseudosubstrate domain (Figure 1). While they share over 80% homology in the amino acid sequence of their kinase domains, as well as a high degree of similarity in the adjacent kinase extension and pseudosubstrate or autoinhibitory domains with conserved sites critical for their activation [16,17], important differences exist in the extreme N- and C-terminal regions. S6K1 possesses a C-terminal PDZ-binding domain, which promotes association with the actin cytoskeleton [28], whereas S6K2 but not S6K1 harbors a functional nuclear localization signal (NLS) and a proline-rich domain, which may promote interaction with the SH3-domain containing proteins at its C-terminus [16] (Figure 1). It is believed that these key differences between S6K1 and S6K2 will result in differential localization and binding partners and hence distinct functions for the two proteins [21].
3. Regulation of S6Ks
3.1. Activation of S6Ks
Growth factor- and hormone-mediated activation of receptor tyrosine kinases promotes phosphatidylinositol 3-kinase (PI3K) activation, which then phosphorylates phosphatidylinositol-4,5-bisphosphate (PIP2) to produce phosphatidylinositol-3,4,5-trisphosphate (PIP3). This leads to the membrane recruitment and activation of pleckstrin homology domain-containing proteins such as phosphoinositide-dependent kinase 1 (PDK1). Activation of PDK1 leads to the phosphorylation and activation of several drivers of cell survival and proliferation such as Akt, which then promotes mTORC1 activation by negatively regulating the tuberous sclerosis complex (TSC), a tumor suppressor complex [29] mutated in hamartomas [30]. Inhibition of the TSC allows activation of the small GTPase ras homolog enriched in brain (RHEB) [31,32], and subsequent mTORC1 activation results in downstream signaling and cap-dependent translation by phosphorylating and inhibiting the eIF4E-binding protein (4E-BP) and activating S6K (Figure 2).
The sensitivity of S6K to the immunosuppressant drug rapamycin implied its regulation by mTOR [33,34,35]. Further studies suggested that the phospho-mimetic mutation of a conserved phosphorylation site in the kinase-extension domain (T389 in S6K1 and T388 in S6K2), known as the hydrophobic motif, by mTOR leads to resistance to rapamycin [36]. Raptor within the mTORC1 complex binds to the tor signaling motif (TOS), a conserved amino acid sequence found in S6K [37], and promotes its interaction with mTOR, which mediates the hydrophobic motif phosphorylation [38]. The activation of S6K is then achieved by PDK-1-mediated phosphorylation at a threonine residue in the activation loop (T229 in S6K1 and T228 in S6K2) within the kinase domain [39]. However, in order for mTORC1 and PDK1 to be able to access their target sites, a series of phosphorylation events at C-terminal serine residues first needs to occur so as to render the pseudosubstrate/autoinhibitory domain inactive and expose the activation loop, making it accessible for PDK1. The C-terminal autoinhibitory domain phosphorylations are believed to be carried out by members of the mitogen-activated protein kinase (MAPK) family [40]. Thus, the current model of S6K activation follows that the initial phosphorylation events in the C-terminal pseudosubstrate domain expose the kinase extension and kinase domains and promote mTORC1-mediated phosphorylation followed by the activating phosphorylation by PDK1 [21,23].
While S6K1 and S6K2 share the majority of conserved phosphorylation sites, they are believed to exhibit differences in their sensitivities to rapamycin and inputs from the MAPK signaling pathway. For example, in H510 lung cancer cells, which are characterized by highly active mitogen-activated protein kinase kinase (MEK) signaling, S6K2 was less responsive to rapamycin but highly sensitive to MEK inhibition [41]. The MAPK-mediated C-terminal phosphorylations appear to be more critical for the activation of S6K2 than that of S6K1 [41,42]. Furthermore, leucine deprivation affected S6K1 but not S6K2 or S6 phosphorylation, suggesting the differential regulation of the two kinases.
Growth factor-mediated activation of S6K1 is followed by its rapid dephosphorylation and desensitization via the serine/threonine protein phosphatase PP2A [43,44,45,46]. In addition to the phosphorylation and activation of S6K, mTOR leads to increases in levels of the serine/threonine protein phosphatase PHLPP (pleckstrin homology domain leucine-rich repeat protein phosphatase) [47], which aids in switching off S6K activity by dephosphorylating the hydrophobic motif [48]. Thus, functional mTOR not only promotes the activation of S6K but also ensures tight regulation of its activity via dephosphorylation.
3.2. Subcellular Localization, Tissue Distribution and Protein Turnover
The presence of a nuclear localization sequence at its C-terminus suggests S6K2 but not S6K1 is nuclear, where its activation can be regulated by a pool of nuclear mTOR [49]. Several phosphorylation events have been linked to the regulation of S6K localization. For example, S6K2 has been shown to shuttle between the nucleus and cytoplasm, which is believed to be regulated by growth factor-induced phosphorylation by protein kinase C (PKC) in its C-terminus close to its nuclear localization signal, thus inhibiting its nuclear translocation. This is believed to maintain a pool of active S6K2 in the cytoplasm [50]. Similarly, the phosphorylation of S6K1 by casein kinase 2 prevents its nuclear translocation [51], which was shown to occur upon growth factor stimulation despite the lack of a nuclear localization signal [51,52]. While these phosphorylation events do not directly correlate with the activity of S6Ks, they are believed to recruit them to specific cellular compartments or serve to bring them into the proximity of their targets, thus determining their functions. S6 kinases have also been shown to undergo tyrosine phosphorylation following their membrane recruitment via a platelet-derived growth factor receptor (PDGFR)-Src pathway [53]. However, the precise function of this phosphorylation event remains unclear.
While S6K1 and S6K2 mRNAs were ubiquitously expressed in all tissues tested, S6K2 mRNA and protein levels did not correlate with each other, prompting the authors to suggest the existence of post-transcriptional fine-tuning of tissue-specificity for S6K2 [54]. Recently, microRNA-mediated regulation of S6K2 has come into light in non-small cell lung cancers, where it was shown to be targeted by miR-193a-3p [55].
S6Ks are also regulated via protein degradation and stabilization. Both S6K1 and S6K2 have been reported to be ubiquitinated and degraded via the proteasome. While the Roc1 ubiquitin ligase was shown to ubiquitinate S6K1, the identity of the ubiquitin ligase mediating the proteasomal degradation of S6K2 remains unknown [56,57]. Furthermore, S6Ks have been shown to be acetylated by p300 in vitro and the inhibition of histone deacetylase in cells increased their levels, suggesting that S6K acetylation promotes their stabilization [58].
Thus, while the core activation mechanisms are conserved between S6K1 and S6K2, there exist important differences in their sensitivities to upstream signaling pathways, localization and modes of protein turnover.
4. Cellular Functions of S6Ks
Initial studies on the physiological role of S6K came from Drosophila, which possesses a single S6K (dS6K) gene [59]. The disruption of this gene decreases the probability of survival to adulthood with a marked decrease in body size, which was associated with a decrease in cell size rather than total cell numbers. This suggests a role for dS6K in regulating cell growth in individuals that reach adulthood [59].
Similar to Drosophila, S6K1−/− mice exhibit defects such as small size, hypoinsulinemia and glucose intolerance associated with decreased pancreatic beta cell size [60]. It has been observed that there is an upregulation of S6K2 expression in tissues from S6K1 knockout mice, which compensates for the decrease in rpS6 phosphorylation in these mice [61]. Conversely, rpS6 phosphorylation is abrogated in S6K2−/− mice, suggesting that physiologically S6K2 is the principal kinase for rpS6. While S6K1/S6K2 double knockout mice exhibit perinatal lethality, S6K2−/− mice survive to adulthood with no apparent phenotype [61].
What then is the physiological role of the highly conserved rpS6 phosphorylation? The mitogen-inducible phosphorylation of rpS6 occurs at five C-terminal serine residues [62] and is mediated by several distinct kinases [63]. In vivo knock-in mouse models that harbor a non-phosphorylatable mutant of rpS6 showed small size, hypoinsulinemia, decreased beta cell size and muscle weakness—phenotypes similar to those of S6K1 knockout mice [64,65]—which is counterintuitive since S6K2 seems to be the primary kinase mediating rpS6 phosphorylation [64]. Several explanations have been put forth to resolve this discrepancy. For example, given their distinct localization patterns, a pool of rpS6 that is accessible only by S6K1 may be required for mediating cell growth. Similarly, S6K1 but not S6K2 may be activated during a particular developmental stage when rpS6 phosphorylation is required [21]. Further studies in mice deficient for rpS6 phosphorylation revealed that this highly conserved phosphorylation event was dispensable for the translation of 5′ TOP mRNAs, an event long considered to depend on it [64].
Although originally identified as the kinase mediating rpS6 phosphorylation, several other cellular substrates of S6K, specifically S6K1, have since been reported that play important roles in gene transcription, protein translation, insulin resistance and cell survival. For example, S6K1 has been reported to phosphorylate and regulate components of the translation apparatus, such as eIF4B [66] and eEF2K [67], and regulators of protein synthesis, such as programmed cell death 4 (PDCD4), which inhibits the translation machinery [68]. It is believed to regulate cell survival via the phosphorylation and regulation of murine double minute 2 (MDM2) [69], a negative regulator of p53, and BAD [70], a cell death-promoting protein.
While glucose intolerance has been observed in S6K1−/− mice, it has also been shown to promote the phosphorylation and feedback inhibition of insulin receptor substrate 1 (IRS1) [71,72,73,74]. Insulin- and amino acid-mediated activation of mTOR/S6K via the PI3K pathway leads to a negative feedback loop resulting in the phosphorylation and downregulation of IRS1 by S6K1 and eventually insulin resistance and type 2 diabetes. In addition to diabetes, mTOR/S6K1-mediated feedback inhibition of IRS1 and by extension the oncogenic PI3K/Akt pathway poses drawbacks for cancer therapy and limits the cytotoxic effects of rapamycin-based therapeutic approaches [75]. In addition to its role in the inhibition of IRS1, S6K1 has also been implicated in the phosphorylation and negative regulation of rictor, a component of the mTORC2 complex, which mediates Akt activation, although this conclusion remains controversial [76,77,78,79].
While several studies have documented the cellular functions and substrates of S6K1, little is known about those of S6K2. Given the lack of a phenotype in S6K2−/− mice, the physiological role of S6K2 is poorly understood. Some reports suggest that it could play roles in cell proliferation and gene regulation via interaction with heterogeneous nuclear ribonucleoproteins (hnRNPs) [80], histone H3 [81] and ying-yang-1 (YY1) [82].
5. S6 Kinases and Breast Cancer
5.1. Gene Amplification of S6Ks in Breast Cancer
The first clue regarding the involvement of S6K1 in breast cancer came from the identification that S6K1 (encoded by RPS6KB1) is localized on the chromosomal region 17q23, which is amplified in 20% of primary breast cancers [83]. cDNA microarray analyses showed that S6K1 is amplified and overexpressed in MCF-7 breast cancer cells. Tissue microarray analysis of 668 primary breast tumors showed amplification of S6K1 in 8.8% of primary tumors [83]. Co-amplification of S6K1 and HER-2 located in chromosome 17q12 was associated with poor patient survival [83].
S6K2 encoded by the gene RPS6KB2 was shown to be located on chromosome 11q13, which harbors several key mediators of breast cancer [84]. Perez-Tenorio et al., demonstrated that both RPS6KB1 and RPS6KB2 are often amplified in breast cancer tissues [84]. RPS6KB1 amplification (≥4 copies) has been reported in 10.7% of breast cancers, and gene gains (≥3 copies) have been reported in 21.4% of breast cancers [84]. Furthermore, this has been associated with loco-regional recurrence [85]. While amplification of RPS6KB2 is only associated with 4.3% of breast cancers, a large number of samples (21.3%) exhibit gains, suggesting that RPS6KB2 gain rather than amplification is a major event in breast cancer [21]. A co-amplification of RPS6KB2 and 4EBP1 has been reported, suggesting a synergy between these mTOR targets in breast cancer development and progression [86].
5.2. Expression and Localization of S6Ks in Breast Cancer
Immunohistochemical analysis demonstrated that both S6K1 and S6K2 are overexpressed in breast cancer, with S6K1 being primarily cytosolic and S6K2 predominantly nuclear in localization [87,88]. Furthermore, nuclear S6K2 correlated with staining of proliferation markers such as Ki-67 and proliferating cell nuclear antigen (PCNA), suggesting a role for nuclear S6K2 in breast cancer cell proliferation [87]. Additionally, nuclear accumulation of S6K2 was increased in cells at the periphery of the tumor, suggesting a unique role in breast cancer pathogenesis. However, Bostner et al., reported that high nuclear S6K1 was indicative of reduced benefits from tamoxifen treatment [89]. A recent study suggests that the subcellular distribution of S6K1 depends on the cell density and cell motility [90]. For example, at low cell density S6K1 was predominantly nuclear but it relocalized to the cytoplasm in confluent monolayers. During cell migration, S6K1 translocated to the nucleus and interacted with the transcription factor TBR2 (T-box brain protein 2). This study implicates nucleocytoplasmic shuttling of S6K1 to play an important role in the migration and invasion of breast cancer.
5.3. Function of S6Ks in Breast Cancer
5.3.1. Involvement of S6Ks in Estrogen Receptor (ER)-Positive Breast Cancer
Estrogen receptor-α (ERα)-positive breast cancers account for over half of all breast cancers and hence constitute the major subtype [91]. The canonical or genomic ER signaling is characterized by the binding of estrogen and subsequent activation of ERα, which then translocates to the nucleus and regulates its target genes by either promoting or repressing their transcription [92]. Activation of ERα is associated with its phosphorylation by several different kinases including S6K1 [93,94,95]. Further studies showed that S6K1 and ERα constitute a positive feed-forward loop, where the phosphorylation of ERα by S6K1 promotes its activity, which in turn promotes transcription of RPS6KB1 to mediate breast cancer cell proliferation [96,97]. The insulin-like growth factor (IGF) pathway plays a critical role in breast cancer. It was shown that knockdown/inhibition of S6K1 prevented IGF (insulin-like growth factor)-induced phosphorylation of ERα at Ser167 and transcription of ERα-regulated genes [98]. It has been reported that S6K1 mediates the phosphorylation of histone deacetylase 1 (HDAC1) by mitogens, recruitment of HDAC1 to the ERα promoter and increases in ERα transcription in breast cancer cells [99]. While the role and regulation of S6K1 in breast cancer have been addressed, little is known about the causes and consequences of S6K2 overexpression.
The availability of anti-estrogen therapies suggests a favorable prognosis for patients with ER-positive breast cancers. However, a substantial number of patients fail to respond to therapy [100], and the development of resistance further complicates treatment [91]. Studies in breast cancer tissues reported that RPS6KB2 gains/amplifications correlate with ER-positivity [84]. Also, distant recurrence-free survival was substantially reduced in patients with ER-positive tumors associated with RPS6KB2 gains/amplifications [84]. Furthermore, 11q13 amplifications have been intimately linked to ER-positive breast cancers [86,101,102,103,104] and constitute a high-risk subgroup of ER-positive cancers [101], suggesting that this region may play an important role in the response and resistance of breast cancer cells to death induced by anti-estrogen therapy. S6K2 but not S6K1 is frequently co-expressed with 4E-BP1, and high mRNA levels of S6K2 and/or 4E-BP1 mRNA were associated with poor prognosis and endocrine resistance in randomized Stockholm tamoxifen trials of four different cohorts of breast cancer patients [105]. Kim et al., proposed that phospho-S6K1 status is associated with poor prognosis and endocrine resistance of hormone receptor-positive breast cancers but not hormone receptor-negative breast cancers [106]. This is consistent with two other independent clinical trials demonstrating that S6K1 was associated with reduced response to tamoxifen [89]. While high levels of S6K1 correlated with markers of increased proliferation, HER2 status and cytoplasmic Akt, high levels of S6K2 correlated with low proliferation, ER status and nuclear Akt [89]. However, Ma et al., reported that although the levels of p-mTOR, p-4E-BP1 and p-S6K1 were significantly higher in breast tumor tissues compared to normal tissues, the status of phosphorylated mTOR, 4E-BP1 and S6K1 could not serve as prognostic factors for breast cancer [107]. The disease state (early versus late), node positivity, tumor size and treatment regime may influence the outcome of the biomarker studies.
Whole genome expression profiling of S6K1, S6K2 and 4E-BP1 in breast tumors also suggested distinct roles of S6K1 and S6K2 in breast cancer [26]. Knockdown of S6K2 but not S6K1 caused upregulation of genes associated with metabolism and regulation of cell cycle progression and checkpoints in ER-positive ZR751 cells. Knockdown of S6K1 caused upregulation of S6K2 and vice versa. Moreover, S6K2 knockdown caused an increase in raptor, whereas silencing of both S6K1 and S6K2 caused increases in mTOR and rictor, suggesting a compensatory cross-talk between the mTORC1 and mTORC2 complexes [26].
5.3.2. Involvement of S6Ks in Triple-Negative Breast Cancer
S6K1 has also been associated with triple-negative breast cancer (TNBC). Estrogen-related receptor-α (ERRα), a member of the orphan nuclear receptor family, is closely related to ERα and plays an important role in cellular metabolism [108]. While ERRα has been associated with endocrine resistance, ERRα level could also predict tamoxifen sensitivity in TNBC [109]. TNBC cells express high levels of ERRα, and it was shown to negatively regulate S6K1 expression by directly binding to its promoter [110]. Knockdown/inhibition of ERRα enhanced S6K1 level and sensitized cells to rapamycin or S6K1 inhibitor to inhibit cell proliferation, migration, invasion and metastasis, and decreased the expression of the pro-survival proteins survivin and myeloid cell leukemia 1 (Mcl-1). PF-4708671, a pharmacological inhibitor of S6K1, inhibited cell migration in a highly metastatic variant of MDA-MB-231 cells [111]. In addition, miRNA-15/16 was shown to inhibit cell proliferation, induce apoptosis and inhibit epithelial to mesenchymal transition (EMT) in TNBC MDA-MB-231 cells by targeting RPS6KB1 through binding to its 3′-UTR [112]. The miRNAs miR-96, miR-557 and miR-3182 were also shown to downregulate S6K1 by targeting the 3′-UTR of S6K1 mRNA [113].
Although the involvement of S6K2 in breast cancer metastasis has not been studied yet, S6K2 was shown to be a direct target of miR-193a-3p, which suppresses lung metastasis; downregulation of S6K2 by miR-193a-3p was shown to be a potential mechanism by which miR-193a-3p inhibited migration, invasion and EMT in non-small cell lung cancer [55]. On the other hand, upregulation of S6K2 was implicated in mediating the effects of miR-1273g-3p on cell proliferation, migration and invasion of colorectal cancer cells [114]. Based on analysis of the TCGA dataset, S6K2 but not S6K1 was overexpressed in both ER-positive and TNBC breast tumor tissues [115]. Moreover, overexpression of S6K2 in TNBC cells attenuated cell death by apoptosis [115].
5.3.3. Regulation of Apoptosis by S6Ks
Both S6K1 and S6K2 have been implicated in regulating cell death by apoptosis. While S6K1 inhibitor PF-4708671 alone did not influence the levels of anti-apoptotic proteins, it decreased Mcl-1 and survivin levels when MCF-7 cells were deprived of glucose [116] or treated in combination with tamoxifen [117]. PF-4708671 in combination with Bcl-2 inhibitor ABT263 also decreased survivin levels but increased Mcl-1 levels in BT474 cells [118]. In this study, the effect of S6K1 knockdown on the levels of anti-apoptotic proteins was not examined. Although PF-4708671 is specific towards S6K1 at low concentrations, it could inhibit MSK1, AMPK, RSK as well as S6K2 at high concentrations [119]. Moreover, PF-4708671 could phosphorylate S6K1 at Thr229 and Thr389, resulting in its activation [119].
We have shown that knockdown of S6K2 alone but not S6K1 sensitized cells to apoptotic stimuli by altering the ratio of pro- and anti-apoptotic proteins [27,120]. We made an important observation that S6K2 cooperates with Akt in mediating breast cancer cell survival [27]. While knockdown of S6K1 caused activation of Akt and inhibited cell death by apoptosis, knockdown of S6K2 decreased Akt activation and increased cell death by TNF and TRAIL by enhancing the cleavage of the pro-apoptotic Bcl-2 family protein Bid [27]. S6K2 could also promote breast cancer cell survival via an Akt-independent but JNK (c-Jun N-terminal kinase)-dependent pathway [120]. Knockdown of S6K2 decreased both Bcl-xl and Mcl-1 in T47D cells. While S6K2 appears to regulate Bcl-xl via the tumor suppressor PDCD4 [121], it positively regulated Mcl-1 via a JNK-dependent but GSK3-independent pathway [120].
5.3.4. The Involvement of Long and Short S6K Isoforms in Breast Cancer
There are controversies regarding which isoform of S6K1 is involved in breast cancer. It has been reported that the short kinase-inactive splice variants of S6K1 contribute to breast cancer, whereas the long p85/p70 S6K1 form causes tumor suppression [122]. The short isoforms were overexpressed in breast cancer cells and tissues and exhibited their oncogenic properties partly by causing activation of mTORC1 and increases in 4E-BP1 phosphorylation, cap-dependent translation and Mcl-1 levels. A recent study showed that the long p85 S6K1 but not p70 S6K1 or p56 S6K2 is secreted from cancer cells and can enter surrounding cells to promote breast cancer cell growth, migration and invasion [123]. Moreover, injection of recombinant p85 S6K1 in mice enhanced tumorigenesis and lung metastasis in an MDA-MB-231 tumor xenograft [123].
6. Conclusions
Given the role of mTOR in promoting protein translation, cell growth and proliferation, it is an attractive target for cancer therapy [124]. However, S6K1 and S6K2, the downstream targets of mTORC1, carry out distinct functions. While S6K1 has a more prominent role in regulating cell proliferation, invasion and metastasis, S6K2 appears to have a greater impact on cell death regulation. Rapamycin and its analogs that inhibit mTORC1 are being evaluated for their clinical potential in several cancers, including breast cancer [125,126]. Furthermore, due to the association between RPS6KB1 amplifications and cancer, there has been considerable interest in the development of inhibitors of S6K1, such as LYS6K2 [127] and PF-4708671 [119]. However, Ly2584702 tosylate, an inhibitor of S6K1 that is not an analog of rapamycin, was ineffective as a single agent [128] and was not well tolerated when administered in combination with everolimus and erlotinib [129] in phase I clinical trials. The therapeutic efficacy of mTORC1 and S6K1 inhibitors is also thwarted due to the existence of a negative feedback loop between PI3K/Akt and mTOR/S6K1 signaling [75]. Persistent inhibition of S6K1 leads to the activation of PI3K/Akt, allowing survival of cancer cells [130,131,132,133]. Inhibition of mTORC1 can also cause a compensatory activation of the MAPK pathway [134]. Phosphorylation of Grb10, a substrate for mTORC1, leads to feedback inhibition of both the PI3K/Akt and MAPK pathways [135,136]. These observations dampen the enthusiasm for mTORC1- and S6K1-inhibitor-based approaches in cancer therapy. Currently, combinatorial approaches using dual-specificity inhibitors of PI3K/Akt and mTOR are being evaluated [137]. Furthermore, the observation that S6K1−/− mice are characterized by small size and exhibit hypoinsulinemia suggests that targeting S6K1 for cancer therapy may be associated with significant side effects [60]. The normal development and the lack of an apparent phenotype in S6K2−/− mice suggests that it is a potential target in the treatment of endometrial [138], gastric [139] and breast cancers [84,87,88], which have been shown to have RPS6KB2 amplification or elevated S6K2 expression. Moreover, knockdown of S6K2 resulted in inhibition rather than activation of Akt in breast cancer cells [27]. Thus, there should be an emphasis on the development of S6K2-specific inhibitors. Even though the kinase domains of S6K1 and S6K2 are similar, the unique amino acids at the ATP binding pocket of S6K1 (Tyr) and S6K2 (Cys) may allow development of S6K2-specific inhibitors [26]. Further mechanistic studies dissecting the function of S6K1 and S6K2 at various stages and types of breast cancer are needed in order to properly exploit these two homologs for breast cancer therapy.
Acknowledgments
We apologize if we inadvertently left out any major contribution in this field.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| 4E-BP1 | Eukaryotic translation initiation factor 4E (eIF4E)-binding protein 1 |
| EMT | Epithelial–mesenchymal transition |
| ERα | Estrogen receptor-α |
| ERRα | Estrogen-related receptor-α |
| HDAC | Histone deacetylase |
| hnRNP | Heterogeneous nuclear ribonucleoprotein |
| IGF | Insulin-like growth factor |
| IRS1 | Insulin receptor substrate 1 |
| MAPK | Mitogen-activated protein kinase |
| Mcl-1 | Myeloid cell leukemia 1 |
| MDM2 | Murine double minute 2 |
| MEK | Mitogen-activated protein kinase kinase |
| mTOR | Mechanistic target of rapamycin |
| NLS | Nuclear localization signal |
| PDCD4 | Programmed cell death 4 |
| PDK1 | 3-Phosphoinositide-dependent protein kinase 1 |
| PDZ | Post synaptic density protein; PSD95, Drosophila disc large tumor suppressor; Dlg1, and Zonula occludens-1 protein; zo-1 |
| PHLPP | Pleckstrin homology domain leucine-rich repeat protein phosphatase |
| PI3K | Phosphatidylinositol 3-kinase |
| PKC | Protein kinase C |
| S6K | 40S ribosomal S6 kinase |
| TNBC | Triple-negative breast cancer |
| TOS | Tor signaling motif |
| TSC | Tuberous sclerosis complex |
| YY1 | Ying-yang-1 |
References
- Dey, N.; De, P.; Leyland-Jones, B. PI3K-AKT-mTOR inhibitors in breast cancers: From tumor cell signaling to clinical trials. Pharmacol. Ther. 2017, 175, 91–106. [Google Scholar] [CrossRef]
- Guerrero-Zotano, A.; Mayer, I.A.; Arteaga, C.L. PI3K/AKT/mTOR: Role in breast cancer progression, drug resistance, and treatment. Cancer Metastasis Rev. 2016, 35, 515–524. [Google Scholar] [CrossRef]
- Castaneda, C.A.; Cortes-Funes, H.; Gomez, H.L.; Ciruelos, E.M. The phosphatidyl inositol 3-kinase/AKT signaling pathway in breast cancer. Cancer Metastasis Rev. 2010, 29, 751–759. [Google Scholar] [CrossRef] [PubMed]
- Dillon, R.L.; Muller, W.J. Distinct biological roles for the akt family in mammary tumor progression. Cancer Res. 2010, 70, 4260–4264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wickenden, J.A.; Watson, C.J. Key signalling nodes in mammary gland development and cancer. Signalling downstream of PI3 kinase in mammary epithelium: A play in 3 Akts. Breast Cancer Res. 2010, 12, 202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Cosimo, S.; Baselga, J. Management of breast cancer with targeted agents: Importance of heterogeneity. [corrected]. Nat. Rev. Clin. Oncol. 2010, 7, 139–147. [Google Scholar] [CrossRef]
- Sanchez-Munoz, A.; Perez-Ruiz, E.; Jimenez, B.; Ribelles, N.; Marquez, A.; Garcia-Rios, I.; Alba Conejo, E. Targeted therapy of metastatic breast cancer. Clin. Transl. Oncol. 2009, 11, 643–650. [Google Scholar] [CrossRef]
- Ghayad, S.E.; Cohen, P.A. Inhibitors of the PI3K/Akt/mTOR pathway: New hope for breast cancer patients. Recent Pat. Anticancer Drug Discov. 2010, 5, 29–57. [Google Scholar] [CrossRef]
- Basu, A. Molecular targets of breast cancer: AKTing in concert. Breast Cancer 2008, 2, 11–16. [Google Scholar]
- Araki, K.; Miyoshi, Y. Mechanism of resistance to endocrine therapy in breast cancer: The important role of PI3K/Akt/mTOR in estrogen receptor-positive, HER2-negative breast cancer. Breast Cancer 2018, 25, 392–401. [Google Scholar] [CrossRef]
- Sharma, V.R.; Gupta, G.K.; Sharma, A.K.; Batra, N.; Sharma, D.K.; Joshi, A.; Sharma, A.K. PI3K/Akt/mTOR Intracellular Pathway and Breast Cancer: Factors, Mechanism and Regulation. Curr. Pharm. Des. 2017, 23, 1633–1638. [Google Scholar] [CrossRef]
- Mita, M.M.; Mita, A.; Rowinsky, E.K. Mammalian target of rapamycin: A new molecular target for breast cancer. Clin. Breast Cancer 2003, 4, 126–137. [Google Scholar] [CrossRef] [PubMed]
- Jeno, P.; Ballou, L.M.; Novak-Hofer, I.; Thomas, G. Identification and characterization of a mitogen-activated S6 kinase. Proc. Natl. Acad. Sci. USA 1988, 85, 406–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nemenoff, R.A.; Price, D.J.; Mendelsohn, M.J.; Carter, E.A.; Avruch, J. An S6 kinase activated during liver regeneration is related to the insulin-stimulated S6 kinase in H4 hepatoma cells. J. Biol. Chem. 1988, 263, 19455–19460. [Google Scholar] [PubMed]
- Koh, H.; Jee, K.; Lee, B.; Kim, J.; Kim, D.; Yun, Y.H.; Kim, J.W.; Choi, H.S.; Chung, J. Cloning and characterization of a nuclear S6 kinase, S6 kinase-related kinase (SRK); a novel nuclear target of Akt. Oncogene 1999, 18, 5115–5119. [Google Scholar] [CrossRef] [Green Version]
- Gout, I.; Minami, T.; Hara, K.; Tsujishita, Y.; Filonenko, V.; Waterfield, M.D.; Yonezawa, K. Molecular cloning and characterization of a novel p70 S6 kinase, p70 S6 kinase beta containing a proline-rich region. J. Biol. Chem. 1998, 273, 30061–30064. [Google Scholar] [CrossRef] [Green Version]
- Lee-Fruman, K.K.; Kuo, C.J.; Lippincott, J.; Terada, N.; Blenis, J. Characterization of S6K2, a novel kinase homologous to S6K1. Oncogene 1999, 18, 5108–5114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saitoh, M.; ten Dijke, P.; Miyazono, K.; Ichijo, H. Cloning and characterization of p70(S6K beta) defines a novel family of p70 S6 kinases. Biochem. Biophys. Res. Commun. 1998, 253, 470–476. [Google Scholar] [CrossRef]
- Shima, H.; Pende, M.; Chen, Y.; Fumagalli, S.; Thomas, G.; Kozma, S.C. Disruption of the p70(s6k)/p85(s6k) gene reveals a small mouse phenotype and a new functional S6 kinase. EMBO J. 1998, 17, 6649–6659. [Google Scholar] [CrossRef] [Green Version]
- Grove, J.R.; Banerjee, P.; Balasubramanyam, A.; Coffer, P.J.; Price, D.J.; Avruch, J.; Woodgett, J.R. Cloning and expression of two human p70 S6 kinase polypeptides differing only at their amino termini. Mol. Cell Biol. 1991, 11, 5541–5550. [Google Scholar] [CrossRef] [Green Version]
- Fenton, T.R.; Gout, I.T. Functions and regulation of the 70kDa ribosomal S6 kinases. Int. J. Biochem. Cell Biol. 2011, 43, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Ismail, H.M. Downstream the mTOR: S6 Kinases between Divergence and Redundancy. J. Biochem. Pharmacol. Res. 2013, 1, 94–105. [Google Scholar]
- Pardo, O.E.; Seckl, M.J. S6K2: The Neglected S6 Kinase Family Member. Front. Oncol. 2013, 3, 191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sever, N.I.; Cengiz Sahin, S. S6K2 promises an important therapeutic potential for cancer. Future Oncol. 2019, 15, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Tavares, M.R.; Pavan, I.C.; Amaral, C.L.; Meneguello, L.; Luchessi, A.D.; Simabuco, F.M. The S6K protein family in health and disease. Life Sci. 2015, 131, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, E.; Magic, I.; Bostner, J.; Dyrager, C.; Lysholm, F.; Hallbeck, A.L.; Stal, O.; Lundstrom, P. Revealing Different Roles of the mTOR-Targets S6K1 and S6K2 in Breast Cancer by Expression Profiling and Structural Analysis. PLoS ONE 2015, 10, e0145013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sridharan, S.; Basu, A. S6 kinase 2 promotes breast cancer cell survival via Akt. Cancer Res. 2011, 71, 2590–2599. [Google Scholar] [CrossRef] [Green Version]
- Burnett, P.E.; Blackshaw, S.; Lai, M.M.; Qureshi, I.A.; Burnett, A.F.; Sabatini, D.M.; Snyder, S.H. Neurabin is a synaptic protein linking p70 S6 kinase and the neuronal cytoskeleton. Proc. Natl. Acad. Sci. USA 1998, 95, 8351–8356. [Google Scholar] [CrossRef] [Green Version]
- Sabatini, D.M. mTOR and cancer: Insights into a complex relationship. Nat. Rev. Cancer 2006, 6, 729–734. [Google Scholar] [CrossRef]
- Borkowska, J.; Schwartz, R.A.; Kotulska, K.; Jozwiak, S. Tuberous sclerosis complex: Tumors and tumorigenesis. Int. J. Dermatol. 2011, 50, 13–20. [Google Scholar] [CrossRef]
- Garami, A.; Zwartkruis, F.J.; Nobukuni, T.; Joaquin, M.; Roccio, M.; Stocker, H.; Kozma, S.C.; Hafen, E.; Bos, J.L.; Thomas, G. Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Mol. Cell 2003, 11, 1457–1466. [Google Scholar] [CrossRef] [Green Version]
- Castro, A.F.; Rebhun, J.F.; Clark, G.J.; Quilliam, L.A. Rheb binds tuberous sclerosis complex 2 (TSC2) and promotes S6 kinase activation in a rapamycin- and farnesylation-dependent manner. J. Biol. Chem. 2003, 278, 32493–32496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, J.; Kuo, C.J.; Crabtree, G.R.; Blenis, J. Rapamycin-FKBP specifically blocks growth-dependent activation of and signaling by the 70 kd S6 protein kinases. Cell 1992, 69, 1227–1236. [Google Scholar] [CrossRef]
- Kuo, C.J.; Chung, J.; Fiorentino, D.F.; Flanagan, W.M.; Blenis, J.; Crabtree, G.R. Rapamycin selectively inhibits interleukin-2 activation of p70 S6 kinase. Nature 1992, 358, 70–73. [Google Scholar] [CrossRef] [PubMed]
- Price, D.J.; Grove, J.R.; Calvo, V.; Avruch, J.; Bierer, B.E. Rapamycin-induced inhibition of the 70-kilodalton S6 protein kinase. Science 1992, 257, 973–977. [Google Scholar] [CrossRef] [PubMed]
- Weng, Q.P.; Kozlowski, M.; Belham, C.; Zhang, A.; Comb, M.J.; Avruch, J. Regulation of the p70 S6 kinase by phosphorylation in vivo. Analysis using site-specific anti-phosphopeptide antibodies. J. Biol. Chem. 1998, 273, 16621–16629. [Google Scholar] [CrossRef] [Green Version]
- Schalm, S.S.; Blenis, J. Identification of a conserved motif required for mTOR signaling. Curr. Biol. 2002, 12, 632–639. [Google Scholar] [CrossRef] [Green Version]
- Yonezawa, K.; Tokunaga, C.; Oshiro, N.; Yoshino, K. Raptor, a binding partner of target of rapamycin. Biochem. Biophys. Res. Commun. 2004, 313, 437–441. [Google Scholar] [CrossRef]
- Mora, A.; Komander, D.; van Aalten, D.M.; Alessi, D.R. PDK1, the master regulator of AGC kinase signal transduction. Semin. Cell Dev. Biol. 2004, 15, 161–170. [Google Scholar] [CrossRef] [Green Version]
- Mukhopadhyay, N.K.; Price, D.J.; Kyriakis, J.M.; Pelech, S.; Sanghera, J.; Avruch, J. An array of insulin-activated, proline-directed serine/threonine protein kinases phosphorylate the p70 S6 kinase. J. Biol. Chem. 1992, 267, 3325–3335. [Google Scholar]
- Pardo, O.E.; Arcaro, A.; Salerno, G.; Tetley, T.D.; Valovka, T.; Gout, I.; Seckl, M.J. Novel cross talk between MEK and S6K2 in FGF-2 induced proliferation of SCLC cells. Oncogene 2001, 20, 7658–7667. [Google Scholar] [CrossRef]
- Martin, K.A.; Schalm, S.S.; Romanelli, A.; Keon, K.L.; Blenis, J. Ribosomal S6 kinase 2 inhibition by a potent C-terminal repressor domain is relieved by mitogen-activated protein-extracellular signal-regulated kinase kinase-regulated phosphorylation. J. Biol. Chem. 2001, 276, 7892–7898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bettoun, D.J.; Buck, D.W., 2nd; Lu, J.; Khalifa, B.; Chin, W.W.; Nagpal, S. A vitamin D receptor-Ser/Thr phosphatase-p70 S6 kinase complex and modulation of its enzymatic activities by the ligand. J. Biol. Chem. 2002, 277, 24847–24850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petritsch, C.; Beug, H.; Balmain, A.; Oft, M. TGF-beta inhibits p70 S6 kinase via protein phosphatase 2A to induce G(1) arrest. Genes Dev. 2000, 14, 3093–3101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peterson, R.T.; Desai, B.N.; Hardwick, J.S.; Schreiber, S.L. Protein phosphatase 2A interacts with the 70-kDa S6 kinase and is activated by inhibition of FKBP12-rapamycinassociated protein. Proc. Natl. Acad. Sci. USA 1999, 96, 4438–4442. [Google Scholar] [CrossRef] [Green Version]
- Parrott, L.A.; Templeton, D.J. Osmotic stress inhibits p70/85 S6 kinase through activation of a protein phosphatase. J. Biol. Chem. 1999, 274, 24731–24736. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Stevens, P.D.; Gao, T. mTOR-dependent regulation of PHLPP expression controls the rapamycin sensitivity in cancer cells. J. Biol. Chem. 2011, 286, 6510–6520. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Stevens, P.D.; Li, X.; Schmidt, M.D.; Gao, T. PHLPP-mediated dephosphorylation of S6K1 inhibits protein translation and cell growth. Mol. Cell Biol. 2011, 31, 4917–4927. [Google Scholar] [CrossRef] [Green Version]
- Park, I.H.; Bachmann, R.; Shirazi, H.; Chen, J. Regulation of ribosomal S6 kinase 2 by mammalian target of rapamycin. J. Biol. Chem. 2002, 277, 31423–31429. [Google Scholar] [CrossRef] [Green Version]
- Valovka, T.; Verdier, F.; Cramer, R.; Zhyvoloup, A.; Fenton, T.; Rebholz, H.; Wang, M.L.; Gzhegotsky, M.; Lutsyk, A.; Matsuka, G.; et al. Protein kinase C phosphorylates ribosomal protein S6 kinase betaII and regulates its subcellular localization. Mol. Cell. Biol. 2003, 23, 852–863. [Google Scholar] [CrossRef] [Green Version]
- Panasyuk, G.; Nemazanyy, I.; Zhyvoloup, A.; Bretner, M.; Litchfield, D.W.; Filonenko, V.; Gout, I.T. Nuclear export of S6K1 II is regulated by protein kinase CK2 phosphorylation at Ser-17. J. Biol. Chem. 2006, 281, 31188–31201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosner, M.; Hengstschlager, M. Nucleocytoplasmic localization of p70 S6K1, but not of its isoforms p85 and p31, is regulated by TSC2/mTOR. Oncogene 2011, 30, 4509–4522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rebholz, H.; Panasyuk, G.; Fenton, T.; Nemazanyy, I.; Valovka, T.; Flajolet, M.; Ronnstrand, L.; Stephens, L.; West, A.; Gout, I.T. Receptor association and tyrosine phosphorylation of S6 kinases. FEBS J. 2006, 273, 2023–2036. [Google Scholar] [CrossRef]
- Nardella, C.; Lunardi, A.; Fedele, G.; Clohessy, J.G.; Alimonti, A.; Kozma, S.C.; Thomas, G.; Loda, M.; Pandolfi, P.P. Differential expression of S6K2 dictates tissue-specific requirement for S6K1 in mediating aberrant mTORC1 signaling and tumorigenesis. Cancer Res. 2011, 71, 3669–3675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, T.; Li, J.; Yan, M.; Liu, L.; Lin, H.; Zhao, F.; Sun, L.; Zhang, Y.; Cui, Y.; Zhang, F.; et al. MicroRNA-193a-3p and -5p suppress the metastasis of human non-small-cell lung cancer by downregulating the ERBB4/PIK3R3/mTOR/S6K2 signaling pathway. Oncogene 2015, 34, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Gwalter, J.; Wang, M.L.; Gout, I. The ubiquitination of ribosomal S6 kinases is independent from the mitogen-induced phosphorylation/activation of the kinase. Int. J. Biochem. Cell Biol. 2009, 41, 828–833. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.L.; Panasyuk, G.; Gwalter, J.; Nemazanyy, I.; Fenton, T.; Filonenko, V.; Gout, I. Regulation of ribosomal protein S6 kinases by ubiquitination. Biochem. Biophys. Res. Commun. 2008, 369, 382–387. [Google Scholar] [CrossRef]
- Fenton, T.R.; Gwalter, J.; Ericsson, J.; Gout, I.T. Histone acetyltransferases interact with and acetylate p70 ribosomal S6 kinases in vitro and in vivo. Int. J. Biochem. Cell Biol. 2010, 42, 359–366. [Google Scholar] [CrossRef]
- Montagne, J.; Stewart, M.J.; Stocker, H.; Hafen, E.; Kozma, S.C.; Thomas, G. Drosophila S6 kinase: A regulator of cell size. Science 1999, 285, 2126–2129. [Google Scholar] [CrossRef]
- Pende, M.; Kozma, S.C.; Jaquet, M.; Oorschot, V.; Burcelin, R.; Le Marchand-Brustel, Y.; Klumperman, J.; Thorens, B.; Thomas, G. Hypoinsulinaemia, glucose intolerance and diminished beta-cell size in S6K1-deficient mice. Nature 2000, 408, 994–997. [Google Scholar] [CrossRef]
- Pende, M.; Um, S.H.; Mieulet, V.; Sticker, M.; Goss, V.L.; Mestan, J.; Mueller, M.; Fumagalli, S.; Kozma, S.C.; Thomas, G. S6K1(−/−)/S6K2(−/−) mice exhibit perinatal lethality and rapamycin-sensitive 5’-terminal oligopyrimidine mRNA translation and reveal a mitogen-activated protein kinase-dependent S6 kinase pathway. Mol. Cell Biol. 2004, 24, 3112–3124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krieg, J.; Hofsteenge, J.; Thomas, G. Identification of the 40 S ribosomal protein S6 phosphorylation sites induced by cycloheximide. J. Biol. Chem. 1988, 263, 11473–11477. [Google Scholar] [PubMed]
- Anjum, R.; Blenis, J. The RSK family of kinases: Emerging roles in cellular signalling. Nat. Rev. Mol. Cell Biol. 2008, 9, 747–758. [Google Scholar] [CrossRef] [PubMed]
- Ruvinsky, I.; Sharon, N.; Lerer, T.; Cohen, H.; Stolovich-Rain, M.; Nir, T.; Dor, Y.; Zisman, P.; Meyuhas, O. Ribosomal protein S6 phosphorylation is a determinant of cell size and glucose homeostasis. Genes Dev 2005, 19, 2199–2211. [Google Scholar] [CrossRef] [Green Version]
- Ruvinsky, I.; Katz, M.; Dreazen, A.; Gielchinsky, Y.; Saada, A.; Freedman, N.; Mishani, E.; Zimmerman, G.; Kasir, J.; Meyuhas, O. Mice deficient in ribosomal protein S6 phosphorylation suffer from muscle weakness that reflects a growth defect and energy deficit. PLoS ONE 2009, 4, e5618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raught, B.; Peiretti, F.; Gingras, A.C.; Livingstone, M.; Shahbazian, D.; Mayeur, G.L.; Polakiewicz, R.D.; Sonenberg, N.; Hershey, J.W. Phosphorylation of eucaryotic translation initiation factor 4B Ser422 is modulated by S6 kinases. EMBO J. 2004, 23, 1761–1769. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Li, W.; Williams, M.; Terada, N.; Alessi, D.R.; Proud, C.G. Regulation of elongation factor 2 kinase by p90(RSK1) and p70 S6 kinase. EMBO J 2001, 20, 4370–4379. [Google Scholar] [CrossRef]
- Dorrello, N.V.; Peschiaroli, A.; Guardavaccaro, D.; Colburn, N.H.; Sherman, N.E.; Pagano, M. S6K1- and betaTRCP-mediated degradation of PDCD4 promotes protein translation and cell growth. Science 2006, 314, 467–471. [Google Scholar] [CrossRef]
- Lai, K.P.; Leong, W.F.; Chau, J.F.; Jia, D.; Zeng, L.; Liu, H.; He, L.; Hao, A.; Zhang, H.; Meek, D.; et al. S6K1 is a multifaceted regulator of Mdm2 that connects nutrient status and DNA damage response. EMBO J. 2010, 29, 2994–3006. [Google Scholar] [CrossRef] [Green Version]
- Harada, H.; Andersen, J.S.; Mann, M.; Terada, N.; Korsmeyer, S.J. p70S6 kinase signals cell survival as well as growth, inactivating the pro-apoptotic molecule BAD. Proc. Natl. Acad. Sci. USA 2001, 98, 9666–9670. [Google Scholar] [CrossRef] [Green Version]
- Harrington, L.S.; Findlay, G.M.; Gray, A.; Tolkacheva, T.; Wigfield, S.; Rebholz, H.; Barnett, J.; Leslie, N.R.; Cheng, S.; Shepherd, P.R.; et al. The TSC1-2 tumor suppressor controls insulin-PI3K signaling via regulation of IRS proteins. J. Cell Biol. 2004, 166, 213–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Gao, Z.; Yin, J.; Quon, M.J.; Ye, J. S6K directly phosphorylates IRS-1 on Ser-270 to promote insulin resistance in response to TNF-(alpha) signaling through IKK2. J. Biol. Chem. 2008, 283, 35375–35382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, O.J.; Wang, Z.; Hunter, T. Inappropriate activation of the TSC/Rheb/mTOR/S6K cassette induces IRS1/2 depletion, insulin resistance, and cell survival deficiencies. Curr. Biol. 2004, 14, 1650–1656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tremblay, F.; Brule, S.; Hee Um, S.; Li, Y.; Masuda, K.; Roden, M.; Sun, X.J.; Krebs, M.; Polakiewicz, R.D.; Thomas, G.; et al. Identification of IRS-1 Ser-1101 as a target of S6K1 in nutrient- and obesity-induced insulin resistance. Proc. Natl. Acad. Sci. USA 2007, 104, 14056–14061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernandez-Aya, L.F.; Gonzalez-Angulo, A.M. Targeting the phosphatidylinositol 3-kinase signaling pathway in breast cancer. Oncologist 2011, 16, 404–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akcakanat, A.; Singh, G.; Hung, M.C.; Meric-Bernstam, F. Rapamycin regulates the phosphorylation of rictor. Biochem. Biophys. Res. Commun. 2007, 362, 330–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dibble, C.C.; Asara, J.M.; Manning, B.D. Characterization of Rictor phosphorylation sites reveals direct regulation of mTOR complex 2 by S6K1. Mol. Cell. Biol. 2009, 29, 5657–5670. [Google Scholar] [CrossRef] [Green Version]
- Treins, C.; Warne, P.H.; Magnuson, M.A.; Pende, M.; Downward, J. Rictor is a novel target of p70 S6 kinase-1. Oncogene 2010, 29, 1003–1016. [Google Scholar] [CrossRef] [Green Version]
- Julien, L.A.; Carriere, A.; Moreau, J.; Roux, P.P. mTORC1-activated S6K1 phosphorylates Rictor on threonine 1135 and regulates mTORC2 signaling. Mol. Cell. Biol. 2010, 30, 908–921. [Google Scholar] [CrossRef] [Green Version]
- Goh, E.T.; Pardo, O.E.; Michael, N.; Niewiarowski, A.; Totty, N.; Volkova, D.; Tsaneva, I.R.; Seckl, M.J.; Gout, I. Involvement of heterogeneous ribonucleoprotein F in the regulation of cell proliferation via the mammalian target of rapamycin/S6 kinase 2 pathway. J. Biol. Chem. 2010, 285, 17065–17076. [Google Scholar] [CrossRef] [Green Version]
- Ismail, H.M.S.; Hurd, P.J.; Khalil, M.I.M.; Kouzarides, T.; Bannister, A.; Gout, I. S6 kinase 2 is bound to chromatin-nuclear matrix cellular fractions and is able to phosphorylate histone H3 at threonine 45 in vitro and in vivo. J. Cell. Biochem. 2014, 115, 1048–1062. [Google Scholar] [CrossRef] [PubMed]
- Ismail, H.M.; Myronova, O.; Tsuchiya, Y.; Niewiarowski, A.; Tsaneva, I.; Gout, I. Identification of the general transcription factor Yin Yang 1 as a novel and specific binding partner for S6 kinase 2. Cell Signal. 2013, 25, 1054–1063. [Google Scholar] [CrossRef] [PubMed]
- Barlund, M.; Forozan, F.; Kononen, J.; Bubendorf, L.; Chen, Y.; Bittner, M.L.; Torhorst, J.; Haas, P.; Bucher, C.; Sauter, G.; et al. Detecting activation of ribosomal protein S6 kinase by complementary DNA and tissue microarray analysis. J. Natl. Cancer Inst. 2000, 92, 1252–1259. [Google Scholar] [CrossRef] [PubMed]
- Perez-Tenorio, G.; Karlsson, E.; Waltersson, M.A.; Olsson, B.; Holmlund, B.; Nordenskjold, B.; Fornander, T.; Skoog, L.; Stal, O. Clinical potential of the mTOR targets S6K1 and S6K2 in breast cancer. Breast Cancer Res. Treat 2011, 128, 713–723. [Google Scholar] [CrossRef] [Green Version]
- Van der Hage, J.A.; van den Broek, L.J.; Legrand, C.; Clahsen, P.C.; Bosch, C.J.; Robanus-Maandag, E.C.; van de Velde, C.J.; van de Vijver, M.J. Overexpression of P70 S6 kinase protein is associated with increased risk of locoregional recurrence in node-negative premenopausal early breast cancer patients. Br. J. Cancer 2004, 90, 1543–1550. [Google Scholar] [CrossRef]
- Karlsson, E.; Waltersson, M.A.; Bostner, J.; Perez-Tenorio, G.; Olsson, B.; Hallbeck, A.L.; Stal, O. High-resolution genomic analysis of the 11q13 amplicon in breast cancers identifies synergy with 8p12 amplification, involving the mTOR targets S6K2 and 4EBP1. Genes Chromosom. Cancer 2011, 50, 775–787. [Google Scholar] [CrossRef] [Green Version]
- Lyzogubov, V.; Khozhaenko, Y.; Usenko, V.; Antonjuk, S.; Ovcharenko, G.; Tikhonkova, I.; Filonenko, V. Immunohistochemical analysis of Ki-67, PCNA and S6K1/2 expression in human breast cancer. Exp. Oncol. 2005, 27, 141–144. [Google Scholar]
- Filonenko, V.V.; Tytarenko, R.; Azatjan, S.K.; Savinska, L.O.; Gaydar, Y.A.; Gout, I.T.; Usenko, V.S.; Lyzogubov, V.V. Immunohistochemical analysis of S6K1 and S6K2 localization in human breast tumors. Exp. Oncol. 2004, 26, 294–299. [Google Scholar]
- Bostner, J.; Karlsson, E.; Eding, C.B.; Perez-Tenorio, G.; Franzen, H.; Konstantinell, A.; Fornander, T.; Nordenskjold, B.; Stal, O. S6 kinase signaling: Tamoxifen response and prognostic indication in two breast cancer cohorts. Endocr.-Related Cancer 2015, 22, 331–343. [Google Scholar] [CrossRef] [Green Version]
- Kosach, V.; Shkarina, K.; Kravchenko, A.; Tereshchenko, Y.; Kovalchuk, E.; Skoroda, L.; Krotevych, M.; Khoruzhenko, A. Nucleocytoplasmic distribution of S6K1 depends on the density and motility of MCF-7 cells in vitro. F1000Res 2018, 7, 1332. [Google Scholar] [CrossRef]
- Ali, S.; Coombes, R.C. Estrogen receptor alpha in human breast cancer: Occurrence and significance. J. Mammary Gland Biol. Neoplasia 2000, 5, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Manavathi, B.; Dey, O.; Gajulapalli, V.N.; Bhatia, R.S.; Bugide, S.; Kumar, R. Derailed estrogen signaling and breast cancer: An authentic couple. Endocr. Rev. 2013, 34, 1–32. [Google Scholar] [CrossRef] [Green Version]
- Yamnik, R.L.; Digilova, A.; Davis, D.C.; Brodt, Z.N.; Murphy, C.J.; Holz, M.K. S6 kinase 1 regulates estrogen receptor alpha in control of breast cancer cell proliferation. J. Biol. Chem. 2009, 284, 6361–6369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weitsman, G.E.; Li, L.; Skliris, G.P.; Davie, J.R.; Ung, K.; Niu, Y.; Curtis-Snell, L.; Tomes, L.; Watson, P.H.; Murphy, L.C. Estrogen receptor-alpha phosphorylated at Ser118 is present at the promoters of estrogen-regulated genes and is not altered due to HER-2 overexpression. Cancer Res. 2006, 66, 10162–10170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.P.; Shu, S.K.; Esposito, N.N.; Coppola, D.; Koomen, J.M.; Cheng, J.Q. IKKepsilon phosphorylation of estrogen receptor alpha Ser-167 and contribution to tamoxifen resistance in breast cancer. J. Biol. Chem. 2010, 285, 3676–3684. [Google Scholar] [CrossRef] [Green Version]
- Maruani, D.M.; Spiegel, T.N.; Harris, E.N.; Shachter, A.S.; Unger, H.A.; Herrero-Gonzalez, S.; Holz, M.K. Estrogenic regulation of S6K1 expression creates a positive regulatory loop in control of breast cancer cell proliferation. Oncogene 2012, 31, 5073–5080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holz, M.K. The role of S6K1 in ER-positive breast cancer. Cell Cycle 2012, 11, 3159–3165. [Google Scholar] [CrossRef] [Green Version]
- Becker, M.A.; Ibrahim, Y.H.; Cui, X.; Lee, A.V.; Yee, D. The IGF pathway regulates ERalpha through a S6K1-dependent mechanism in breast cancer cells. Mol. Endocrinol. 2011, 25, 516–528. [Google Scholar] [CrossRef] [Green Version]
- Citro, S.; Miccolo, C.; Meloni, L.; Chiocca, S. PI3K/mTOR mediate mitogen-dependent HDAC1 phosphorylation in breast cancer: A novel regulation of estrogen receptor expression. J. Mol. Cell Biol. 2015, 7, 132–142. [Google Scholar] [CrossRef] [Green Version]
- Early Breast Cancer Trialists’ Collaborative Group. Systemic treatment of early breast cancer by hormonal, cytotoxic, or immune therapy. 133 randomised trials involving 31,000 recurrences and 24,000 deaths among 75,000 women. Lancet 1992, 339, 1–15. [Google Scholar]
- Curtis, C.; Shah, S.P.; Chin, S.F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.; Yuan, Y.; et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 2012, 486, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Lambrechts, D.; Truong, T.; Justenhoven, C.; Humphreys, M.K.; Wang, J.; Hopper, J.L.; Dite, G.S.; Apicella, C.; Southey, M.C.; Schmidt, M.K.; et al. 11q13 is a susceptibility locus for hormone receptor positive breast cancer. Hum. Mutat. 2012, 33, 1123–1132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loo, L.W.; Grove, D.I.; Williams, E.M.; Neal, C.L.; Cousens, L.A.; Schubert, E.L.; Holcomb, I.N.; Massa, H.F.; Glogovac, J.; Li, C.I.; et al. Array comparative genomic hybridization analysis of genomic alterations in breast cancer subtypes. Cancer Res. 2004, 64, 8541–8549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fantl, V.; Richards, M.A.; Smith, R.; Lammie, G.A.; Johnstone, G.; Allen, D.; Gregory, W.; Peters, G.; Dickson, C.; Barnes, D.M. Gene amplification on chromosome band 11q13 and oestrogen receptor status in breast cancer. Eur. J. Cancer 1990, 26, 423–429. [Google Scholar] [CrossRef]
- Karlsson, E.; Perez-Tenorio, G.; Amin, R.; Bostner, J.; Skoog, L.; Fornander, T.; Sgroi, D.C.; Nordenskjold, B.; Hallbeck, A.L.; Stal, O. The mTOR effectors 4EBP1 and S6K2 are frequently coexpressed, and associated with a poor prognosis and endocrine resistance in breast cancer: A retrospective study including patients from the randomised Stockholm tamoxifen trials. Breast Cancer Res. 2013, 15, R96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, E.K.; Kim, H.A.; Koh, J.S.; Kim, M.S.; Kim, K.I.; Lee, J.I.; Moon, N.M.; Ko, E.; Noh, W.C. Phosphorylated S6K1 is a possible marker for endocrine therapy resistance in hormone receptor-positive breast cancer. Breast Cancer Res. Treat. 2011, 126, 93–99. [Google Scholar] [CrossRef]
- Ma, B.L.; Shan, M.H.; Sun, G.; Ren, G.H.; Dong, C.; Yao, X.; Zhou, M. Immunohistochemical analysis of phosphorylated mammalian target of rapamycin and its downstream signaling components in invasive breast cancer. Mol. Med. Rep. 2015, 12, 5246–5254. [Google Scholar] [CrossRef] [Green Version]
- Ranhotra, H.S. The estrogen-related receptors in metabolism and cancer: Newer insights. J. Recept. Signal. Transduct. Res. 2018, 38, 95–100. [Google Scholar] [CrossRef]
- Manna, S.; Bostner, J.; Sun, Y.; Miller, L.D.; Alayev, A.; Schwartz, N.S.; Lager, E.; Fornander, T.; Nordenskjold, B.; Yu, J.J.; et al. ERRalpha Is a Marker of Tamoxifen Response and Survival in Triple-Negative Breast Cancer. Clin. Cancer Res. 2016, 22, 1421–1431. [Google Scholar] [CrossRef] [Green Version]
- Berman, A.Y.; Manna, S.; Schwartz, N.S.; Katz, Y.E.; Sun, Y.; Behrmann, C.A.; Yu, J.J.; Plas, D.R.; Alayev, A.; Holz, M.K. ERRalpha regulates the growth of triple-negative breast cancer cells via S6K1-dependent mechanism. Signal. Transduct. Target Ther. 2017, 2. [Google Scholar] [CrossRef] [Green Version]
- Khotskaya, Y.B.; Goverdhan, A.; Shen, J.; Ponz-Sarvise, M.; Chang, S.S.; Hsu, M.C.; Wei, Y.; Xia, W.; Yu, D.; Hung, M.C. S6K1 promotes invasiveness of breast cancer cells in a model of metastasis of triple-negative breast cancer. Am. J. Transl. Res. 2014, 6, 361–376. [Google Scholar] [PubMed]
- Janaki Ramaiah, M.; Lavanya, A.; Honarpisheh, M.; Zarea, M.; Bhadra, U.; Bhadra, M.P. MiR-15/16 complex targets p70S6 kinase 1 and controls cell proliferation in MDA-MB-231 breast cancer cells. Gene 2014, 552, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Razaviyan, J.; Hadavi, R.; Tavakoli, R.; Kamani, F.; Paknejad, M.; Mohammadi-Yeganeh, S. Expression of miRNAs Targeting mTOR and S6K1 Genes of mTOR Signaling Pathway Including miR-96, miR-557, and miR-3182 in Triple-Negative Breast Cancer. Appl. Biochem. Biotechnol. 2018, 186, 1074–1089. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Qian, X.; Zhu, M.; Li, A.; Fang, M.; Zhu, Y.; Zhang, J. miR1273g3p promotes proliferation, migration and invasion of LoVo cells via cannabinoid receptor 1 through activation of ERBB4/PIK3R3/mTOR/S6K2 signaling pathway. Mol. Med. Rep. 2018, 17, 4619–4626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sridharan, S.; Xuan, Z.; Basu, A. Ribosomal S6 Kinase 2 Promotes Survival of Triple- Negative Breast Cancer Cells to Apoptotic Stimuli. Cancer Studies Therap. 2019, 4, 1–6. [Google Scholar]
- Choi, H.N.; Jin, H.O.; Kim, J.H.; Hong, S.E.; Kim, H.A.; Kim, E.K.; Lee, J.K.; Park, I.C.; Noh, W.C. Inhibition of S6K1 enhances glucose deprivation-induced cell death via downregulation of anti-apoptotic proteins in MCF-7 breast cancer cells. Biochem. Biophys. Res. Commun. 2013, 432, 123–128. [Google Scholar] [CrossRef]
- Hong, S.E.; Kim, E.K.; Jin, H.O.; Kim, H.A.; Lee, J.K.; Koh, J.S.; Seol, H.; Kim, J.I.; Park, I.C.; Noh, W.C. S6K1 inhibition enhances tamoxifen-induced cell death in MCF-7 cells through translational inhibition of Mcl-1 and survivin. Cell Biol. Toxicol. 2013, 29, 273–282. [Google Scholar] [CrossRef]
- Park, J.A.; Jin, H.O.; Lee, H.N.; Kim, J.H.; Park, I.C.; Noh, W.C.; Chang, Y.H.; Hong, Y.J.; Kim, K.C.; Lee, J.K. S6K1 inhibition enhances the apoptotic cell death of breast cancer cells in response to Bcl-2/Bcl-xL inhibition by the downregulation of survivin. Oncol. Lett. 2015, 10, 829–834. [Google Scholar] [CrossRef] [Green Version]
- Pearce, L.R.; Alton, G.R.; Richter, D.T.; Kath, J.C.; Lingardo, L.; Chapman, J.; Hwang, C.; Alessi, D.R. Characterization of PF-4708671, a novel and highly specific inhibitor of p70 ribosomal S6 kinase (S6K1). Biochem. J. 2010, 431, 245–255. [Google Scholar] [CrossRef] [Green Version]
- Basu, A.; Sridharan, S. Regulation of anti-apoptotic Bcl-2 family protein Mcl-1 by S6 kinase 2. PLoS ONE 2017, 12, e0173854. [Google Scholar] [CrossRef]
- Liwak, U.; Thakor, N.; Jordan, L.E.; Roy, R.; Lewis, S.M.; Pardo, O.E.; Seckl, M.; Holcik, M. Tumor suppressor PDCD4 represses internal ribosome entry site-mediated translation of antiapoptotic proteins and is regulated by S6 kinase 2. Mol. Cell. Biol. 2012, 32, 1818–1829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben-Hur, V.; Denichenko, P.; Siegfried, Z.; Maimon, A.; Krainer, A.; Davidson, B.; Karni, R. S6K1 alternative splicing modulates its oncogenic activity and regulates mTORC1. Cell Rep. 2013, 3, 103–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Guo, J.; Qin, X.; Wang, B.; Zhang, L.; Wang, Y.; Gan, W.; Pandolfi, P.P.; Chen, W.; Wei, W. The p85 isoform of the kinase S6K1 functions as a secreted oncoprotein to facilitate cell migration and tumor growth. Sci. Signal. 2018, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fasolo, A.; Sessa, C. Targeting mTOR pathways in human malignancies. Curr. Pharm. Des. 2012, 18, 2766–2777. [Google Scholar] [CrossRef]
- Liu, J.; Li, H.Q.; Zhou, F.X.; Yu, J.W.; Sun, L.; Han, Z.H. Targeting the mTOR pathway in breast cancer. Tumour Biol. 2017, 39. [Google Scholar] [CrossRef] [Green Version]
- Piccart, M.; Hortobagyi, G.N.; Campone, M.; Pritchard, K.I.; Lebrun, F.; Ito, Y.; Noguchi, S.; Perez, A.; Rugo, H.S.; Deleu, I.; et al. Everolimus plus exemestane for hormone-receptor-positive, human epidermal growth factor receptor-2-negative advanced breast cancer: Overall survival results from BOLERO-2dagger. Ann. Oncol. 2014, 25, 2357–2362. [Google Scholar] [CrossRef]
- Li, S.; Brown, M.S.; Goldstein, J.L. Bifurcation of insulin signaling pathway in rat liver: mTORC1 required for stimulation of lipogenesis, but not inhibition of gluconeogenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 3441–3446. [Google Scholar] [CrossRef] [Green Version]
- Tolcher, A.; Goldman, J.; Patnaik, A.; Papadopoulos, K.P.; Westwood, P.; Kelly, C.S.; Bumgardner, W.; Sams, L.; Geeganage, S.; Wang, T.; et al. A phase I trial of LY2584702 tosylate, a p70 S6 kinase inhibitor, in patients with advanced solid tumours. Eur. J. Cancer 2014, 50, 867–875. [Google Scholar] [CrossRef]
- Hollebecque, A.; Houede, N.; Cohen, E.E.; Massard, C.; Italiano, A.; Westwood, P.; Bumgardner, W.; Miller, J.; Brail, L.H.; Benhadji, K.A.; et al. A phase Ib trial of LY2584702 tosylate, a p70 S6 inhibitor, in combination with erlotinib or everolimus in patients with solid tumours. Eur. J. Cancer 2014, 50, 876–884. [Google Scholar] [CrossRef]
- O’Reilly, K.E.; Rojo, F.; She, Q.B.; Solit, D.; Mills, G.B.; Smith, D.; Lane, H.; Hofmann, F.; Hicklin, D.J.; Ludwig, D.L.; et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006, 66, 1500–1508. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.Y.; Rosenberg, L.M.; Wang, X.; Zhou, Z.; Yue, P.; Fu, H.; Khuri, F.R. Activation of Akt and eIF4E survival pathways by rapamycin-mediated mammalian target of rapamycin inhibition. Cancer Res. 2005, 65, 7052–7058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, X.; Harkavy, B.; Shen, N.; Grohar, P.; Helman, L.J. Rapamycin induces feedback activation of Akt signaling through an IGF-1R-dependent mechanism. Oncogene 2007, 26, 1932–1940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Yue, P.; Kim, Y.A.; Fu, H.; Khuri, F.R.; Sun, S.Y. Enhancing mammalian target of rapamycin (mTOR)-targeted cancer therapy by preventing mTOR/raptor inhibition-initiated, mTOR/rictor-independent Akt activation. Cancer Res. 2008, 68, 7409–7418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carracedo, A.; Ma, L.; Teruya-Feldstein, J.; Rojo, F.; Salmena, L.; Alimonti, A.; Egia, A.; Sasaki, A.T.; Thomas, G.; Kozma, S.C.; et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J. Clin. Investig. 2008, 118, 3065–3074. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.P.; Kang, S.A.; Rameseder, J.; Zhang, Y.; Ottina, K.A.; Lim, D.; Peterson, T.R.; Choi, Y.; Gray, N.S.; Yaffe, M.B.; et al. The mTOR-regulated phosphoproteome reveals a mechanism of mTORC1-mediated inhibition of growth factor signaling. Science 2011, 332, 1317–1322. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Yoon, S.O.; Poulogiannis, G.; Yang, Q.; Ma, X.M.; Villen, J.; Kubica, N.; Hoffman, G.R.; Cantley, L.C.; Gygi, S.P.; et al. Phosphoproteomic analysis identifies Grb10 as an mTORC1 substrate that negatively regulates insulin signaling. Science 2011, 332, 1322–1326. [Google Scholar] [CrossRef] [Green Version]
- Sheppard, K.; Kinross, K.M.; Solomon, B.; Pearson, R.B.; Phillips, W.A. Targeting PI3 kinase/AKT/mTOR signaling in cancer. Crit. Rev. Oncog. 2012, 17, 69–95. [Google Scholar] [CrossRef]
- Lyzogubov, V.V.; Lytvyn, D.I.; Dudchenko, T.M.; Lubchenko, N.V.; Pogrybniy, P.V.; Nespryadko, S.V.; Vinnitska, A.B.; Usenko, V.S.; Gout, I.T.; Filonenko, V.V. Immunohistochemical analysis of S6K1 and S6K2 expression in endometrial adenocarcinomas. Exp. Oncol. 2004, 26, 287–293. [Google Scholar]
- Yoshida, S.; Matsumoto, K.; Arao, T.; Taniguchi, H.; Goto, I.; Hanafusa, T.; Nishio, K.; Yamada, Y. Gene amplification of ribosomal protein S6 kinase-1 and -2 in gastric cancer. Anticancer Res. 2013, 33, 469–475. [Google Scholar]
Figure 1.
Modular structure of S6Ks. S6K1 and S6K2 share significant homology in their kinase domains. However, the shorter isoforms of both S6K1 and S6K2, which are the predominant forms, exhibit substantial divergence in the extreme N- and C-terminal regions.
Figure 1.
Modular structure of S6Ks. S6K1 and S6K2 share significant homology in their kinase domains. However, the shorter isoforms of both S6K1 and S6K2, which are the predominant forms, exhibit substantial divergence in the extreme N- and C-terminal regions.

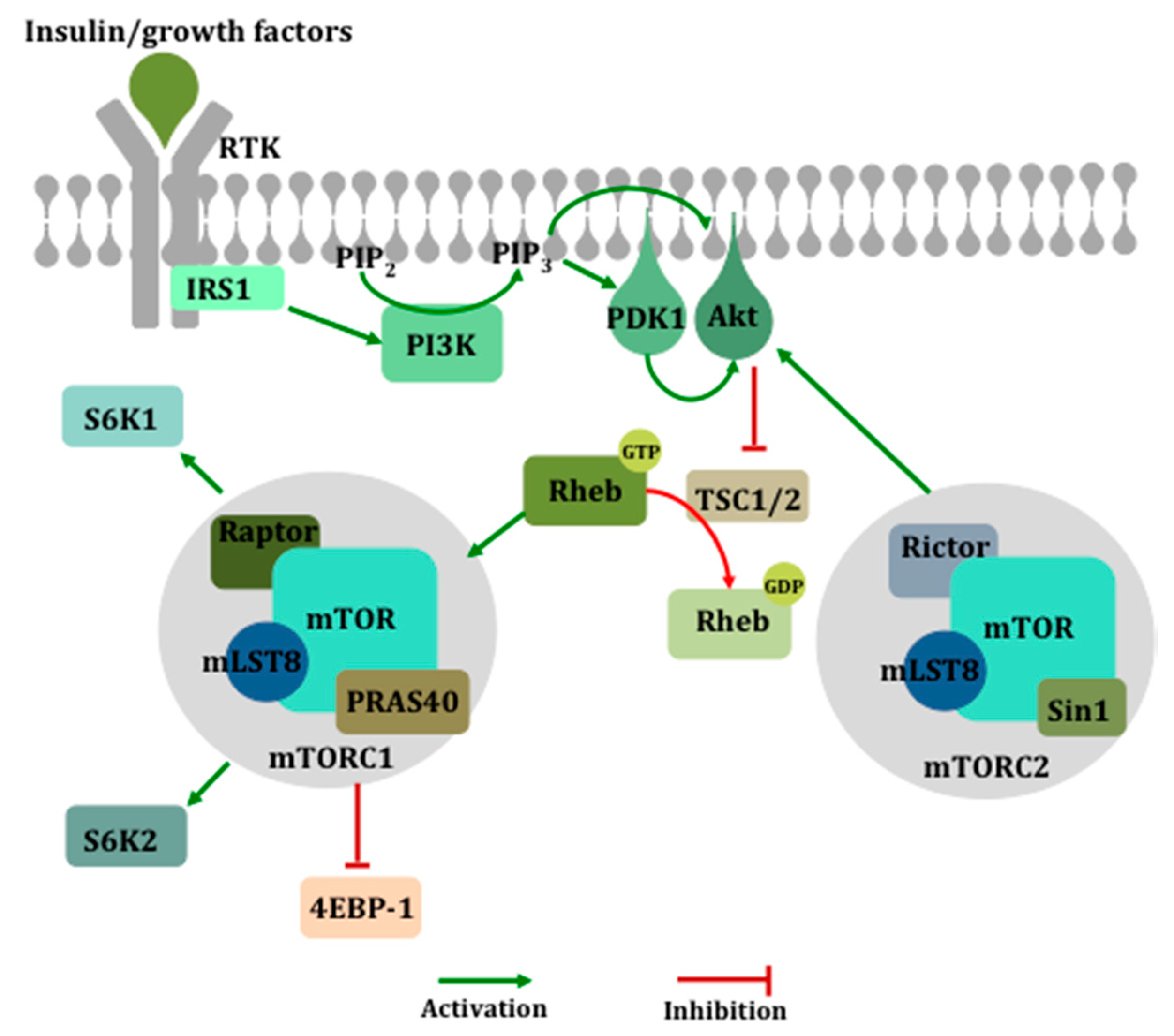
Figure 2.
The activation of the mechanistic target of rapamycin (mTOR). Growth factor-mediated activation of the phosphatidylinositol 3-kinase (PI3K) pathway leads to the membrane recruitment and activation of phosphoinositide-dependent kinase 1 (PDK1) and Akt, which then phosphorylates and inactivates the tuberous sclerosis complex (TSC1/2), a negative regulator of ras homolog enriched in brain (Rheb), ultimately resulting in the activation of mTOR within complex 1. mTORC1 mediates its downstream effects primarily via the inhibition of eukaryotic translation initiation factor 4E (eIF4E)-binding protein 1 (4E-BP1) and the activation of S6 kinase (S6K).
Figure 2.
The activation of the mechanistic target of rapamycin (mTOR). Growth factor-mediated activation of the phosphatidylinositol 3-kinase (PI3K) pathway leads to the membrane recruitment and activation of phosphoinositide-dependent kinase 1 (PDK1) and Akt, which then phosphorylates and inactivates the tuberous sclerosis complex (TSC1/2), a negative regulator of ras homolog enriched in brain (Rheb), ultimately resulting in the activation of mTOR within complex 1. mTORC1 mediates its downstream effects primarily via the inhibition of eukaryotic translation initiation factor 4E (eIF4E)-binding protein 1 (4E-BP1) and the activation of S6 kinase (S6K).


{kind=link}
{kind=link}
Table 1.
Genes and isoforms of the 40S ribosomal S6 kinases (S6Ks).
| Gene | Chromosome | Isoforms | Size |
|---|---|---|---|
| RPS6KB1 | 17 | S6KαI | p85 |
| S6KαII * | p70 | ||
| RPS6KB2 | 11 | S6KβI | p56 |
| S6KβII * | p54 |
* Predominant isoform.
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Sridharan, S.; Basu, A. Distinct Roles of mTOR Targets S6K1 and S6K2 in Breast Cancer. Int. J. Mol. Sci. 2020, 21, 1199. https://doi.org/10.3390/ijms21041199
AMA Style
Sridharan S, Basu A. Distinct Roles of mTOR Targets S6K1 and S6K2 in Breast Cancer. International Journal of Molecular Sciences. 2020; 21(4):1199. https://doi.org/10.3390/ijms21041199
Chicago/Turabian StyleSridharan, Savitha, and Alakananda Basu. 2020. "Distinct Roles of mTOR Targets S6K1 and S6K2 in Breast Cancer" International Journal of Molecular Sciences 21, no. 4: 1199. https://doi.org/10.3390/ijms21041199
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.