A Tale of Two Families: Whole Genome and Segmental Duplications Underlie Glutamine Synthetase and Phosphoenolpyruvate Carboxylase Diversity in Narrow-Leafed Lupin (Lupinus angustifolius L.)

 , , , and
, , , and
Abstract
1. Introduction
2. Results and Discussion
2.1. Narrow-Leafed Lupin GS and PEPC are Encoded by Multigene Families
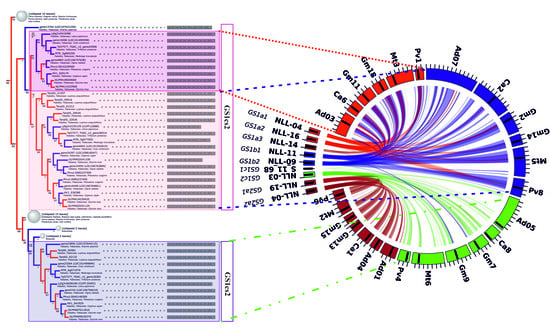
2.2. GS and PEPC Gene Variants are Localized in Distinct Narrow-leafed Lupin Genome Regions
2.3. GS and PEPC Gene Variants Present a Conserved Sequence Structure among All L. angustifolius Homologs and Other Legume Species
2.4. The Initial GS and PEPC Complement was Subsequently Duplicated in a Lineage-Specific Manner and Can be Traced to the Common Ancestor of Legumes
2.5. Compared to GS Genes, the History of Coding Sequences of PEPC Genes More Closely Recapitulates the History of Species
2.6. L. angustifolius Genome Regions Carrying GS and PEPC Genes Arise from Duplication/Triplication with Additional Complex Chromosome Rearrangements
2.7. The Major Events Promoting the Evolution of GS and PEPC Genes in Legumes were Whole-Genome Duplications
2.8. The Majority of Positively Selected GS and PEPC Genes are Duplicates
3. Material and Methods
3.1. Research Material
3.2. Identifying GS and PEPC in the L. angustifolius Genome
3.3. Estimating GS and PEPC Sequence Variant Numbers
3.4. Characterizing GS1, GS2, and PEPC Gene Variants, as well as Their Corresponding L. angustifolius Genome Regions
3.5. Positioning GS1, GS2, and PEPC in NLL Pseudochromosomes
3.6. Describing Local Genome Rearrangements Harboring GS and PEPC Loci
3.7. Phylogenetic Reconstruction of the Plant Species Tree
3.8. Determining GS1, GS2, and PEPC Gene Families Evolutionary Patterns
3.9. Selection Pressure Analysis
4. Conclusions
- GS and PEPC genes were shown to have had a complex history, with bacterial-type PEPCs emerging as those best suited for future phylogenetic inquiries into relationships between divergent legumes.
- Legume GS and PEPC genes evolved by both ancestral legume-wide and more recent lineage-specific WGDs. Descendants of these duplications have been retained in the majority of lineages and have sustained typical gene structures, implying differences in carbon/nitrogen metabolism due to regulatory rather than mechanistic changes.
- Legume PEPC and GS gene sequences were highly conserved by significant purifying selection. Tentative traces of positive selection can only be inferred in several branches and point to single residues, outside of the core set involved in ligand binding.
- Monocot family members of the GS gene family might be more ancient than dicot ones, stemming from the selective culling of duplicates predating the separation of both lineages.
- The general patterns of lineage-specific duplications suggest that sub-functionalization and/or regulatory rewiring played a large role in shaping the extant carbon and nitrogen primary metabolic pathways in some lineages (L. angustifolius, L. japonicus, and G. max).
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ACCase | cytosolic and plastid acetyl-coenzyme A carboxylases |
| AFLP | amplified fragment length polymorphism |
| BAC | bacterial artificial chromosome |
| BTPC | bacterial-type PEPC |
| CHI | chalcone isomerase |
| EST | expressed sequence tag |
| GS | glutamine synthetase |
| IFSs | isoflavone synthetases |
| ITAP | intron targeted amplified polymorphism |
| LTRs | long terminal repeats |
| MFLP | molecular fragment length polymorphism |
| NLL | narrow-leafed lupin linkage group |
| PEBPs | phosphatidylethanolamine binding proteins |
| PEPC | phosphoenolpyruvate carboxylase |
| PTPC | plant-type PEPC |
| RFLP | restriction fragment length polymorphism |
| RADs | restriction site associated DNA markers |
| SSR | single sequence repeat |
| TEs | transposable elements |
| WGD | whole genome duplication |
References
- Bertioli, D.J.; Moretzsohn, M.C.; Madsen, L.H.; Sandal, N.; Leal-Bertioli, S.C.; Guimaraes, P.M.; Hougaard, B.K.; Fredslund, J.; Schauser, L.; Nielsen, A.M.; et al. An analysis of synteny of Arachis with Lotus and Medicago sheds new light on the structure stability and evolution of legume genomes. BMC Genom. 2009, 10, 45. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, D.; de Queiroz, L.P.; Pennington, R.T.; de Lima, H.C.; Fonty, E.; Wojciechowski, M.F.; Lavin, M. Revisiting the phylogeny of papilionoid legumes: New insights from comprehensively sampled early-branching lineages. Am. J. Bot. 2012, 99, 1991–2013. [Google Scholar] [CrossRef]
- Cannon, S.B.; McKain, M.R.; Harkess, A.; Nelson, M.N.; Dash, S.; Deyholos, M.K.; Peng, Y.; Joyce, B.; Stewart, C.N., Jr.; Rolf, M.; et al. Multiple polyploidy events in the early radiation of nodulating and nonnodulating legumes. Mol. Biol. Evol. 2015, 32, 193–210. [Google Scholar] [CrossRef] [PubMed]
- Doyle, J.J.; Luckow, M.A. The rest of the iceberg. Legume diversity and evolution in a phylogenetic context. Plant Physiol. 2003, 131, 900–910. [Google Scholar] [CrossRef]
- Lewis, G.; Schrire, B.; Mackind, B.; Lock, M. Legumes of the World; Royal Botanic Gardens Kew: London, UK, 2005. [Google Scholar]
- Bertioli, D.J.; Cannon, S.B.; Froenicke, L.; Huang, G.; Farmer, A.D.; Cannon, E.K.; Liu, X.; Gao, D.; Clevenger, J.; Dash, S.; et al. The genome sequences of Arachis duranensis and Arachis ipaensis the diploid ancestors of cultivated peanut. Nat. Genet. 2016, 48, 438–446. [Google Scholar] [CrossRef]
- Varshney, R.K.; Chen, W.; Li, Y.; Bharti, A.K.; Saxena, R.K.; Schlueter, J.A.; Donoghue, M.T.A.; Azam, S.; Fan, G.; Whaley, A.M.; et al. Draft genome sequence of pigeonpea (Cajanus cajan) an orphan legume crop of resource-poor farmers. Nat. Biotechnol. 2012, 30, 83–89. [Google Scholar] [CrossRef]
- Varshney, R.K.; Song, C.; Saxena, R.K.; Azam, S.; Yu, S.; Sharpe, A.G.; Cannon, S.; Baek, J.; Rosen, B.D.; Tar’an, B.; et al. Draft genome sequence of chickpea (Cicer arietinum) provides a resource for trait improvement. Nat. Biotechnol. 2013, 31, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Schmutz, J.; Cannon, S.B.; Schlueter, J.; Ma, J.; Mitros, T.; Nelson, W.; Hyten, D.L.; Song, Q.; Thelen, J.J.; Cheng, J.; et al. Genome sequence of the palaeopolyploid soybean. Nature 2010, 463, 178–183. [Google Scholar] [CrossRef]
- Sato, S.; Nakamura, Y.; Kaneko, T.; Asamizu, E.; Kato, T.; Nakao, M.; Sasamoto, S.; Watanabe, A.; Ono, A.; Kawashima, K.; et al. Genome structure of the legume Lotus japonicus. DNA Res. 2008, 15, 227–239. [Google Scholar] [CrossRef]
- Hane, J.K.; Ming, Y.; Kamphuis, L.G.; Nelson, M.N.; Garg, G.; Atkins, C.A.; Bayer, P.E.; Bravo, A.; Bringans, S.; Cannon, S.; et al. A comprehensive draft genome sequence for lupin (Lupinus angustifolius) an emerging health food: insights into plant-microbe interactions and legume evolution. Plant Biotechnol. J. 2017, 15, 318–330. [Google Scholar] [CrossRef]
- Young, N.D.; Debelle, F.; Oldroyd, G.E.; Geurts, R.; Cannon, S.B.; Udvardi, M.K.; Benedito, V.A.; Mayer, K.F.; Gouzy, J.; Schoof, H.; et al. The Medicago genome provides insight into the evolution of rhizobial symbioses. Nature 2011, 480, 520–524. [Google Scholar] [CrossRef] [PubMed]
- Schmutz, J.; McClean, P.E.; Mamidi, S.; Wu, G.A.; Cannon, S.B.; Grimwood, J.; Jenkins, J.; Shu, S.; Song, Q.; Chavarro, C.; et al. A reference genome for common bean and genome-wide analysis of dual domestications. Nat. Genet. 2014, 46, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.J.; Kim, S.K.; Kim, M.Y.; Lestari, P.; Kim, K.H.; Ha, B.K.; Jun, T.H.; Hwang, W.J.; Lee, T.; Lee, J.; et al. Genome sequence of mungbean and insights into evolution within Vigna species. Nat. Commun. 2014, 5, 5443. [Google Scholar] [CrossRef]
- Abbo, S.; Miller, T.E.; Reader, S.M.; Dunford, R.P.; King, I.P. Detection of ribosomal DNA sites in lentil and chickpea by fluorescent in situ hybridization. Genome 1994, 37, 713–716. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Tao, Y.; Zheng, Z.; Shao, D.; Li, Z.; Sweetingham, M.W.; Buirchell, B.J.; Li, C. Rapid development of molecular markers by next-generation sequencing linked to a gene conferring phomopsis stem blight disease resistance for marker-assisted selection in lupin (Lupinus angustifolius L.) breeding. Theor. Appl. Genet. 2013, 126, 511–522. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.N.; Moolhuijzen, P.M.; Boersma, J.G.; Chudy, M.; Lesniewska, K.; Bellgard, M.; Oliver, R.P.; Swiecicki, W.; Wolko, B.; Cowling, W.A.; et al. Aligning a new reference genetic map of Lupinus angustifolius with the genome sequence of the model legume Lotus japonicus. DNA Res. 2010, 17, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Boersma, J.G.; You, M.; Buirchell, B.J.; Sweetingham, M.W. Development and implementation of a sequence-specific PCR marker linked to a gene conferring resistance to anthracnose disease in narrow-leafed lupin (Lupinus angustifolius L.). Mol. Breed. 2004, 14, 145–151. [Google Scholar] [CrossRef]
- Książkiewicz, M.; Wyrwa, K.; Szczepaniak, A.; Rychel, S.; Majcherkiewicz, K.; Przysiecka, L.; Karlowski, W.; Wolko, B.; Naganowska, B. Comparative genomics of Lupinus angustifolius gene-rich regions: BAC library exploration genetic mapping and cytogenetics. BMC Genom. 2013, 14, 79. [Google Scholar] [CrossRef]
- Nelson, M.N.; Phan, H.T.; Ellwood, S.R.; Moolhuijzen, P.M.; Hane, J.; Williams, A.; O’Lone, C.E.; Fosu-Nyarko, J.; Scobie, M.; Cakir, M.; et al. The first gene-based map of Lupinus angustifolius L.—location of domestication genes and conserved synteny with Medicago truncatula. Theor. Appl. Genet. 2006, 113, 225–238. [Google Scholar] [CrossRef]
- Kamphuis, L.G.; Hane, J.K.; Nelson, M.N.; Gao, L.; Atkins, C.A.; Singh, K.B. Transcriptome sequencing of different narrow-leafed lupin tissue types provides a comprehensive uni-gene assembly and extensive gene-based molecular markers. Plant Biotechnol. J. 2015, 13, 14–25. [Google Scholar] [CrossRef]
- Zhou, G.; Jian, J.; Wang, P.; Li, C.; Tao, Y.; Li, X.; Renshaw, D.; Clements, J.; Sweetingham, M.W.; Yang, H. Construction of an ultra-high density consensus genetic map and enhancement of the physical map from genome sequencing in Lupinus angustifolius. Theor. Appl. Genet. 2018, 131, 209–223. [Google Scholar] [CrossRef]
- Li, H.; Renshaw, D.; Yang, H.; Yan, G. Development of a co-dominant DNA marker tightly linked to gene tardus conferring reduced pod shattering in narrow-leafed lupin (Lupinus angustifolius L.). Euphytica 2010, 176, 49–58. [Google Scholar] [CrossRef]
- Boersma, J.G.; Buirchel, B.J.; Sivasithamparam, K.; Yang, H. Development of two sequence-specific PCR markers linked to the le gene that reduces pod shattering in narrow-leafed Lupin (Lupinus angustifolius L.). Genet. Mol. Biol. 2007, 30, 623–629. [Google Scholar] [CrossRef]
- Nelson, M.N.; Ksiazkiewicz, M.; Rychel, S.; Besharat, N.; Taylor, C.M.; Wyrwa, K.; Jost, R.; Erskine, W.; Cowling, W.A.; Berger, J.D.; et al. The loss of vernalization requirement in narrow-leafed lupin is associated with a deletion in the promoter and de-repressed expression of a Flowering Locus T (FT) homologue. New Phytol. 2017, 213, 220–232. [Google Scholar] [CrossRef] [PubMed]
- You, M.; Boersma, J.G.; Buirchell, B.J.; Sweetingham, M.W.; Siddique, K.H.; Yang, H. A PCR-based molecular marker applicable for marker-assisted selection for anthracnose disease resistance in lupin breeding. Cell. Mol. Biol. Lett. 2005, 10, 123–134. [Google Scholar] [PubMed]
- Książkiewicz, M.; Yang, H. Molecular Marker Resources Supporting the Australian Lupin Breeding Program. In Compendium of Plant Genomes, The Lupin Genome; Karam, S., Kamphuis, L., Nelson, M., Eds.; Springer Nature Switzerland AG: Cham, Switzerland, 2020. [Google Scholar]
- Kasprzak, A.; Safar, J.; Janda, J.; Dolezel, J.; Wolko, B.; Naganowska, B. The bacterial artificial chromosome (BAC) library of the narrow-leafed lupin (Lupinus angustifolius L.). Cell. Mol. Biol. Lett. 2006, 11, 396–407. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.L.; Hane, J.K.; Kamphuis, L.G.; Foley, R.; Shi, B.J.; Atkins, C.A.; Singh, K.B. Development of genomic resources for the narrow-leafed lupin (Lupinus angustifolius): construction of a bacterial artificial chromosome (BAC) library and BAC-end sequencing. BMC Genom. 2011, 12, 521. [Google Scholar] [CrossRef]
- Książkiewicz, M.; Rychel, S.; Nelson, M.N.; Wyrwa, K.; Naganowska, B.; Wolko, B. Expansion of the phosphatidylethanolamine binding protein family in legumes: a case study of Lupinus angustifolius L. FLOWERING LOCUS T homologs LanFTc1 and LanFTc2. BMC Genom. 2016, 17, 820. [Google Scholar] [CrossRef]
- Wyrwa, K.; Ksiazkiewicz, M.; Szczepaniak, A.; Susek, K.; Podkowinski, J.; Naganowska, B. Integration of Lupinus angustifolius L. (narrow-leafed lupin) genome maps and comparative mapping within legumes. Chromosome Res. 2016, 24, 355–378. [Google Scholar] [CrossRef]
- Susek, K.; Bielski, W.K.; Hasterok, R.; Naganowska, B.; Wolko, B. A First Glimpse of Wild Lupin Karyotype Variation As Revealed by Comparative Cytogenetic Mapping. Front. Plant Sci. 2016, 7, 1152. [Google Scholar] [CrossRef]
- Susek, K.; Naganowska, B. Cytomolecular Insight Into Lupinus Genomes. In Compendium of Plant Genomes, The Lupin Genome; Karam, S., Kamphuis, L., Nelson, M., Eds.; Springer Nature Switzerland AG: Cham, Switzerland, 2020. [Google Scholar]
- Książkiewicz, M.; Zielezinski, A.; Wyrwa, K.; Szczepaniak, A.; Rychel, S.; Karlowski, W.; Wolko, B.; Naganowska, B. Remnants of the Legume Ancestral Genome Preserved in Gene-Rich Regions: Insights from Physical Genetic and Comparative Mapping. Plant Mol. Biol. Rep. 2015, 33, 84–101. [Google Scholar] [CrossRef] [PubMed]
- Cannon, S.B. Chromosomal Structure History and Genomic Synteny Relationships in Lupinus. In Compendium of Plant Genomes, The Lupin Genome; Karam, S., Kamphuis, L., Nelson, M., Eds.; Springer Nature Switzerland AG: Cham, Switzerland, 2020. [Google Scholar]
- Kroc, M.; Koczyk, G.; Święcicki, W.; Kilian, A.; Nelson, M.N. New evidence of ancestral polyploidy in the Genistoid legume Lupinus angustifolius L. (narrow-leafed lupin). Theor. Appl. Genet. 2014, 127, 1237–1249. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhou, Z.; Liu, Y.; Liu, T.; Li, Q.; Ji, Y.; Li, C.; Fang, C.; Wang, M.; Wu, M.; et al. Functional evolution of phosphatidylethanolamine binding proteins in soybean and Arabidopsis. Plant Cell 2015, 27, 323–336. [Google Scholar] [CrossRef] [PubMed]
- De Bodt, S.; Theissen, G.; Van de Peer, Y. Promoter analysis of MADS-box genes in eudicots through phylogenetic footprinting. Mol. Biol. Evol. 2006, 23, 1293–1303. [Google Scholar] [CrossRef] [PubMed]
- Maere, S.; De Bodt, S.; Raes, J.; Casneuf, T.; Van Montagu, M.; Kuiper, M.; Van de Peer, Y. Modeling gene and genome duplications in eukaryotes. Proc. Natl. Acad. Sci. USA 2005, 102, 5454–5459. [Google Scholar] [CrossRef] [PubMed]
- Hahn, M.W. Bias in phylogenetic tree reconciliation methods: implications for vertebrate genome evolution. Genome Biol. 2007, 8, R141. [Google Scholar] [CrossRef]
- Freeling, M. Bias in plant gene content following different sorts of duplication: tandem whole-genome segmental or by transposition. Annu. Rev. Plant Biol. 2009, 60, 433–453. [Google Scholar] [CrossRef]
- Tautz, D.; Domazet-Loso, T. The evolutionary origin of orphan genes. Nat. Rev. Genet. 2011, 12, 692–702. [Google Scholar] [CrossRef]
- De Smet, R.; Van de Peer, Y. Redundancy and rewiring of genetic networks following genome-wide duplication events. Curr. Opin. Plant Biol. 2012, 15, 168–176. [Google Scholar] [CrossRef]
- Gladieux, P.; Ropars, J.; Badouin, H.; Branca, A.; Aguileta, G.; de Vienne, D.M.; Rodriguez de la Vega, R.C.; Branco, S.; Giraud, T. Fungal evolutionary genomics provides insight into the mechanisms of adaptive divergence in eukaryotes. Mol. Ecol. 2014, 23, 753–773. [Google Scholar] [CrossRef]
- Page, R.D.; Charleston, M.A. From gene to organismal phylogeny: reconciled trees and the gene tree/species tree problem. Mol. Phylogenet. Evol. 1997, 7, 231–240. [Google Scholar] [CrossRef]
- Koczyk, G.; Dawidziuk, A.; Popiel, D. The Distant Siblings-A Phylogenomic Roadmap Illuminates the Origins of Extant Diversity in Fungal Aromatic Polyketide Biosynthesis. Genome Biol. Evol. 2015, 7, 3132–3154. [Google Scholar] [CrossRef] [PubMed]
- Fedorowicz-Strońska, O.; Koczyk, G.; Kaczmarek, M.; Krajewski, P.; Sadowski, J. Genome-wide identification, characterisation and expression profiles of calcium-dependent protein kinase genes in barley (Hordeum vulgare L.). J. Appl. Genet. 2017, 58, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Kumada, Y.; Benson, D.R.; Hillemann, D.; Hosted, T.J.; Rochefort, D.A.; Thompson, C.J.; Wohlleben, W.; Tateno, Y. Evolution of the glutamine synthetase gene one of the oldest existing and functioning genes. Proc. Natl. Acad. Sci. USA 1993, 90, 3009–3013. [Google Scholar] [CrossRef] [PubMed]
- Betti, M.; Garcia-Calderon, M.; Perez-Delgado, C.M.; Credali, A.; Estivill, G.; Galvan, F.; Vega, J.M.; Marquez, A.J. Glutamine synthetase in legumes: recent advances in enzyme structure and functional genomics. Int. J. Mol. Sci. 2012, 13, 7994–8024. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, B.; Park, J.; Plaxton, W.C. The remarkable diversity of plant PEPC (phosphoenolpyruvate carboxylase): recent insights into the physiological functions and post-translational controls of non-photosynthetic PEPCs. Biochem. J. 2011, 436, 15–34. [Google Scholar] [CrossRef]
- Sheen, J. C4 Gene Expression. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1999, 50, 187–217. [Google Scholar] [CrossRef]
- Yang, H.; Tao, Y.; Zheng, Z.; Zhang, Q.; Zhou, G.; Sweetingham, M.W.; Howieson, J.G.; Li, C. Draft genome sequence, and a sequence-defined genetic linkage map of the legume crop species Lupinus angustifolius L. PLoS ONE 2013, 8, e64799. [Google Scholar] [CrossRef]
- Choi, S.; Creelman, R.A.; Mullet, J.E.; Wing, R.A. Construction and characterization of a bacterial artificial chromosome library of Arabidopsis thaliana. Plant Mol. Biol. Rep. 1995, 13, 124–128. [Google Scholar] [CrossRef]
- Schulte, D.; Ariyadasa, R.; Shi, B.; Fleury, D.; Saski, C.; Atkins, M.; deJong, P.; Wu, C.C.; Graner, A.; Langridge, P.; et al. BAC library resources for map-based cloning and physical map construction in barley (Hordeum vulgare L.). BMC Genom. 2011, 12, 247. [Google Scholar] [CrossRef]
- Yang, X.; Makaroff, C.A.; Ma, H. The Arabidopsis MALE MEIOCYTE DEATH1 gene encodes a PHD-finger protein that is required for male meiosis. Plant Cell. 2003, 15, 1281–1295. [Google Scholar] [CrossRef] [PubMed]
- Gebhardt, C.; Oliver, J.E.; Forde, B.G.; Saarelainen, R.; Miflin, B.J. Primary structure and differential expression of glutamine synthetase genes in nodules roots and leaves of Phaseolus vulgaris. EMBO J. 1986, 5, 1429–1435. [Google Scholar] [CrossRef] [PubMed]
- Tingey, S.V.; Walker, E.L.; Coruzzi, G.M. Glutamine synthetase genes of pea encode distinct polypeptides which are differentially expressed in leaves roots and nodules. EMBO J. 1987, 6, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Stanford, A.C.; Larsen, K.; Barker, D.G.; Cullimore, J.V. Differential expression within the glutamine synthetase gene family of the model legume Medicago truncatula. Plant Physiol. 1993, 103, 73–81. [Google Scholar] [CrossRef]
- Temple, S.J.; Heard, J.; Ganter, G.; Dunn, K.; Sengupta-Gopalan, C. Characterization of a nodule-enhanced glutamine synthetase from alfalfa: nucleotide sequence in situ localization and transcript analysis. Mol. Plant Microbe Interact. 1995, 8, 218–227. [Google Scholar] [CrossRef]
- Morey, K.J.; Ortega, J.L.; Sengupta-Gopalan, C. Cytosolic glutamine synthetase in soybean is encoded by a multigene family and the members are regulated in an organ-specific and developmental manner. Plant Physiol. 2002, 128, 182–193. [Google Scholar] [CrossRef]
- Susek, K.; Bielski, W.; Czyz, K.B.; Hasterok, R.; Jackson, S.A.; Wolko, B.; Naganowska, B. Impact of Chromosomal Rearrangements on the Interpretation of Lupin Karyotype Evolution. Genes 2019, 10. [Google Scholar] [CrossRef]
- Przysiecka, L.; Ksiazkiewicz, M.; Wolko, B.; Naganowska, B. Structure expression profile and phylogenetic inference of chalcone isomerase-like genes from the narrow-leafed lupin (Lupinus angustifolius L.) genome. Front. Plant Sci. 2015, 6, 268. [Google Scholar] [CrossRef]
- Narożna, D.; Ksiazkiewicz, M.; Przysiecka, L.; Kroliczak, J.; Wolko, B.; Naganowska, B.; Madrzak, C.J. Legume isoflavone synthase genes have evolved by whole-genome and local duplications yielding transcriptionally active paralogs. Plant Sci. 2017, 264, 149–167. [Google Scholar] [CrossRef]
- Szczepaniak, A.; Książkiewicz, M.; Podkowiński, J.; Czyż, K.B.; Figlerowicz, M.; Naganowska, B. Legume Cytosolic and Plastid Acetyl-Coenzyme-A Carboxylase Genes Differ by Evolutionary Patterns and Selection Pressure Schemes Acting before and after Whole-Genome Duplications. Genes 2018, 9. [Google Scholar] [CrossRef]
- Ainouche, A. The Repetitive Content in Lupin Genomes. In Compendium of Plant Genomes, The Lupin Genome; Karam, S., Kamphuis, L., Nelson, M., Eds.; Springer Nature Switzerland AG: Cham, Switzerland, 2020. [Google Scholar]
- Ma, B.; Kuang, L.; Xin, Y.; He, N. New Insights into Long Terminal Repeat Retrotransposons in Mulberry Species. Genes 2019, 10. [Google Scholar] [CrossRef]
- Lockton, S.; Gaut, B. The evolution of transposable elements in natural populations of self-fertilizing Arabidopsis thaliana and its outcrossing relative Arabidopsis lyrata. BMC Evol. Biol. 2010, 10. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, K.; Abe, J.; Kanazawa, A. Chromosomal distribution of soybean retrotransposon SORE-1 suggests its recent preferential insertion into euchromatic regions. Chromosome Res. 2018, 26, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, L.G.; Deyholos, M.K. Identification characterization and distribution of transposable elements in the flax (Linum usitatissimum L.) genome. BMC Genom. 2012, 13, 644. [Google Scholar] [CrossRef] [PubMed]
- Seabra, A.R.; Vieira, C.P.; Cullimore, J.V.; Carvalho, H.G. Medicago truncatula contains a second gene encoding a plastid located glutamine synthetase exclusively expressed in developing seeds. BMC Plant Biol. 2010, 10, 183. [Google Scholar] [CrossRef] [PubMed]
- Forde, B.G.; Day, H.M.; Turton, J.F.; Shen, W.J.; Cullimore, J.V.; Oliver, J.E. Two glutamine synthetase genes from Phaseolus vulgaris L. display contrasting developmental and spatial patterns of expression in transgenic Lotus corniculatus plants. Plant Cell 1989, 1, 391–401. [Google Scholar] [CrossRef]
- Tang, H.; Wang, X.; Bowers, J.E.; Ming, R.; Alam, M.; Paterson, A.H. Unraveling ancient hexaploidy through multiply-aligned angiosperm gene maps. Genome Res. 2008, 18, 1944–1954. [Google Scholar] [CrossRef]
- Wang, B.; Zhang, Y.; Wei, P.; Sun, M.; Ma, X.; Zhu, X. Identification of nuclear low-copy genes and their phylogenetic utility in rosids. Genome 2014, 57, 547–554. [Google Scholar] [CrossRef]
- Cannon, S.B.; Sterck, L.; Rombauts, S.; Sato, S.; Cheung, F.; Gouzy, J.; Wang, X.; Mudge, J.; Vasdewani, J.; Schiex, T.; et al. Legume genome evolution viewed through the Medicago truncatula and Lotus japonicus genomes. Proc. Natl. Acad. Sci. USA 2006, 103, 14959–14964. [Google Scholar] [CrossRef]
- Cannon, S.B.; Ilut, D.; Farmer, A.D.; Maki, S.L.; May, G.D.; Singer, S.R.; Doyle, J.J. Polyploidy did not predate the evolution of nodulation in all legumes. PLoS ONE 2010, 5, e11630. [Google Scholar] [CrossRef]
- Zimmer, E.A.; Wen, J. Using nuclear gene data for plant phylogenetics: progress and prospects. Mol. Phylogenetics Evol. 2012, 65, 774–785. [Google Scholar] [CrossRef] [PubMed]
- Doyle, J.J. Evolution of higher plant glutamine synthetase genes: tissue specifity as a criterior for predicting orhology. Mol. Biol. Evol. 1991, 8, 366–377. [Google Scholar]
- Deng, H.; Zhang, L.S.; Zhang, G.Q.; Zheng, B.Q.; Liu, Z.J.; Wang, Y. Evolutionary history of PEPC genes in green plants: Implications for the evolution of CAM in orchids. Mol. Phylogenetics Evol. 2016, 94, 559–564. [Google Scholar] [CrossRef] [PubMed]
- Lavin, M.; Herendeen, P.S.; Wojciechowski, M.F. Evolutionary rates analysis of Leguminosae implicates a rapid diversification of lineages during the tertiary. Syst. Biol. 2005, 54, 575–594. [Google Scholar] [CrossRef] [PubMed]
- Schlueter, J.A.; Dixon, P.; Granger, C.; Grant, D.; Clark, L.; Doyle, J.J.; Shoemaker, R.C. Mining EST databases to resolve evolutionary events in major crop species. Genome 2004, 47, 868–876. [Google Scholar] [CrossRef]
- Pfeil, B.E.; Schlueter, J.A.; Shoemaker, R.C.; Doyle, J.J. Placing paleopolyploidy in relation to taxon divergence: A phylogenetic analysis in legumes using 39 gene families. Syst. Biol. 2005, 54, 441–454. [Google Scholar] [CrossRef]
- Rizzon, C.; Ponger, L.; Gaut, B.S. Striking similarities in the genomic distribution of tandemly arrayed genes in Arabidopsis and rice. PLoS Comput. Biol. 2006, 2, e115. [Google Scholar] [CrossRef]
- Blanc, G.; Wolfe, K.H. Widespread paleopolyploidy in model plant species inferred from age distributions of duplicate genes. Plant Cell 2004, 16, 1667–1678. [Google Scholar] [CrossRef]
- Xu, C.; Nadon, B.D.; Kim, K.D.; Jackson, S.A. Genetic and epigenetic divergence of duplicate genes in two legume species. Plant Cell Environ. 2018, 41, 2033–2044. [Google Scholar] [CrossRef]
- Plewiński, P.; Książkiewicz, M.; Rychel-Bielska, S.; Rudy, E.; Wolko, B. Candidate domestication-related genes revealed by expression quantitative trait loci mapping of narrow-leafed lupin (Lupinus angustifolius L.). Int. J. Mol. Sci. 2019, 20, 5670. [Google Scholar] [CrossRef]
- Torreira, E.; Seabra, A.R.; Marriott, H.; Zhou, M.; Llorca, O.; Robinson, C.V.; Carvalho, H.G.; Fernandez-Tornero, C.; Pereira, P.J. The structures of cytosolic and plastid-located glutamine synthetases from Medicago truncatula reveal a common and dynamic architecture. Acta Crystallogr. Sect. D Biol. Crystallogr. 2014, 70, 981–993. [Google Scholar] [CrossRef]
- Cork, J.M.; Purugganan, M.D. The evolution of molecular genetic pathways and networks. Bioessays 2004, 26, 479–484. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Rodriguez, J.; Wagner, A. Metabolic Determinants of Enzyme Evolution in a Genome-Scale Bacterial Metabolic Network. Genome Biol. Evol. 2018, 10, 3076–3088. [Google Scholar] [CrossRef] [PubMed]
- Maeda, H.A. Evolutionary Diversification of Primary Metabolism and Its Contribution to Plant Chemical Diversity. Front. Plant Sci. 2019, 10, 881. [Google Scholar] [CrossRef] [PubMed]
- Moore, B.M.; Wang, P.; Fan, P.; Leong, B.; Schenck, C.A.; Lloyd, J.P.; Lehti-Shiu, M.D.; Last, R.L.; Pichersky, E.; Shiu, S.H. Robust predictions of specialized metabolism genes through machine learning. Proc. Natl. Acad. Sci. USA 2019, 116, 2344–2353. [Google Scholar] [CrossRef] [PubMed]
- Mett, V.; Mett, V.L.; Reynolds, P.H. The aspartate aminotransferase-P2 gene from Lupinus angustifolius. Plant Physiol. 1994, 106, 1683–1684. [Google Scholar] [CrossRef][Green Version]
- Salamov, A.A.; Solovyev, V.V. Ab initio gene finding in Drosophila genomic DNA. Genome Res. 2000, 10, 516–522. [Google Scholar] [CrossRef]
- Kohany, O.; Gentles, A.J.; Hankus, L.; Jurka, J. Annotation submission and screening of repetitive elements in Repbase: RepbaseSubmitter and Censor. BMC Bioinform. 2006, 7, 474. [Google Scholar] [CrossRef]
- Lyons, E.; Pedersen, B.; Kane, J.; Alam, M.; Ming, R.; Tang, H.; Wang, X.; Bowers, J.; Paterson, A.; Lisch, D.; et al. Finding and comparing syntenic regions among Arabidopsis and the outgroups papaya poplar and grape: CoGe with rosids. Plant Physiol. 2008, 148, 1772–1781. [Google Scholar] [CrossRef]
- Revanna, K.V.; Chiu, C.C.; Bierschank, E.; Dong, Q. GSV: a web-based genome synteny viewer for customized data. BMC Bioinform. 2011, 12, 316. [Google Scholar] [CrossRef]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: an information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [PubMed]
- Goodstein, D.M.; Shu, S.; Howson, R.; Neupane, R.; Hayes, R.D.; Fazo, J.; Mitros, T.; Dirks, W.; Hellsten, U.; Putnam, N.; et al. Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res. 2012, 40, D1178–D1186. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference sequence (RefSeq) database at NCBI: current status taxonomic expansion and functional annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef]
- Bolser, D.M.; Staines, D.M.; Perry, E.; Kersey, P.J. Ensembl Plants: Integrating Tools for Visualizing Mining and Analyzing Plant Genomic Data. Methods Mol. Biol. 2017, 1533, 1–31. [Google Scholar] [CrossRef] [PubMed]
- Aubry, S.; Kelly, S.; Kumpers, B.M.; Smith-Unna, R.D.; Hibberd, J.M. Deep evolutionary comparison of gene expression identifies parallel recruitment of trans-factors in two independent origins of C4 photosynthesis. PLoS Genet. 2014, 10, e1004365. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Capella-Gutierrez, S.; Silla-Martinez, J.M.; Gabaldon, T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Minh, B.Q.; Nguyen, M.A.; von Haeseler, A. Ultrafast approximation for phylogenetic bootstrap. Mol. Biol. Evol. 2013, 30, 1188–1195. [Google Scholar] [CrossRef]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [PubMed]
- Enright, A.J.; Van Dongen, S.; Ouzounis, C.A. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002, 30, 1575–1584. [Google Scholar] [CrossRef] [PubMed]
- Rousseeuw, P. Silhouettes: A graphical aid to the interpretation and validation of cluster analysis. J. Comput. Appl. Math. 1987, 20, 53–65. [Google Scholar] [CrossRef]
- Jehl, P.; Sievers, F.; Higgins, D.G. OD-seq: outlier detection in multiple sequence alignments. BMC Bioinform. 2015, 16, 269. [Google Scholar] [CrossRef] [PubMed]
- Thykjaer, T.; Danielsen, D.; She, Q.; Stougaard, J. Organization and expression of genes in the genomic region surrounding the glutamine synthetase gene Gln1 from Lotus japonicus. Mol. Genet. Genom. 1997, 255, 628–636. [Google Scholar] [CrossRef]
- Schneider, A.; Cannarozzi, G.M.; Gonnet, G.H. Empirical codon substitution matrix. BMC Bioinform. 2005, 6, 134. [Google Scholar] [CrossRef] [PubMed]
- Soubrier, J.; Steel, M.; Lee, M.S.; Der Sarkissian, C.; Guindon, S.; Ho, S.Y.; Cooper, A. The influence of rate heterogeneity among sites on the time dependence of molecular rates. Mol. Biol. Evol. 2012, 29, 3345–3358. [Google Scholar] [CrossRef]
- Bansal, M.S.; Kellis, M.; Kordi, M.; Kundu, S. RANGER-DTL 2.0: rigorous reconstruction of gene-family evolution by duplication transfer and loss. Bioinformatics 2018, 34, 3214–3216. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Dopazo, J.; Gabaldon, T. ETE: a python Environment for Tree Exploration. BMC Bioinformatics 2010, 11, 24. [Google Scholar] [CrossRef]
- Librado, P.; Rozas, J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef]
- Yang, Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Wong, W.S.; Nielsen, R. Bayes empirical bayes inference of amino acid sites under positive selection. Mol. Biol. Evol. 2005, 22, 1107–1118. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Variant | Gene ID | BAC nb | Scaffold nb | NLL nb | Cyto marker | GC% | % RE | RE (bp) | RE type | CDS nb | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Lupin Express ID | GenBank ID | ||||||||||
| GS1a1 | Lup21297 | gene6261 | 047P22 | 4_25 | 4 | 047P22_5 | 33.1 | 8.58 | 8584 | Ty1/Copia | 12 |
| GS1a2 | Lup001512 | gene27466 | 087N22 | 106 | 16 | 087N22_2 | 32.94 | 15.63 | 15635 | TY1/Copia; Gypsy/DIRS1; DNA transposons | 10 |
| GS1a3 | Lup009916 | gene24502 | - | 192 | 14 | - | 36.11 | 10.54 | 9282 | Ty1/Copia; Gypsy/DIRS1; DNA transpozons | 17 |
| GS1b1 | Lup029429 | gene 19431 | 036L23 | 73 | 11 | 036L23_3 | 33 | 0 | 0 | - | 15 |
| GS1b2 | Lup032636 | gene17555 | 059J08 | 94_15 | 9 | 059J08_3 | 32.43 | 0.17 | 174 | Ty1/Copia | 16 |
| GS1c1 | Lup002132 | gene34907 | - | 11_68 | UN | - | 30.56 | 9.19 | 2621 | Ty1/Copia; DNA transposons | 3 |
| GS1c2 | Lup04581 | gene4422 | - | 13 | 3 | - | 31.89 | 7.2 | 7202 | Ty1/Copia; DNA transposons | 15 |
| GS2a1 | Lup023221 | gene31805 | - | 45_213 | 19 | - | 33.88 | 9.96 | 9963 | Ty1/Copia; Gypsy/DIRS1; DNA transpozons | 12 |
| GS2a2 | no | gene6462 | - | 186 | 4 | - | 32.66 | 7.73 | 7732 | Ty1/Copia; Gypsy/DIRS1; DNA transpozons | 12 |
| PEPC1a | Lup022696 | gene23490 | 064J15 | 437 | 13 | - | 34.25 | 8.85 | 8852 | Ty1/Copia; Gypsy/DIRS1 | 13 |
| PEPC1b | Lup029825 | gene15450 | - | 74_10 | 8 | - | 32.28 | 1.88 | 1879 | Ty1/Copia | 13 |
| PEPC1c | Lup015178 | gene12998 | - | 274 | 7 | - | 32.36 | 1.17 | 1169 | Ty1/Copia | 14 |
| PEPC2a | Lup002214 | gene31196 | 067C07 | 110_41 | 19 | 067C07_2 | 32.41 | 3.63 | 3634 | Ty1/Copia; Gypsy/DIRS1 | 14 |
| PEPC2b | Lup26946 | gene9184 | 131K15 | 59_19 | 5 | 131K15_5_3 | 33.21 | 6.49 | 6748 | Ty1/Copia; Gypsy/DIRS1 | 14 |
| PEPC3a | Lup031846 | gene18605 | - | 9_1 | 10 | - | 33.97 | 5.17 | 1628 | Ty1/Copia | 3 |
| PEPC3b | Lup016482 | gene7147 | - | 296 | 4 | - | 32.76 | 15.64 | 15641 | Ty1/Copia; DNA transposon | 5 |
| PEPC4 | Lup002996 | no | - | 12_32 | 7 | - | 33.76 | 8.64 | 8644 | Ty1/Copia; Gypsy/DIRS1 | 16 |
| PEPC5 | Lup031638 | - | 88_60 | 20 | - | 32.73 | 8.93 | 8933 | Ty1/Copia; Gypsy/DIRS1; DNA transposons | 10 | |
| GS Subset | Legume Clade | Taxon | Locus Tag (NCBI: Gene Locus ID 1) |
|---|---|---|---|
| GS2 | dalbergioids | Arachis ipaensis | gene15537 (LOC107638560), gene3699 (LOC107637831) |
| genistoids | Lupinus angustifolius | TanjilG_23221, gene6462 (LOC109345303) | |
| IRLC | Cicer arietinum | gene633 (LOC101511058) | |
| Medicago truncatula | MTR_2g021242, MTR_2g021255 | ||
| Trifolium pratense | Tp57577_TGAC_v2_gene28916 | ||
| milletioids | Cajanus cajan | KK1_005408 | |
| Glycine max | GLYMA13G28180, GLYMA15G10890 | ||
| Phaseolus vulgaris | Phvul.006G155800 | ||
| Vigna radiata | gene21293 (LOC106775732) | ||
| robinioids | Lotus japonicus | Lj6g3v1887790 (CUFF.60993), Lj6g3v1887800, Lj6g3v1953860 | |
| GS1cs1 | dalbergioids | Arachis ipaensis | gene13764 (LOC107631250) |
| genistoids | Lupinus angustifolius | TanjilG_32636, TanjilG_29429, TanjilG_09916, TanjilG_01512, TanjilG_21297 | |
| IRLC | Cicer arietinum | gene15008 (LOC101499598), gene4692 (LOC101502819) | |
| Medicago truncatula | MTR_3g065250, MTR_5g077950 | ||
| Trifolium pratense | Tp57577_TGAC_v2_gene24906, Tp57577_TGAC_v2_gene30014 | ||
| milletioids | Cajanus cajan | gene24397 (LOC109818547), KK1_036386, KK1_020174 | |
| Glycine max | GLYMA02G41106, GLYMA02G41120, GLYMA11G33560, GLYMA14G39420, GLYMA18G04660 | ||
| Phaseolus vulgaris | Phvul.001G229500, Phvul.008G237400, Phvul.008G237500 | ||
| Vigna radiata | gene10448 (LOC106764801), gene10450 (LOC106763809), gene4883 (LOC106757638) | ||
| Lotus japonicus | Lj0g3v0335159 (CUFF.22888), Lj6g3v0410480 | ||
| GS1cs2 | dalbergioids | Arachis ipaensis | gene23856 (LOC107644115) |
| genistoids | Lupinus angustifolius | TanjilG_02132, TanjilG_04581 | |
| IRLC | Cicer arietinum | gene23264 (LOC101499849) | |
| Medicago truncatula | MTR_6g071070 | ||
| Trifolium pratense | Tp57577_TGAC_v2_gene18383 | ||
| milletioids | Cajanus cajan | KK1_041929 | |
| Glycine max | GLYMA07G11810, GLYMA09G30370 | ||
| Phaseolus vulgaris | Phvul.004G148300 | ||
| Vigna radiata | gene742 (LOC106756019) | ||
| robinioids | Lotus japonicus | Lj2g3v0658180 (CUFF.35493) |
| PEPC Subset | Legume Clade | Taxon | Locus Tag (NCBI:Gene Locus ID 1) |
|---|---|---|---|
| PEPC1a | dalbergioids | Arachis ipaensis | gene10946 (LOC107630016), gene5131 (LOC107624747) |
| genistoids | Lupinus angustifolius | TanjilG_02996, TanjilG_31846, TanjilG_16482 | |
| IRLC | Cicer arietinum | gene1498 (LOC101500264), gene16990 (LOC101510288) | |
| Medicago truncatula | MTR_2g092930, MTR_4g079860 | ||
| Trifolium pratense | Tp57577_TGAC_v2_gene11496 | ||
| milletioids | Cajanus cajan | KK1_024667, KK1_032556 | |
| Glycine max | GLYMA06G43050, GLYMA12G33820, GLYMA13G36670 | ||
| Phaseolus vulgaris | Phvul.005G095300, Phvul.011G130400 | ||
| Vigna radiata | gene23996 (LOC106778590), gene26799 (LOC106753186) | ||
| PEPC1b | dalbergioids | Arachis ipaensis | gene11232 (LOC107630060), gene37010 (LOC107612799) |
| genistoids | Lupinus angustifolius | TanjilG_15178, TanjilG_29825, TanjilG_22696, TanjilG_02214, TanjilG_26946 | |
| IRLC | Cicer arietinum | gene26512 (LOC101514127), gene3089 (LOC101497901) | |
| Medicago truncatula | MTR_2g076670, MTR_8g463920 | ||
| Trifolium pratense | Tp57577_TGAC_v2_gene19367 | ||
| milletioids | Cajanus cajan | KK1_014855, KK1_026796 | |
| Glycine max | GLYMA06G33380, GLYMA12G35840 (PPC1), GLYMA13G34560, GLYMA20G09810 (PPC16) | ||
| Phaseolus vulgaris | Phvul.005G066400, Phvul.011G160200 | ||
| Vigna radiata | gene3386 (LOC106756025), gene9625 (LOC106760805) | ||
| robinioids | Lotus japonicus | Lj3g3v0428380 (CUFF.40719), Lj3g3v0428390, Lj3g3v1061390 | |
| PEPC2 | dalbergioids | Arachis ipaensis | gene23112 (LOC107641982), gene43520 (LOC107617655) |
| genistoids | Lupinus angustifolius | TanjilG_31638 | |
| IRLC | Cicer arietinum | gene4202 (LOC101494422), gene9231 (LOC101496857) | |
| Medicago truncatula | MTR_0002s0890 | ||
| milletioids | Cajanus cajan | KK1_025033, KK1_045915 | |
| Glycine max | GLYMA01G22840, GLYMA10G34970 | ||
| Phaseolus vulgaris | Phvul.003G024800, Phvul.007G101300 | ||
| Vigna radiata | gene17891 (LOC106770762), gene23485 (LOC106777342) | ||
| robinioids | Lotus japonicus | Lj0g3v0165109 (CUFF.10370) |
| Gene | Accession | Mean Expression in RIL Population | Min Expression Value in RIL Population | Max Expression Value in RIL Population | Expression SD |
|---|---|---|---|---|---|
| GS1a1 | Lup021297 | 43.1 | 20.4 | 74.1 | 16.4 |
| GS1a2 | Lup001512 | 13.8 | 4.9 | 32.2 | 5.2 |
| GS1a3 | Lup009916 | 11.5 | 3.6 | 43.7 | 5.0 |
| GS1b1 | Lup029429 | 0.3 | 0.0 | 1.4 | 0.3 |
| GS1b2 | Lup032636 | 2.6 | 0.4 | 6.0 | 1.2 |
| GS1c1 | Lup002132 | 0.1 | 0.0 | 0.9 | 0.2 |
| GS1c2 | Lup004581 | 187.5 | 117.6 | 426.6 | 52.4 |
| GS2a1 | Lup023221 | 516.2 | 365.3 | 739.7 | 80.3 |
| GS2a2 | - | - | - | - | - |
| PEPC1a | Lup022696 | 17.0 | 8.1 | 31.1 | 4.0 |
| PEPC1b | Lup029825 | 0.5 | 0.0 | 2.4 | 0.5 |
| PEPC1c | Lup015178 | 65.0 | 44.5 | 87.4 | 8.7 |
| PEPC2a | Lup002214 | 0.0 | 0.0 | 0.5 | 0.1 |
| PEPC2b | Lup026946 | 0.1 | 0.0 | 0.9 | 0.2 |
| PEPC3a | Lup031846 | 10.5 | 4.7 | 16.1 | 2.5 |
| PEPC3b | Lup016482 | 51.0 | 33.0 | 94.2 | 11.2 |
| PEPC4 | Lup002996 | 1.7 | 0.0 | 4.1 | 0.9 |
| PEPC5 | Lup031638 | 11.9 | 1.7 | 28.7 | 4.8 |
| HEL | Lup023733 | 3.0 | 0.4 | 7.4 | 1.2 |
| TUB | Lup021845 | 78.4 | 35.3 | 113.1 | 15.2 |
| Probe Name | PCR Primer Sequence | Length (bp) | T* |
|---|---|---|---|
| GS | GS_F: GTTGGTCCCTCTGTTGGAATCTCTG GS_R: ATAAGCAGCAATGTGCTCATTGTGTCTC | 571 | 56 |
| PEPC | PEPC_F: AAAGATGTTAGGAATCTTCACATGCTGCAAGA PEPC_R: GGGGCATATTCACTTGTTGGGTTCAGT | 643 | 58 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Czyż, K.B.; Książkiewicz, M.; Koczyk, G.; Szczepaniak, A.; Podkowiński, J.; Naganowska, B. A Tale of Two Families: Whole Genome and Segmental Duplications Underlie Glutamine Synthetase and Phosphoenolpyruvate Carboxylase Diversity in Narrow-Leafed Lupin (Lupinus angustifolius L.). Int. J. Mol. Sci. 2020, 21, 2580. https://doi.org/10.3390/ijms21072580
Czyż KB, Książkiewicz M, Koczyk G, Szczepaniak A, Podkowiński J, Naganowska B. A Tale of Two Families: Whole Genome and Segmental Duplications Underlie Glutamine Synthetase and Phosphoenolpyruvate Carboxylase Diversity in Narrow-Leafed Lupin (Lupinus angustifolius L.). International Journal of Molecular Sciences. 2020; 21(7):2580. https://doi.org/10.3390/ijms21072580
Chicago/Turabian StyleCzyż, Katarzyna B., Michał Książkiewicz, Grzegorz Koczyk, Anna Szczepaniak, Jan Podkowiński, and Barbara Naganowska. 2020. "A Tale of Two Families: Whole Genome and Segmental Duplications Underlie Glutamine Synthetase and Phosphoenolpyruvate Carboxylase Diversity in Narrow-Leafed Lupin (Lupinus angustifolius L.)" International Journal of Molecular Sciences 21, no. 7: 2580. https://doi.org/10.3390/ijms21072580
APA StyleCzyż, K. B., Książkiewicz, M., Koczyk, G., Szczepaniak, A., Podkowiński, J., & Naganowska, B. (2020). A Tale of Two Families: Whole Genome and Segmental Duplications Underlie Glutamine Synthetase and Phosphoenolpyruvate Carboxylase Diversity in Narrow-Leafed Lupin (Lupinus angustifolius L.). International Journal of Molecular Sciences, 21(7), 2580. https://doi.org/10.3390/ijms21072580