
The Absence of Extracellular Cold-Inducible RNA-Binding Protein (eCIRP) Promotes Pro-Angiogenic Microenvironmental Conditions and Angiogenesis in Muscle Tissue Ischemia

 , ,
, , 
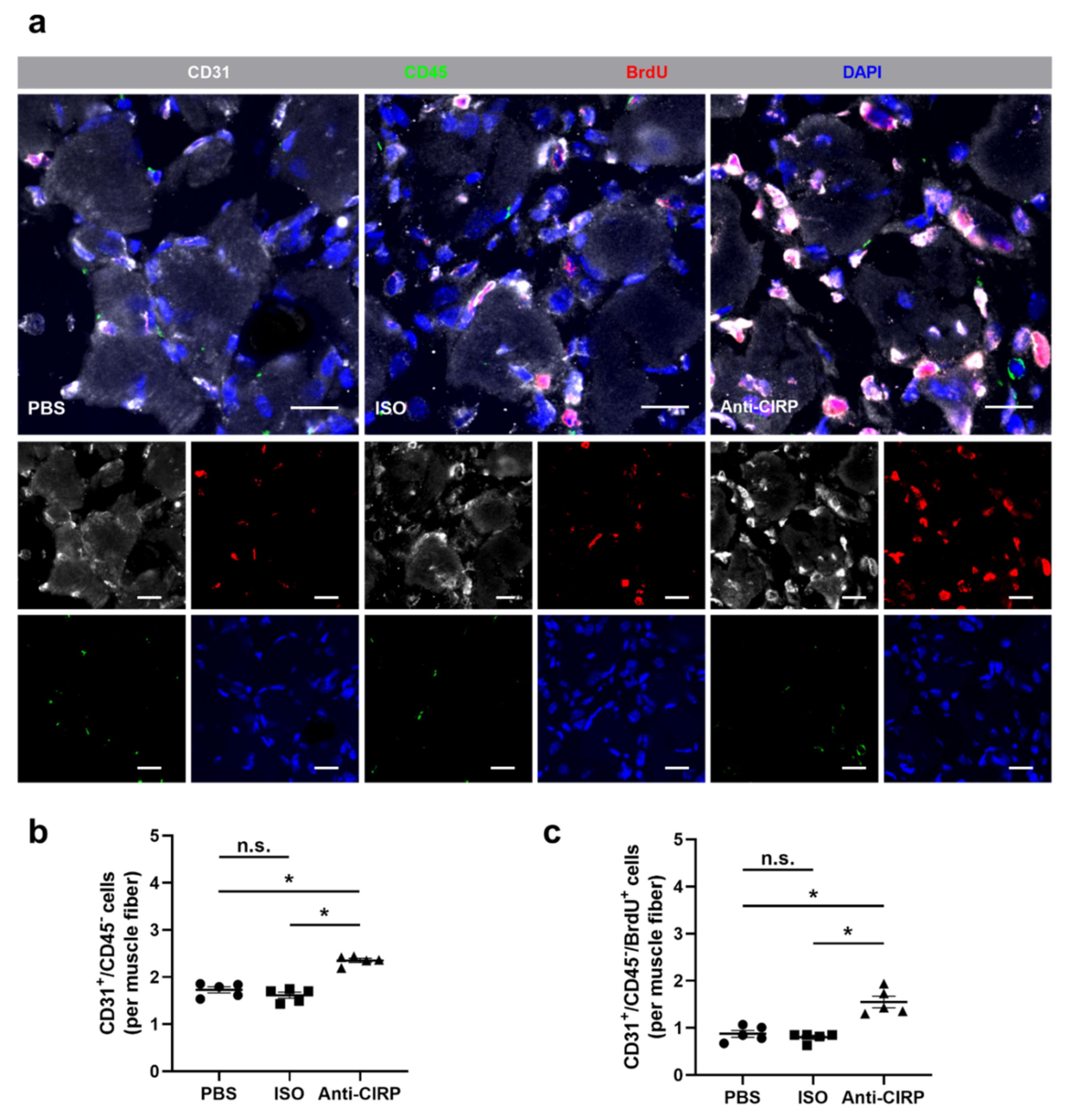{kind=link}
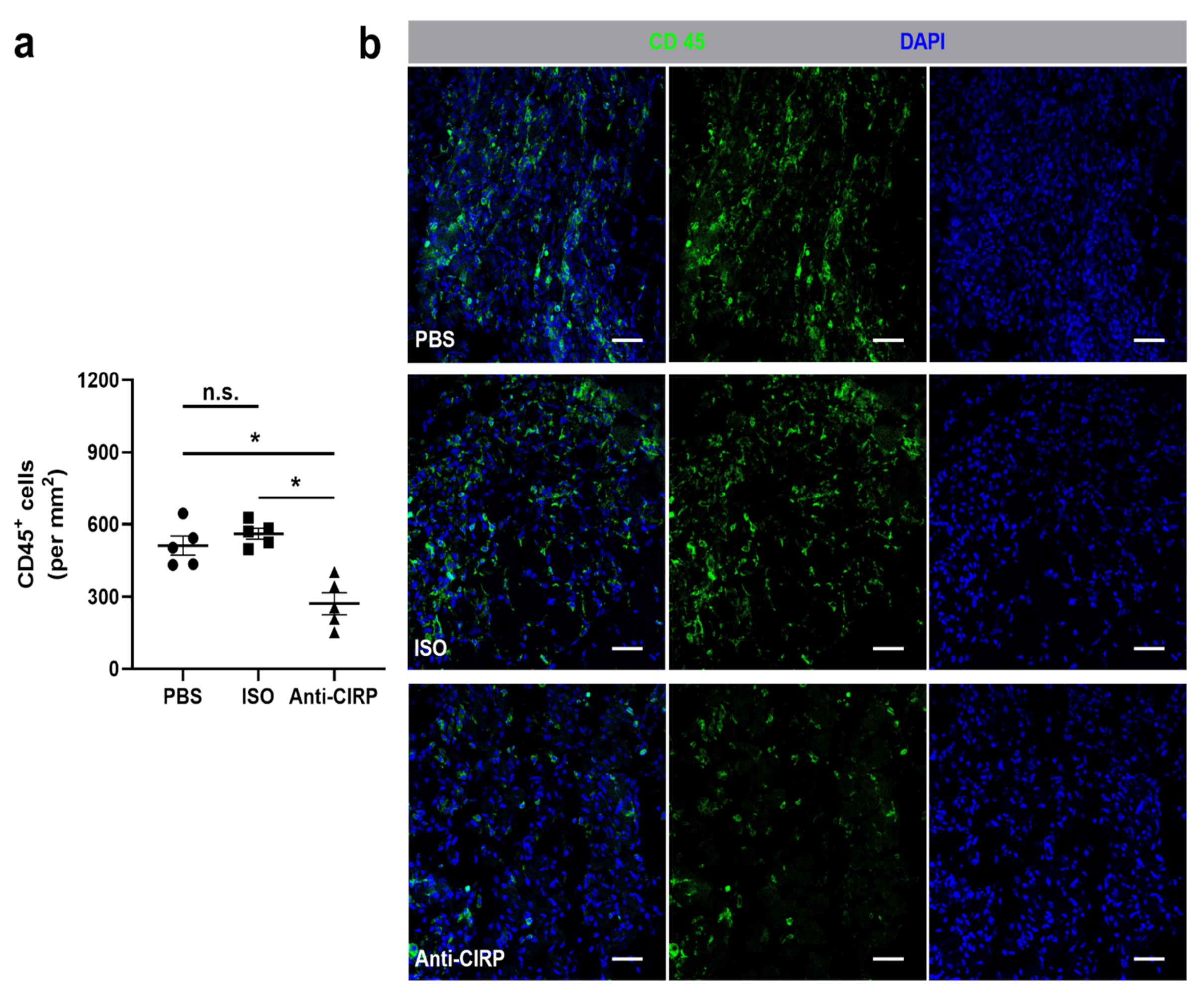{kind=link}
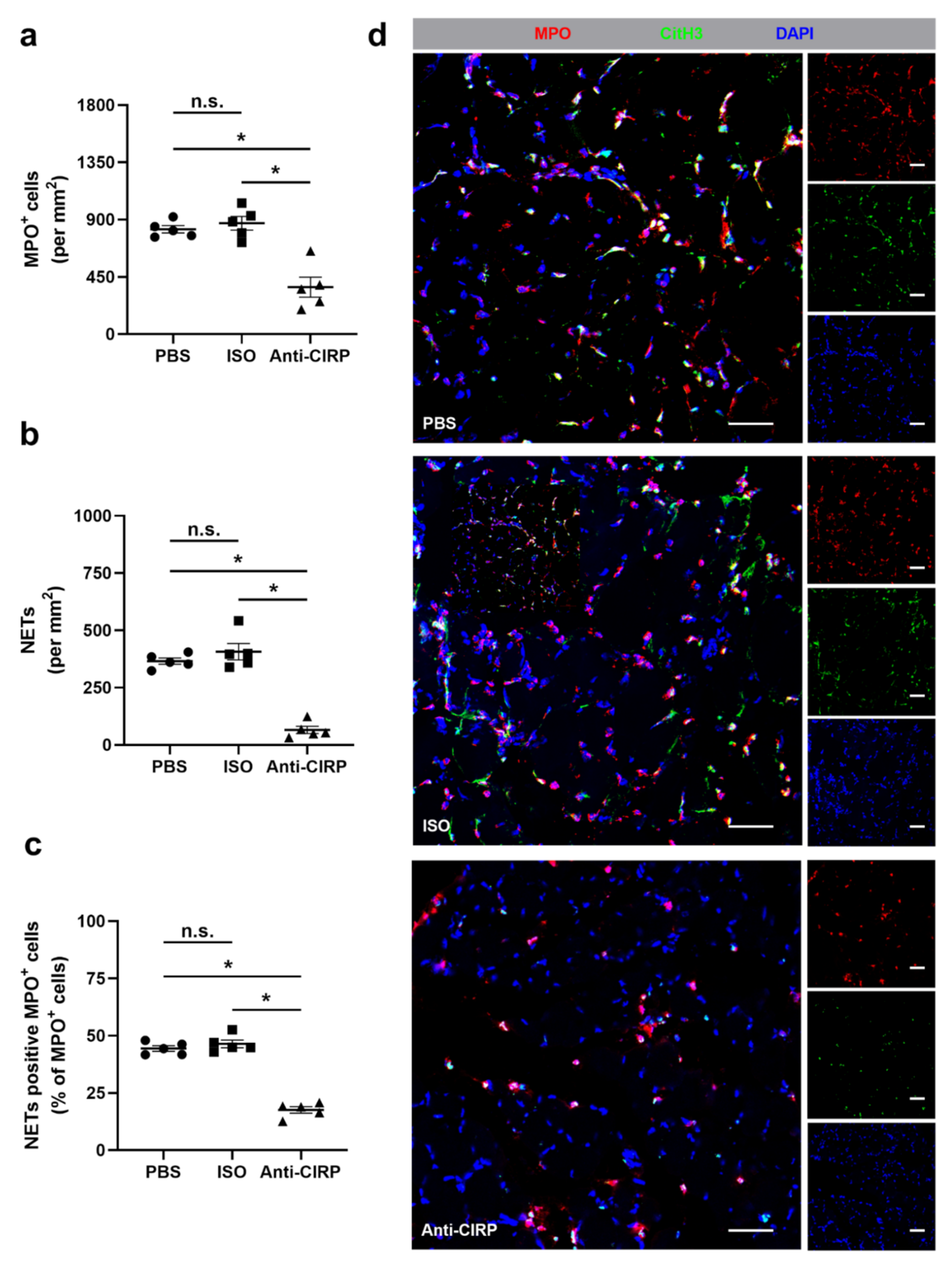{kind=link}
{kind=link}
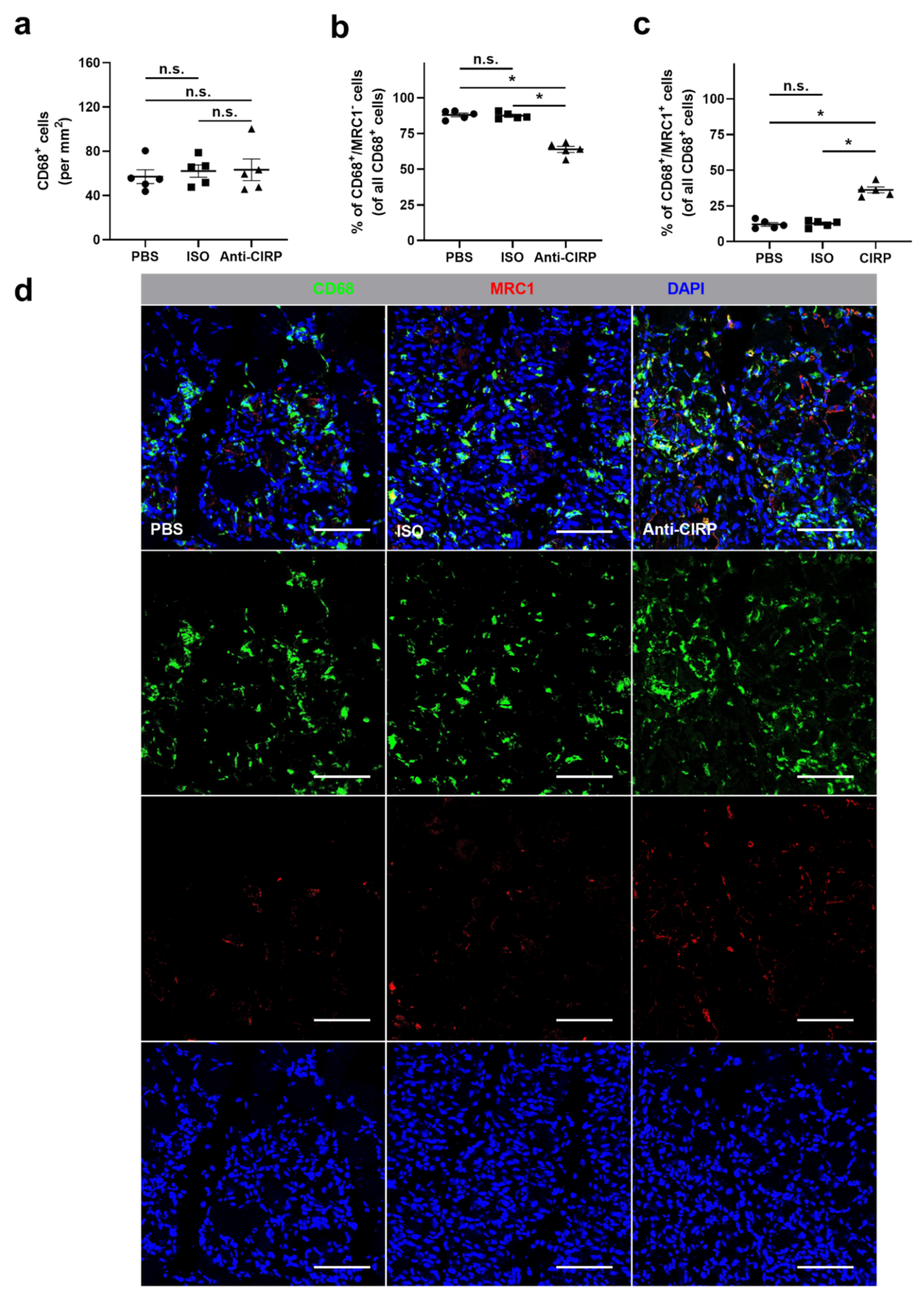{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
3. Discussion
4. Materials and Methods
4.1. Animals and Treatments
4.2. Femoral Artery Ligation and Tissue Processing
4.3. Immunohistology
4.4. Cell Culture and Proliferation Assays
4.5. Quantitative Real-Time PCR (qPCR)
4.6. Statistical Analysis
4.7. Illustration
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nishiyama, H.; Itoh, K.; Kaneko, Y.; Kishishita, M.; Yoshida, O.; Fujita, J. A Glycine-rich RNA-binding Protein Mediating Cold-inducible Suppression of Mammalian Cell Growth. J. Cell Biol. 1997, 137, 899–908. [Google Scholar] [CrossRef] [PubMed]
- Thandapani, P.; O’Connor, T.R.; Bailey, T.L.; Richard, S. Defining the RGG/RG Motif. Mol. Cell 2013, 50, 613–623. [Google Scholar] [CrossRef] [Green Version]
- De Leeuw, F.; Zhang, T.; Wauquier, C.; Huez, G.; Kruys, V.; Gueydan, C. The cold-inducible RNA-binding protein migrates from the nucleus to cytoplasmic stress granules by a methylation-dependent mechanism and acts as a translational repressor. Exp. Cell Res. 2007, 313, 4130–4144. [Google Scholar] [CrossRef]
- Yang, R.; Zhan, M.; Nalabothula, N.R.; Yang, Q.; Indig, F.E.; Carrier, F. Functional Significance for a Heterogenous Ribonucleoprotein A18 Signature RNA Motif in the 3′-Untranslated Region of Ataxia Telangiectasia Mutated and Rad3-related (ATR) Transcript. J. Biol. Chem. 2010, 285, 8887–8893. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Liu, X.; Li, B.; Zhang, Q.; Wang, J.; Zhang, W.; Luo, W.; Chen, J. Cold Inducible RNA Binding Protein Is Involved in Chronic Hypoxia Induced Neuron Apoptosis by Down-Regulating HIF-1α Expression and Regulated By microRNA-23a. Int. J. Biol. Sci. 2017, 13, 518–531. [Google Scholar] [CrossRef] [Green Version]
- Sumitomo, Y.; Higashitsuji, H.; Higashitsuji, H.; Liu, Y.; Fujita, T.; Sakurai, T.; Candeias, M.M.; Itoh, K.; Chiba, T.; Fujita, J. Identification of a novel enhancer that binds Sp1 and contributes to induction of cold-inducible RNA-binding protein (cirp) expression in mammalian cells. BMC Biotechnol. 2012, 12, 72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, R.; Weber, D.J.; Carrier, F. Post-transcriptional regulation of thioredoxin by the stress inducible heterogenous ribonu-cleoprotein A18. Nucleic Acids Res. 2006, 34, 1224–1236. [Google Scholar] [CrossRef] [Green Version]
- Qiang, X.; Yang, W.L.; Wu, R.; Zhou, M.; Jacob, A.; Dong, W.; Kuncewitch, M.; Ji, Y.; Yang, H.; Wang, H.; et al. Cold-inducible RNA-binding protein (CIRP) triggers inflammatory responses in hemorrhagic shock and sepsis. Nat. Med. 2013, 19, 1489–1495. [Google Scholar] [CrossRef] [Green Version]
- Godwin, A.; Yang, W.-L.; Sharma, A.; Khader, A.; Wang, Z.; Zhang, F.; Nicastro, J.; Coppa, G.F.; Wang, P. Blocking Cold-Inducible RNA-Binding Protein Protects Liver From Ischemia-Reperfusion Injury. Shock 2015, 43, 24–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGinn, J.T.; Aziz, M.; Zhang, F.; Yang, W.-L.; Nicastro, J.M.; Coppa, G.F.; Wang, P. Cold-inducible RNA-binding protein-derived peptide C23 attenuates inflammation and tissue injury in a murine model of intestinal ischemia-reperfusion. Surgery 2018, 164, 1191–1197. [Google Scholar] [CrossRef]
- Cen, C.; Yang, W.-L.; Yen, H.-T.; Nicastro, J.M.; Coppa, G.F.; Wang, P. Deficiency of cold-inducible ribonucleic acid-binding protein reduces renal injury after ischemia-reperfusion. Surgery 2016, 160, 473–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, M.; Yang, W.-L.; Ji, Y.; Qiang, X.; Wang, P. Cold-inducible RNA-binding protein mediates neuroinflammation in cerebral ischemia. Biochim. Biophys. Acta 2014, 1840, 2253–2261. [Google Scholar] [CrossRef] [Green Version]
- Pittman, K.; Kubes, P. Damage-Associated Molecular Patterns Control Neutrophil Recruitment. J. Innate Immun. 2013, 5, 315–323. [Google Scholar] [CrossRef]
- Denning, N.-L.; Aziz, M.; Murao, A.; Gurien, S.D.; Ochani, M.; Prince, J.M.; Wang, P. Extracellular CIRP as an endogenous TREM-1 ligand to fuel inflammation in sepsis. JCI Insight 2020, 5. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, T.; Kashida, H.; Watanabe, T.; Hagiwara, S.; Mizushima, T.; Iijima, H.; Nishida, N.; Higashitsuji, H.; Fujita, J.; Kudo, M. Stress Response Protein Cirp Links Inflammation and Tumorigenesis in Colitis-Associated Cancer. Cancer Res. 2014, 74, 6119–6128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakurai, T.; Kashida, H.; Komeda, Y.; Nagai, T.; Hagiwara, S.; Watanabe, T.; Kitano, M.; Nishida, N.; Fujita, J.; Kudo, M. Stress Response Protein RBM3 Promotes the Development of Colitis-associated Cancer. Inflamm. Bowel Dis. 2017, 23, 57–65. [Google Scholar] [CrossRef]
- McGinn, J.; Zhang, F.; Aziz, M.; Yang, W.L.; Nicastro, J.; Coppa, G.F.; Wang, P. The Protective Effect of a Short Peptide Derived from Cold-Inducible RNA-Binding Protein in Renal Ischemia-Reperfusion Injury. Shock 2018, 49, 269–276. [Google Scholar] [CrossRef]
- Zhang, F.; Brenner, M.; Yang, W.-L.; Wang, P. A cold-inducible RNA-binding protein (CIRP)-derived peptide attenuates inflammation and organ injury in septic mice. Sci. Rep. 2018, 8, 3052. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.-L.; Sharma, A.; Wang, Z.; Li, Z.; Fan, J.; Wang, P. Cold-inducible RNA-binding protein causes endothelial dysfunction via activation of Nlrp3 inflammasome. Sci. Rep. 2016, 6, 26571. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Dong, H.; Zhong, Y.; Huang, J.; Lv, J.; Li, J. The Cold-Inducible RNA-Binding Protein (CIRP) Level in Peripheral Blood Predicts Sepsis Outcome. PLoS ONE 2015, 10, e0137721. [Google Scholar] [CrossRef]
- Chang, S.-H.; Hla, T. Gene regulation by RNA binding proteins and microRNAs in angiogenesis. Trends Mol. Med. 2011, 17, 650–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerstberger, S.; Hafner, M.; Tuschl, T. A census of human RNA-binding proteins. Nat. Rev. Genet. 2014, 15, 829–845. [Google Scholar] [CrossRef] [PubMed]
- Lukong, K.E.; Chang, K.-W.; Khandjian, E.W.; Richard, S. RNA-binding proteins in human genetic disease. Trends Genet. 2008, 24, 416–425. [Google Scholar] [CrossRef]
- Adams, R.H.; Alitalo, K. Molecular regulation of angiogenesis and lymphangiogenesis. Nat. Rev. Mol. Cell Biol. 2007, 8, 464–478. [Google Scholar] [CrossRef]
- Egginton, S.; Zhou, A.-L.; Brown, M.D.; Hudlická, O. Unorthodox angiogenesis in skeletal muscle. Cardiovasc. Res. 2001, 49, 634–646. [Google Scholar] [CrossRef] [Green Version]
- Tonnesen, M.G.; Feng, X.; Clark, R.A. Angiogenesis in wound healing. J. Investig. Dermatol. Symp. Proc. 2000, 5, 40–46. [Google Scholar] [CrossRef] [Green Version]
- Karizbodagh, M.P.; Rashidi, B.; Sahebkar, A.; Masoudifar, A.; Mirzaei, H. Implantation Window and Angiogenesis. J. Cell. Biochem. 2017, 118, 4141–4151. [Google Scholar] [CrossRef]
- Gerhardt, H.; Golding, M.; Fruttiger, M.; Ruhrberg, C.; Lundkvist, A.; Abramsson, A.; Jeltsch, M.; Mitchell, C.; Alitalo, K.; Shima, D.; et al. VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J. Cell Biol. 2003, 161, 1163–1177. [Google Scholar] [CrossRef]
- Ferrara, N.; Henzel, W.J. Pituitary follicular cells secrete a novel heparin-binding growth factor specific for vascular endothelial cells. Biochem. Biophys. Res. Commun. 1989, 161, 851–858. [Google Scholar] [CrossRef]
- Tirpe, A.A.; Gulei, D.; Ciortea, S.M.; Crivii, C.; Berindan-Neagoe, I. Hypoxia: Overview on Hypoxia-Mediated Mechanisms with a Focus on the Role of HIF Genes. Int. J. Mol. Sci. 2019, 20, 6140. [Google Scholar] [CrossRef] [Green Version]
- Shima, D.T.; Adamis, A.P.; Ferrara, N.; Yeo, K.-T.; Yeo, T.-K.; Allende, R.; Folkman, J.; D’Amore, P. Hypoxic Induction of Endothelial Cell Growth Factors in Retinal Cells: Identification and Characterization of Vascular Endothelial Growth Factor (VEGF) as the Mitogen. Mol. Med. 1995, 1, 182–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmeliet, P. Mechanisms of angiogenesis and arteriogenesis. Nat. Med. 2000, 6, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Mentzer, S.J.; Konerding, M.A. Intussusceptive angiogenesis: Expansion and remodeling of microvascular networks. Angiogenesis 2014, 17, 499–509. [Google Scholar] [CrossRef] [Green Version]
- Demir, R.; Yaba, A.; Huppertz, B. Vasculogenesis and angiogenesis in the endometrium during menstrual cycle and implantation. Acta Histochem. 2010, 112, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat. Med. 1995, 1, 27–31. [Google Scholar] [CrossRef]
- Priya, S.K.; Nagare, R.; Sneha, V.; Sidhanth, C.; Bindhya, S.; Manasa, P.; Ganesan, T. Tumour angiogenesis—Origin of blood vessels. Int. J. Cancer 2016, 139, 729–735. [Google Scholar] [CrossRef]
- Weckbach, L.T.; Preissner, K.T.; Deindl, E. The Role of Midkine in Arteriogenesis, Involving Mechanosensing, Endothelial Cell Proliferation, and Vasodilation. Int. J. Mol. Sci. 2018, 19, 2559. [Google Scholar] [CrossRef] [Green Version]
- Deindl, E.; Zaruba, M.M.; Brunner, S.; Huber, B.; Mehl, U.; Assmann, G.; Hoefer, I.E.; Mueller-Hoecker, J.; Franz, W.M. G-CSF administration after myocardial infarction in mice attenuates late ischemic cardiomyopathy by enhanced arteriogenesis. FASEB J. 2006, 20, 956–958. [Google Scholar] [CrossRef] [Green Version]
- Deindl, E.; Schaper, W. The Art of Arteriogenesis. Cell Biophys. 2005, 43, 1–15. [Google Scholar] [CrossRef]
- Faber, J.E.; Chilian, W.M.; Deindl, E.; van Royen, N.; Simons, M. A Brief Etymology of the Collateral Circulation. Arter. Thromb. Vasc. Biol. 2014, 34, 1854–1859. [Google Scholar] [CrossRef] [Green Version]
- Rizzi, A.; Benagiano, V.; Ribatti, D. Angiogenesis versus arteriogenesis. Rom. J. Morphol. Embryol. Rev. Roum. Morphol. Embryol. 2017, 58, 15–19. [Google Scholar]
- Heil, M.; Eitenmüller, I.; Schmitz-Rixen, T.; Schaper, W. Arteriogenesis versus angiogenesis: Similarities and differences. J. Cell. Mol. Med. 2006, 10, 45–55. [Google Scholar] [CrossRef] [Green Version]
- Scapini, P.; Morini, M.; Tecchio, C.; Minghelli, S.; Di Carlo, E.; Tanghetti, E.; Albini, A.; Lowell, C.; Berton, G.; Noonan, D.M.; et al. CXCL1/macrophage inflammatory protein-2-induced angiogenesis in vivo is mediated by neutrophil-derived vascular endothelial growth factor-A. J. Immunol. 2004, 172, 5034–5040. [Google Scholar] [CrossRef] [Green Version]
- Gaudry, M.; Brégerie, O.; Andrieu, V.; El Benna, J.; Pocidalo, M.-A.; Hakim, J. Intracellular Pool of Vascular Endothelial Growth Factor in Human Neutrophils. Blood 1997, 90, 4153–4161. [Google Scholar] [CrossRef]
- Gong, Y.; Koh, D.-R. Neutrophils promote inflammatory angiogenesis via release of preformed VEGF in an in vivo corneal model. Cell Tissue Res. 2010, 339, 437–448. [Google Scholar] [CrossRef]
- Ardi, V.C.; Kupriyanova, T.A.; Deryugina, E.I.; Quigley, J.P. Human neutrophils uniquely release TIMP-free MMP-9 to provide a potent catalytic stimulator of angiogenesis. Proc. Natl. Acad. Sci. USA 2007, 104, 20262–20267. [Google Scholar] [CrossRef] [Green Version]
- Du Cheyne, C.; Tay, H.; De Spiegelaere, W. The complex TIE between macrophages and angiogenesis. Anat. Histol. Embryol. 2019. [Google Scholar] [CrossRef]
- Aldabbous, L.; Abdul-Salam, V.; McKinnon, T.; Duluc, L.; Pepke-Zaba, J.; Southwood, M.; Ainscough, A.J.; Hadinnapola, C.; Wilkins, M.R.; Toshner, M.; et al. Neutrophil Extracellular Traps Promote Angiogenesis: Evidence from Vascular Pathology in Pulmonary Hypertension. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 2078–2087. [Google Scholar] [CrossRef] [Green Version]
- Takizawa, S.; Murao, A.; Ochani, M.; Aziz, M.; Wang, P. Frontline Science: Extracellular CIRP generates a proinflammatory Ly6G + CD11b hi subset of low-density neutrophils in sepsis. J. Leukoc. Biol. 2020, 109, 1019–1032. [Google Scholar] [CrossRef]
- Murao, A.; Arif, A.; Brenner, M.; Denning, N.-L.; Jin, H.; Takizawa, S.; Nicastro, B.; Wang, P.; Aziz, M. Extracellular CIRP and TREM-1 axis promotes ICAM-1-Rho-mediated NETosis in sepsis. FASEB J. 2020. [Google Scholar] [CrossRef]
- Ode, Y.; Aziz, M.; Jin, H.; Arif, A.; Nicastro, J.G.; Wang, P. Cold-inducible RNA-binding Protein Induces Neutrophil Extracellular Traps in the Lungs during Sepsis. Sci. Rep. 2019, 9, 6252. [Google Scholar] [CrossRef]
- Ode, Y.; Aziz, M.; Wang, P. CIRP increases ICAM-1(+) phenotype of neutrophils exhibiting elevated iNOS and NETs in sepsis. J. Leukoc. Biol. 2018, 103, 693–707. [Google Scholar] [CrossRef]
- Rohrbach, A.S.; Slade, D.J.; Thompson, P.R.; Mowen, K.A. Activation of PAD4 in NET formation. Front. Immunol. 2012, 3, 360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Idrovo, J.P.; Jacob, A.; Yang, W.L.; Wang, Z.; Yen, H.T.; Nicastro, J.; Coppa, G.F.; Wang, P. A deficiency in cold-inducible RNA-binding protein accelerates the inflammation phase and improves wound healing. Int. J. Mol. Med. 2016, 37, 423–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welten, S.M.; Bastiaansen, A.J.; de Jong, R.C.; de Vries, M.R.; Peters, E.A.; Boonstra, M.C.; Sheikh, S.P.; La Monica, N.; Kandimalla, E.R.; Quax, P.H.; et al. Inhibition of 14q32 MicroRNAs miR-329, miR-487b, miR-494, and miR-495 Increases Neovascularization and Blood Flow Recovery after Ischemia. Circ. Res. 2014, 115, 696–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Downie Ruiz Velasco, A.; Welten, S.M.J.; Goossens, E.A.C.; Quax, P.H.A.; Rappsilber, J.; Michlewski, G.; Nossent, A.Y. Posttranscriptional Regulation of 14q32 MicroRNAs by the CIRBP and HADHB during Vascular Regeneration after Ischemia. Mol. Ther. Nucleic Acids 2019, 14, 329–338. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Luo, Y.; Duan, H.; Xing, S.; Zhang, J.; Lu, D.; Feng, J.; Yang, D.; Song, L.; Yan, X. MicroRNA 329 Suppresses Angiogenesis by Targeting CD146. Mol. Cell. Biol. 2013, 33, 3689–3699. [Google Scholar] [CrossRef] [Green Version]
- Jiang, T.; Zhuang, J.; Duan, H.; Luo, Y.; Zeng, Q.; Fan, K.; Yan, H.; Lu, D.; Ye, Z.; Hao, J.; et al. CD146 is a coreceptor for VEGFR-2 in tumor angiogenesis. Blood 2012, 120, 2330–2339. [Google Scholar] [CrossRef] [Green Version]
- Kübler, M.; Beck, S.; Fischer, S.; Götz, P.; Kumaraswami, K.; Ishikawa-Ankerhold, H.; Lasch, M.; Deindl, E. Absence of Cold-Inducible RNA-Binding Protein (CIRP) Promotes Angiogenesis and Regeneration of Ischemic Tissue by Inducing M2-like Macrophage Polarization. Biomedicines 2021, 9, 395. [Google Scholar] [CrossRef] [PubMed]
- Limbourg, A.; Korff, T.; Napp, L.C.; Schaper, W.; Drexler, H.; Limbourg, F. Evaluation of postnatal arteriogenesis and angiogenesis in a mouse model of hind-limb ischemia. Nat. Protoc. 2009, 4, 1737–1748. [Google Scholar] [CrossRef] [PubMed]
- Baum, O.; Olfert, I.M.; Egginton, S.; Hellsten, Y. Advances and challenges in skeletal muscle angiogenesis. Am. J. Physiol. Heart Circ. Physiol. 2016, 310, H326–H336. [Google Scholar] [CrossRef]
- Chen, K.; Murao, A.; Arif, A.; Takizawa, S.; Jin, H.; Jiang, J.; Aziz, M.; Wang, P. Inhibition of Efferocytosis by Extracellular CIRP–Induced Neutrophil Extracellular Traps. J. Immunol. 2020, 206, 797–806. [Google Scholar] [CrossRef] [PubMed]
- Aziz, M.; Brenner, M.; Wang, P. Extracellular CIRP (eCIRP) and inflammation. J. Leukoc. Biol. 2019, 106, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Zhong, P.; Huang, H. Recent progress in the research of cold-inducible RNA-binding protein. Future Sci. OA 2017, 3, FSO246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seignez, C.; Phillipson, M. The multitasking neutrophils and their involvement in angiogenesis. Curr. Opin. Hematol. 2017, 24, 3–8. [Google Scholar] [CrossRef]
- Wang, J. Neutrophils in tissue injury and repair. Cell Tissue Res. 2018, 371, 531–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castanheira, F.V.S.; Kubes, P. Neutrophils and NETs in modulating acute and chronic inflammation. Blood 2019, 133, 2178–2185. [Google Scholar] [CrossRef]
- Preissner, K.T.; Fischer, S.; Deindl, E. Extracellular RNA as a Versatile DAMP and Alarm Signal That Influences Leukocyte Recruitment in Inflammation and Infection. Front. Cell Dev. Biol. 2020, 8. [Google Scholar] [CrossRef]
- Wang, J.; Hossain, M.; Thanabalasuriar, A.; Gunzer, M.; Meininger, C.; Kubes, P. Visualizing the function and fate of neutrophils in sterile injury and repair. Science 2017, 358, 111–116. [Google Scholar] [CrossRef] [Green Version]
- Christoffersson, G.; Henriksnäs, J.; Johansson, L.; Rolny, C.; Ahlström, H.; Caballero-Corbalan, J.; Segersvärd, R.; Permert, J.; Korsgren, O.; Carlsson, P.O.; et al. Clinical and experimental pancreatic islet transplantation to striated muscle: Establishment of a vascular system similar to that in native islets. Diabetes 2010, 59, 2569–2578. [Google Scholar] [CrossRef] [Green Version]
- Christoffersson, G.; Vågesjö, E.; Vandooren, J.; Lidén, M.; Massena, S.; Reinert, R.; Brissova, M.; Powers, A.C.; Opdenakker, G.; Phillipson, M. VEGF-A recruits a proangiogenic MMP-9–delivering neutrophil subset that induces angiogenesis in transplanted hypoxic tissue. Blood 2012, 120, 4653–4662. [Google Scholar] [CrossRef]
- Kolaczkowska, E.; Kubes, P. Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 2013, 13, 159–175. [Google Scholar] [CrossRef] [PubMed]
- Pizza, F.X.; Peterson, J.M.; Baas, J.H.; Koh, T.J. Neutrophils contribute to muscle injury and impair its resolution after lengthening contractions in mice. J. Physiol. 2005, 562 Pt 3, 899–913. [Google Scholar] [CrossRef]
- Toumi, H.; F’Guyer, S.; Best, T.M. The role of neutrophils in injury and repair following muscle stretch. J. Anat. 2006, 208, 459–470. [Google Scholar] [CrossRef]
- Teixeira, C.; Zamuner, S.; Zuliani, J.P.; Fernandes, C.M.; Cruz-Hofling, M.A.; Fernandes, I.; Chaves, F.; Gutiérrez, J.M. Neutrophils do not contribute to local tissue damage, but play a key role in skeletal muscle regeneration, in mice injected withBothrops aspersnake venom. Muscle Nerve 2003, 28, 449–459. [Google Scholar] [CrossRef]
- De Oliveira, S.; Rosowski, E.E.; Huttenlocher, A. Neutrophil migration in infection and wound repair: Going forward in reverse. Nat. Rev. Immunol. 2016, 16, 378–391. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Aziz, M.; Ode, Y.; Wang, P. CIRP Induces Neutrophil Reverse Transendothelial Migration in Sepsis. Shock 2019, 51, 548–556. [Google Scholar] [CrossRef] [PubMed]
- Denning, N.-L.; Aziz, M.; Gurien, S.D.; Wang, P. DAMPs and NETs in Sepsis. Front. Immunol. 2019, 10, 2536. [Google Scholar] [CrossRef]
- Lefrancais, E.; Mallavia, B.; Zhuo, H.; Calfee, C.S.; Looney, M.R. Maladaptive role of neutrophil extracellular traps in pathogen-induced lung injury. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- Wong, S.L.; Demers, M.; Martinod, K.; Gallant, M.; Wang, Y.; Goldfine, A.B.; Kahn, C.R.; Wagner, D.D. Diabetes primes neutrophils to undergo NETosis, which impairs wound healing. Nat. Med. 2015, 21, 815–819. [Google Scholar] [CrossRef] [Green Version]
- Saffarzadeh, M.; Juenemann, C.; Queisser, M.A.; Lochnit, G.; Barreto, G.; Galuska, S.P.; Lohmeyer, J.; Preissner, K.T. Neutrophil Extracellular Traps Directly Induce Epithelial and Endothelial Cell Death: A Predominant Role of Histones. PLoS ONE 2012, 7, e32366. [Google Scholar] [CrossRef] [PubMed]
- Slaba, I.; Wang, J.; Kolaczkowska, E.; McDonald, B.; Lee, W.-Y.; Kubes, P. Imaging the dynamic platelet-neutrophil response in sterile liver injury and repair in mice. Hepatology 2015, 62, 1593–1605. [Google Scholar] [CrossRef] [Green Version]
- Mutua, V.; Gershwin, L.J. A Review of Neutrophil Extracellular Traps (NETs) in Disease: Potential Anti-NETs Therapeutics. Clin. Rev. Allergy Immunol. 2020, 61, 194–211. [Google Scholar] [CrossRef] [PubMed]
- Greenlee-Wacker, M.C. Clearance of apoptotic neutrophils and resolution of inflammation. Immunol. Rev. 2016, 273, 357–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duffield, J.S.; Forbes, S.; Constandinou, C.M.; Clay, S.; Partolina, M.; Vuthoori, S.; Wu, S.; Lang, R.; Iredale, J.P. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J. Clin. Investig. 2005, 115, 56–65. [Google Scholar] [CrossRef] [Green Version]
- Lucas, T.; Waisman, A.; Ranjan, R.; Roes, J.; Krieg, T.; Müller, W.; Roers, A.; Eming, S.A. Differential Roles of Macrophages in Diverse Phases of Skin Repair. J. Immunol. 2010, 184, 3964–3977. [Google Scholar] [CrossRef] [Green Version]
- Hong, H.; Tian, X.Y. The Role of Macrophages in Vascular Repair and Regeneration after Ischemic Injury. Int. J. Mol. Sci. 2020, 21, 6328. [Google Scholar] [CrossRef]
- Murray, P.J. Macrophage Polarization. Annu. Rev. Physiol. 2017, 79, 541–566. [Google Scholar] [CrossRef]
- Gordon, S.; Taylor, P. Monocyte and macrophage heterogeneity. Nat. Rev. Immunol. 2005, 5, 953–964. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A.; Vannella, K.M. Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity 2016, 44, 450–462. [Google Scholar] [CrossRef] [Green Version]
- Gurevich, D.; Severn, C.; Twomey, C.; Greenhough, A.; Cash, J.; Toye, A.M.; Mellor, H.; Martin, P. Live imaging of wound angiogenesis reveals macrophage orchestrated vessel sprouting and regression. EMBO J. 2018, 37. [Google Scholar] [CrossRef]
- Zhang, J.; Muri, J.; Fitzgerald, G.; Gorski, T.; Gianni-Barrera, R.; Masschelein, E.; D’Hulst, G.; Gilardoni, P.; Turiel, G.; Fan, Z.; et al. Endothelial Lactate Controls Muscle Regeneration from Ischemia by Inducing M2-like Macrophage Polarization. Cell Metab. 2020, 31, 1136–1153.e7. [Google Scholar] [CrossRef]
- Willenborg, S.; Lucas, T.; Van Loo, G.; Knipper, J.; Krieg, T.; Haase, I.; Brachvogel, B.; Hammerschmidt, M.; Nagy, A.; Ferrara, N.; et al. CCR2 recruits an inflammatory macrophage subpopulation critical for angiogenesis in tissue repair. Blood 2012, 120, 613–625. [Google Scholar] [CrossRef] [Green Version]
- Dort, J.; Fabre, P.; Molina, T.; Dumont, N.A. Macrophages Are Key Regulators of Stem Cells during Skeletal Muscle Regeneration and Diseases. Stem Cells Int. 2019, 2019, 4761427. [Google Scholar] [CrossRef]
- Zajac, E.; Schweighofer, B.; Kupriyanova, T.A.; Juncker-Jensen, A.; Minder, P.; Quigley, J.P.; Deryugina, E.I. Angiogenic capacity of M1- and M2-polarized macrophages is determined by the levels of TIMP-1 complexed with their secreted proMMP-9. Blood 2013, 122, 4054–4067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, S.; Martinez, F.O. Alternative Activation of Macrophages: Mechanism and Functions. Immunity 2010, 32, 593–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deshmane, S.L.; Kremlev, S.; Amini, S.; Sawaya, B.E. Monocyte Chemoattractant Protein-1 (MCP-1): An Overview. J. Interf. Cytokine Res. 2009, 29, 313–326. [Google Scholar] [CrossRef]
- Nio, Y.; Yamauchi, T.; Okada-Iwabu, M.; Funata, M.; Yamaguchi, M.; Ueki, K.; Kadowaki, T. Monocyte chemoattractant protein-1 (MCP-1) deficiency enhances alternatively activated M2 macrophages and ameliorates insulin resistance and fatty liver in lipoatrophic diabetic A-ZIP transgenic mice. Diabetologia 2012, 55, 3350–3358. [Google Scholar] [CrossRef] [Green Version]
- Fang, Z.; Wu, D.; Deng, J.; Yang, Q.; Zhang, X.; Chen, J.; Wang, S.; Hu, S.; Hou, W.; Ning, S.; et al. An MD2-perturbing peptide has therapeutic effects in rodent and rhesus monkey models of stroke. Sci. Transl. Med. 2021, 13, eabb6716. [Google Scholar] [CrossRef]
- Troidl, K.; Schubert, C.; Vlacil, A.-K.; Chennupati, R.; Koch, S.; Schütt, J.; Oberoi, R.; Schaper, W.; Schmitz-Rixen, T.; Schieffer, B.; et al. The Lipopeptide MALP-2 Promotes Collateral Growth. Cells 2020, 9, 997. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kübler, M.; Beck, S.; Peffenköver, L.L.; Götz, P.; Ishikawa-Ankerhold, H.; Preissner, K.T.; Fischer, S.; Lasch, M.; Deindl, E. The Absence of Extracellular Cold-Inducible RNA-Binding Protein (eCIRP) Promotes Pro-Angiogenic Microenvironmental Conditions and Angiogenesis in Muscle Tissue Ischemia. Int. J. Mol. Sci. 2021, 22, 9484. https://doi.org/10.3390/ijms22179484
Kübler M, Beck S, Peffenköver LL, Götz P, Ishikawa-Ankerhold H, Preissner KT, Fischer S, Lasch M, Deindl E. The Absence of Extracellular Cold-Inducible RNA-Binding Protein (eCIRP) Promotes Pro-Angiogenic Microenvironmental Conditions and Angiogenesis in Muscle Tissue Ischemia. International Journal of Molecular Sciences. 2021; 22(17):9484. https://doi.org/10.3390/ijms22179484
Chicago/Turabian StyleKübler, Matthias, Sebastian Beck, Lisa Lilian Peffenköver, Philipp Götz, Hellen Ishikawa-Ankerhold, Klaus T. Preissner, Silvia Fischer, Manuel Lasch, and Elisabeth Deindl. 2021. "The Absence of Extracellular Cold-Inducible RNA-Binding Protein (eCIRP) Promotes Pro-Angiogenic Microenvironmental Conditions and Angiogenesis in Muscle Tissue Ischemia" International Journal of Molecular Sciences 22, no. 17: 9484. https://doi.org/10.3390/ijms22179484