
Myelinosome Organelles in the Retina of R6/1 Huntington Disease (HD) Mice: Ubiquitous Distribution and Possible Role in Disease Spreading

 , ,
, ,
Abstract
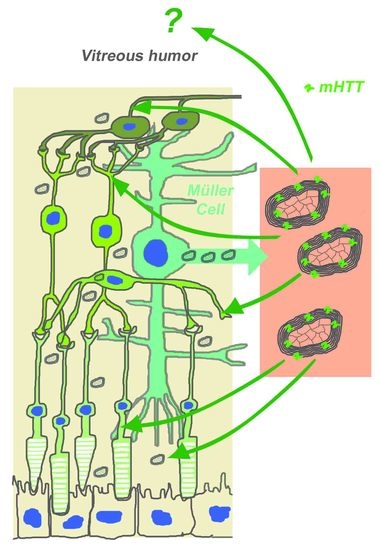
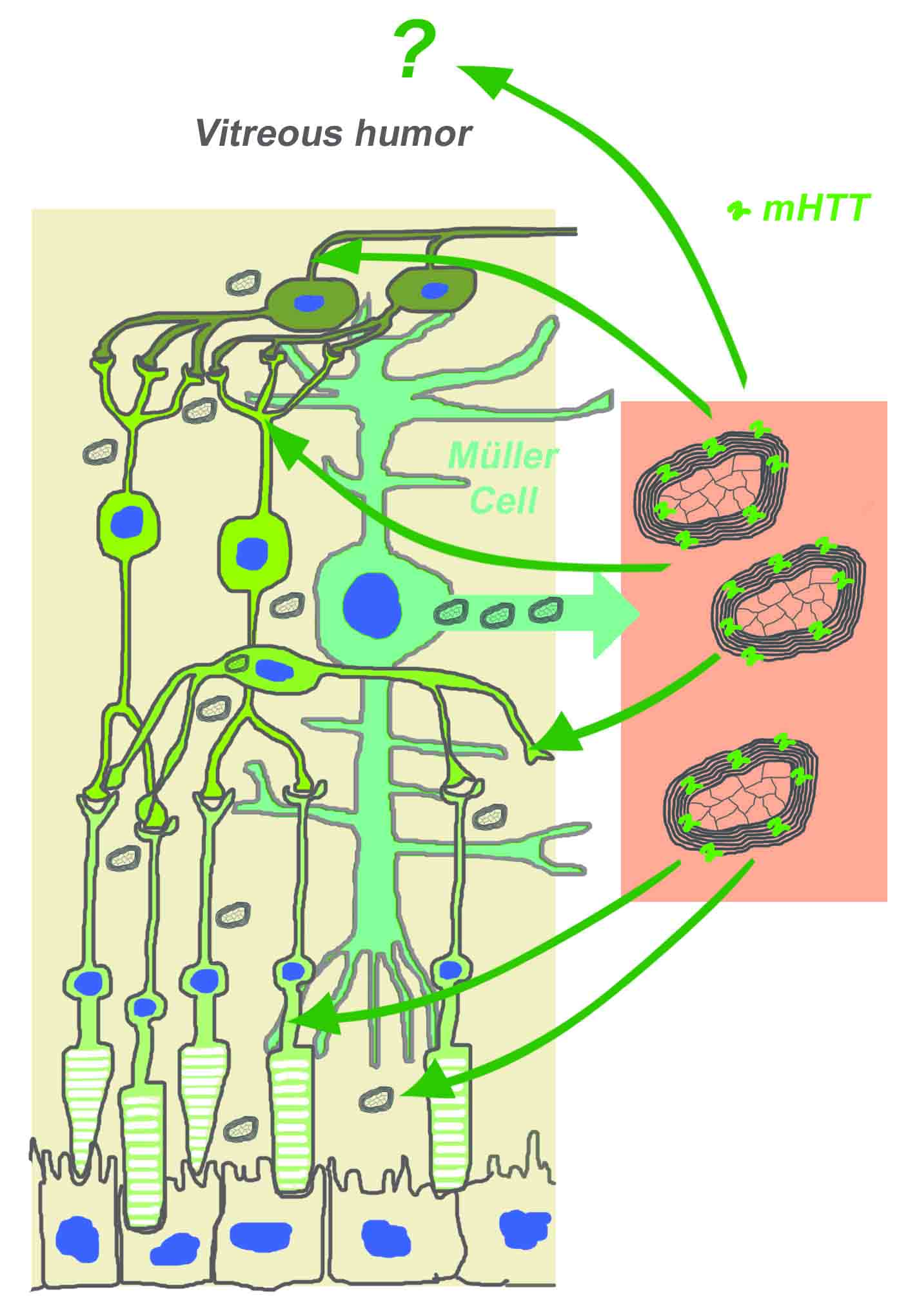
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Transgenic R6/1 Mice Retina Undergoes Remodeling without Cell Loss
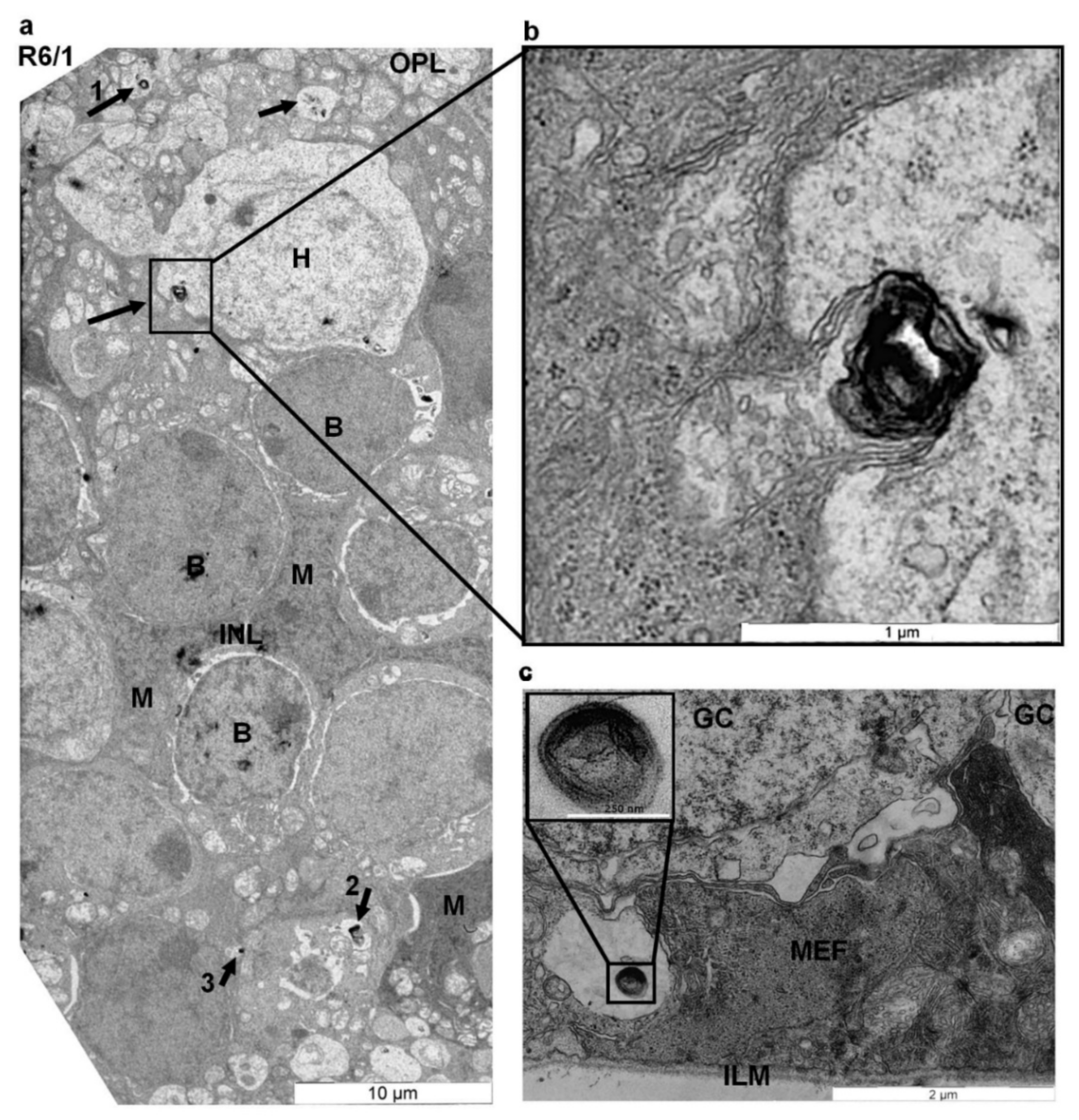
2.2. Myelinosome Organelles Are Present in All Layers of R6/1 Mice Retina
2.3. Myelinosomes Are Released from MIO-M1 Müller Cells Transfected with EGFP-mHTT-exon1
2.4. Myelinosomes Are Incorporated into Human Neuroblastoma SH-SY5Y Cells
2.5. Macropinocytosis Does Not Support the Incorporation of Myelinosomes into Human Neuroblastoma SH-SY5Y Cells
2.6. Incorporation of Myelinosomes into Human Neuroblastoma SH-SY5Y Cells Is Inhibited by Synthetic Drug MDL 28170
3. Discussion
4. Materials and Methods
4.1. Animals and Cells
4.2. Chemicals and Plasmids
4.3. Transient Transfection
4.4. Differential Centrifugation of Culture Media
4.5. Treatment of Cells with Myelinosomes
4.6. Treatment of Cells with Inhibitors of Macropinocytosis and Membrane Fusion
4.7. Western Blotting
4.8. Immunocytochemistry
4.9. Confocal Microscopy, Videomicroscopy, and Image Analysis
4.10. Electron Microscopy Studies
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- London, A.; Benhar, I.; Schwartz, M. The retina as a window to the brain-from eye research to CNS disorders. Nat. Rev. Neurol. 2013, 91, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Toldi, J.; Vécsei, L. Exploring the Etiological Links behind Neurodegenerative Diseases: Inflammatory Cytokines and Bioactive Kynurenines. Int. J. Mol. Sci. 2020, 21, 2431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tai, Y.F.; Pavese, N.; Gerhard, A.; Tabrizi, S.J.; Barker, R.A.; Brooks, D.J.; Piccini, P. Imaging microglial activation in Huntington’s disease. Brain Res. Bull. 2007, 72, 148–151. [Google Scholar] [CrossRef] [PubMed]
- Silvestroni, A.; Faull, R.L.; Strand, A.D.; Möller, T. Distinct neuroinflammatory profile in post-mortem human Huntington’s disease. Neuroreport 2009, 20, 1098–1103. [Google Scholar] [CrossRef]
- Ross, C.A.; Tabrizi, S.J. Huntington’s disease: From molecular pathogenesis to clinical treatment. Lancet Neurol. 2011, 10, 83–98. [Google Scholar] [CrossRef]
- Soulet, D.; Cicchetti, F. The role of immunity in Huntington’s disease. Mol. Psychiatry 2011, 16, 889–902. [Google Scholar] [CrossRef]
- Júlio, F.; Blanco, R.; Casanova, J.P.; D’Alessio, B.; De Schepper, B.; De Sousa, D.; De Sousa, P.; Ferreira, C.; Gommans, H.; Haselberg, R.; et al. Perceptions about Research Participation among Individuals at Risk and Individuals with Premanifest Huntington’s Disease: A Survey Conducted by the European Huntington Association. J. Pers. Med. 2021, 11, 815. [Google Scholar] [CrossRef]
- Frank, S. Treatment of Huntington’s disease. Neurotherapeutics 2014, 11, 153–160. [Google Scholar] [CrossRef] [Green Version]
- Bachoud-Lévi, A.C.; Ferreira, J.; Massart, R.; Youssov, K.; Rosser, A.; Busse, M.; Craufurd, D.; Reilmann, R.; De Michele, G.; Rae, D.; et al. International Guidelines for the Treatment of Huntington’s Disease. Front. Neurol. 2019, 10, 710. [Google Scholar] [CrossRef] [Green Version]
- Kay, C.; Hayden, M.R.; Leavitt, B.R. Epidemiology of Huntington Disease. Handb. Clin. Neurol. 2017, 144, 31–46. [Google Scholar] [CrossRef]
- Török, N.; Tanaka, M.; Vécsei, L. Searching for Peripheral Biomarkers in Neurodegenerative Diseases: The Tryptophan-Kynurenine Metabolic Pathway. Int. J. Mol. Sci. 2020, 21, 9338. [Google Scholar] [CrossRef]
- Paulson, H. Repeat expansion diseases. Handb. Clin. Neurol. 2018, 147, 105–123. [Google Scholar] [CrossRef]
- Andrade, M.A.; Bork, P. HEAT repeats in the Huntington’s disease protein. Nat. Genet. 1995, 11, 115–116. [Google Scholar] [CrossRef]
- The Huntington’s Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 1993, 72, 971–983. [Google Scholar] [CrossRef]
- Yamanishi, E.; Hasegawa, K.; Fujita, K.; Ichinose, S.; Yagishita, S.; Murata, M.; Tagawa, K.; Akashi, T.; Eishi, Y.; Okazawa, H. A novel form of necrosis, TRIAD, occurs in human Huntington’s disease. Acta Neuropathol. Commun. 2017, 5, 19. [Google Scholar] [CrossRef] [Green Version]
- Pecho-Vrieseling, E.; Rieker, C.; Fuchs, S.; Bleckmann, D.; Esposito, M.S.; Botta, P.; Goldstein, C.; Bernhard, M.; Galimberti, I.; Müller, M.; et al. Transneuronal propagation of mutant huntingtin contributes to non-cell autonomous pathology in neurons. Nat. Neurosci. 2014, 17, 1064–1072. [Google Scholar] [CrossRef]
- Kersten, H.M.; Danesh-Meyer, H.V.; Kilfoyle, D.H.; Roxburgh, R.H. Optical coherence tomography findings in Huntington’s disease: A potential bio-marker of disease progression. J. Neurol. 2015, 262, 2457–2465. [Google Scholar] [CrossRef]
- Andrade, C.; Beato, J.; Monteiro, A.; Costa, A.; Penas, S.; Guimarães, J.; Reis, F.F.; Garrett, C. Spectral-Domain Optical Coherence Tomography as a Potential Biomarker in Huntington’s Disease. Mov. Disord. 2016, 31, 377–383. [Google Scholar] [CrossRef]
- Dhalla, A.; Pallikadavath, S.; Hutchinson, C.V. Visual Dysfunction in Huntington’s Disease: A Systematic Review. J. Huntingtons Dis. 2019, 8, 233–242. [Google Scholar] [CrossRef] [Green Version]
- Paulus, W.; Schwarz, G.; Werner, A.; Lange, H.; Bayer, A.; Hofschuster, M.; Müller, N.; Zrenner, E. Impairment of retinal increment thresholds 515 in Huntington’s disease. Ann. Neurol. 1993, 34, 574–578. [Google Scholar] [CrossRef]
- Jackson, G.R.; Salecker, I.; Dong, X.; Yao, X.; Arnheim, N.; Faber, P.W.; MacDonald, M.E.; Zipursky, S.L. Polyglutamine-expanded human huntingtin transgenes induce degeneration of Drosophila photoreceptor neurons. Neuron 1998, 21, 633–642. [Google Scholar] [CrossRef] [Green Version]
- Helmlinger, D.; Yvert, G.; Picaud, S.; Merienne, K.; Sahel, J.; Mandel, J.L.; Devys, D. Progressive retinal degeneration and dysfunction in R6 Huntington’s disease mice. Hum. Mol. Genet. 2002, 11, 3351–3359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrasch-Parwez, E.; Habbes, H.W.; Weickert, S.; Löbbecke-Schumacher, M.; Striedinger, K.; Wieczorek, S.; Dermietzel, R.; Epplen, J.T. Fine-structural analysis and connexin expression in the retina of a transgenic model of Huntington’s disease. J. Comp. Neurol. 2004, 479, 181–197. [Google Scholar] [CrossRef] [PubMed]
- Petrasch-Parwez, E.; Saft, C.; Schlichting, A.; Andrich, J.; Napirei, M.; Arning, L.; Wieczorek, S.; Dermietzel, R.; Epplen, J.T. Is the retina affected in Huntington disease? Acta Neuropathol. 2005, 110, 523–525. [Google Scholar] [CrossRef]
- Batcha, A.H.; Greferath, U.; Jobling, A.I.; Vessey, K.A.; Ward, M.M.; Nithianantharajah, J.; Hannan, A.J.; Kalloniatis, M.; Fletcher, E.L. Retinal dysfunction, photoreceptor protein dysregulation and neuronal remodelling in the R6/1 mouse model of Huntington’s disease. Neurobiol. Dis. 2012, 45, 887–896. [Google Scholar] [CrossRef]
- Ragauskas, S.; Leinonen, H.; Puranen, J.; Rönkkö, S.; Nymark, S.; Gurevicius, K.; Lipponen, A.; Kontkanen, O.; Puoliväli, J.; Tanila, H.; et al. Early retinal function deficit without prominent morphological changes in the R6/2 mouse model of Huntington’s disease. PLoS ONE 2014, 9, e113317. [Google Scholar] [CrossRef]
- Mangiarini, L.; Sathasivam, K.; Seller, M.; Cozens, B.; Harper, A.; Hetherington, C.; Lawton, M.; Trottier, Y.; Lehrach, H.; Davies, S.W.; et al. Exon1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 1996, 87, 493–506. [Google Scholar] [CrossRef] [Green Version]
- Li, J.Y.; Popovic, N.; Brundin, P. The use of the R6 transgenic mouse models of Huntington’s disease in attempts to develop novel therapeutic strategies. NeuroRX 2005, 2, 447–464. [Google Scholar] [CrossRef] [Green Version]
- Pouladi, M.A.; Morton, A.J.; Hayden, M.R. Choosing an animal model for the study of Huntington’s disease. Nat. Rev. Neurosci. 2013, 14, 708–721. [Google Scholar] [CrossRef]
- Nakanishi, Y.; Nagaosa, K.; Shiratsuchi, A. Phagocytic removal of cells that have become unwanted: Implications for animal development and tissue homeostasis. Dev. Growth Differ. 2011, 53, 149–160. [Google Scholar] [CrossRef] [Green Version]
- Penberthy, K.K.; Lysiak, J.J.; Ravichandran, K.S. Rethinking Phagocytes: Clues from the Retina and Testes. Trends Cell. Biol. 2018, 28, 317–327. [Google Scholar] [CrossRef]
- Yefimova, M.G.; Ravel, C.; Rolland, A.D.; Bourmeyster, N.; Jégou, B. MERTK-Mediated LC3-Associated Phagocytosis (LAP) of Apoptotic Substrates in Blood-Separated Tissues: Retina, Testis, Ovarian Follicles. Cells 2021, 10, 1443. [Google Scholar] [CrossRef]
- Campbell, M.; Humphries, P. The blood-retina barrier: Tight junctions and barrier modulation. Adv. Exp. Med. Biol. 2012, 763, 70–84. [Google Scholar]
- Mruk, D.D.; Cheng, C.Y. The Mammalian Blood-Testis Barrier: Its Biology and Regulation. Endocr. Rev. 2015, 36, 564–591. [Google Scholar] [CrossRef]
- Taylor, A.W. Ocular immune privilege. Eye 2009, 23, 1885–1889. [Google Scholar] [CrossRef] [Green Version]
- Fijak, M.; Meinhardt, A. The testis in immune privilege. Immunol. Rev. 2006, 213, 66–81. [Google Scholar] [CrossRef]
- Tosini, G.; Pozdeyev, N.; Sakamoto, K.; Iuvone, P.M. The circadian clock system in the mammalian retina. BioEssays 2008, 30, 624–633. [Google Scholar] [CrossRef] [Green Version]
- Jégou, B. The Sertoli-germ cell communication network in mammals. Int. Rev. Cytol. 1993, 147, 25–96. [Google Scholar]
- Yefimova, M.G.; Messaddeq, N.; Harnois, T.; Meunier, A.C.; Clarhaut, J.; Noblanc, A.; Weickert, J.L.; Cantereau, A.; Philippe, M.; Bourmeyster, N.; et al. A chimerical phagocytosis model reveals the recruitment by Sertoli cells of autophagy for the degradation of ingested illegitimate substrates. Autophagy 2013, 9, 653–666. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.Y.; Zhao, H.; Martinez, J.; Doggett, T.A.; Kolesnikov, A.V.; Tang, P.H.; Ablonczy, Z.; Chan, C.C.; Zhou, Z.; Green, D.R.; et al. Noncanonical autophagy promotes the visual cycle. Cell 2013, 154, 365–376. [Google Scholar] [CrossRef] [Green Version]
- Fliesler, S.J.; Anderson, R.E. Chemistry and metabolism of lipids in the vertebrate retina. Prog. Lipid. Res. 1983, 22, 79–131. [Google Scholar] [CrossRef]
- Wathes, D.C.; Abayasekara, D.R.; Aitken, R.J. Polyunsaturated fatty acids in male and female reproduction. Biol. Reprod. 2007, 77, 190–201. [Google Scholar] [CrossRef] [PubMed]
- Yefimova, M.G.; Béré, E.; Cantereau-Becq, A.; Harnois, T.; Meunier, A.C.; Messaddeq, N.; Becq, F.; Trottier, Y.; Bourmeyster, N. Myelinosomes act as natural secretory organelles in Sertoli cells to prevent accumulation of aggregate-prone mutant Huntingtin and CFTR. Hum. Mol. Genet. 2016, 25, 4170–4185. [Google Scholar] [CrossRef] [PubMed]
- Yefimova, M.; Ravel, C.; Neyroud, A.S.; Béré, E.; Bourmeyster, N. Myelinosomes: A new pathway of protein quality control. Med. Sci. 2020, 36, 1012–1017. [Google Scholar] [CrossRef]
- Ghadially, F.N. Ultrastructural Pathology of the Cell and Matrix, 4th ed.; Ghadially, Butterworth–Heinemann: Boston, MA, USA, 1997; pp. 646–659. [Google Scholar]
- Romanelli, E.; Merkler, D.; Mezydlo, A.; Weil, M.T.; Weber, M.S.; Nikić, I.; Potz, S.; Meinl, E.; Matznick, F.E.H.; Kreutzfeldt, M.; et al. Myelinosome formation represents an early stage of oligodendrocyte damage in multiple sclerosis and its animal model. Nat. Commun. 2016, 7, 13275. [Google Scholar] [CrossRef] [Green Version]
- Matthews, G.; Fuchs, P. The diverse roles of ribbon synapses in sensory neurotransmission. Nat. Rev. Neurosci. 2010, 11, 812–822. [Google Scholar] [CrossRef]
- Kerr, J.F.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef] [Green Version]
- Remington, L.A. Clinical Anatomy and Physiology of the Visual System, 3rd ed.; Remington, Butterworth-Heinemann: Boston, MA, USA, 2012; pp. 61–92. [Google Scholar]
- Jeon, C.J.; Strettoi, E.; Masland, R.H. The major cell populations of the mouse retina. J. Neurosci. 1998, 18, 8936–8946. [Google Scholar] [CrossRef] [Green Version]
- Limb, G.A.; Salt, T.E.; Munro, P.M.; Moss, S.E.; Khaw, P.T. In vitro characterization of a spontaneously immortalized human Müller cell line (MIO-M1). Investig. Ophthalmol. Vis. Sci. 2002, 43, 864–869. [Google Scholar]
- Li, S.H.; Li, X.J. Huntingtin-protein interactions and the pathogenesis of Huntington’s disease. Trends Genet. 2004, 20, 146–154. [Google Scholar] [CrossRef]
- Nakamura, K.C.; Kameda, H.; Koshimizu, Y.; Yanagawa, Y.; Kaneko, T. Production and histological application of affinity-purified antibodies to heat-denatured green fluorescent protein. J. Histochem. Cytochem. 2008, 56, 647–657. [Google Scholar] [CrossRef] [Green Version]
- Toimela, T.; Mäenpää, H.; Mannerström, M.; Tähti, H. Development of an in vitro blood-brain barrier model-cytotoxicity of mercury and aluminum. Toxicol. Appl. Pharmacol. 2004, 195, 73–82. [Google Scholar] [CrossRef]
- Chen, D.W.; Foldvari, M. In vitro bioassay model for screening non-viral neurotrophic factor gene delivery systems for glaucoma treatment. Drug Deliv. Transl. Res. 2016, 6, 676–685. [Google Scholar] [CrossRef]
- Juenemann, K.; Wiemhoefer, A.; Reits, E.A. Detection of ubiquitinated huntingtin species in intracellular aggregates. Front. Mol. Neurosci. 2015, 8, 1. [Google Scholar] [CrossRef]
- Herndon, E.S.; Hladik, C.L.; Shang, P.; Burns, D.K.; Raisanen, J.; White, C.L. 3rd Neuroanatomic profile of polyglutamine immunoreactivity in Huntington disease brains. J. Neuropathol. Exp. Neurol. 2009, 68, 250–261. [Google Scholar] [CrossRef] [Green Version]
- Klein, F.A.; Zeder-Lutz, G.; Cousido-Siah, A.; Mitschler, A.; Katz, A.; Eberling, P.; Mandel, J.L.; Podjarny, A.; Trottier, Y. Linear and extended: A common polyglutamine conformation recognized by the three antibodies MW1, 1C2 and 3B5H10. Hum. Mol. Genet. 2013, 22, 4215–4223. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.E.; Hosp, F.; Frottin, F.; Ge, H.; Mann, M.; Hayer-Hartl, M.; Hartl, F.U. Soluble Oligomers of PolyQ-Expanded Huntingtin Target a Multiplicity of Key Cellular Factors. Mol. Cell 2016, 63, 951–964. [Google Scholar] [CrossRef] [Green Version]
- Nara, A.; Aki, T.; Funakoshi, T.; Unuma, K.; Uemura, K. Hyperstimulation of macropinocytosis leads to lysosomal dysfunction during exposure to methamphetamine in SH-SY5Y cells. Brain Res. 2012, 1466, 1–14. [Google Scholar] [CrossRef]
- Peng, H.; Park, J.K.; Lavker, R.M. Autophagy and Macropinocytosis: Keeping an Eye on the Corneal/Limbal Epithelia. Investig. Ophthalmol. Vis. Sci. 2017, 58, 416–423. [Google Scholar] [CrossRef] [Green Version]
- Koivusalo, M.; Welch, C.; Hayashi, H.; Scott, C.C.; Kim, M.; Alexander, T.; Touret, N.; Hahn, K.M.; Grinstein, S. Amiloride inhibits macropinocytosis by lowering submembranous pH and preventing Rac1 and Cdc42 signaling. J. Cell Biol. 2010, 188, 547–563. [Google Scholar] [CrossRef] [Green Version]
- Grimmer, S.; Van Deurs, B.; Sandvig, K. Membrane ruffling and macropinocytosis in A431 cells require cholesterol. J. Cell Sci. 2002, 115, 2953–2962. [Google Scholar] [CrossRef] [PubMed]
- Kasson, P.M.; Pande, V.S. Control of membrane fusion mechanism by lipid composition: Predictions from ensemble molecular dynamics. PLoS Comput. Biol. 2007, 3, e220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domanska, M.K.; Kiessling, V.; Stein, A.; Fasshauer, D.; Tamm, L.K. Single vesicle millisecond fusion kinetics reveals number of SNARE complexes optimal for fast SNARE-mediated membrane fusion. J. Biol. Chem. 2009, 284, 32158–32166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwak, K.B.; Kambayashi, J.; Kang, M.S.; Ha, D.B.; Chung, C.H. Cell-penetrating inhibitors of calpain block both membrane fusion and filamin cleavage in chick embryonic myoblasts. FEBS Lett. 1993, 323, 151–154. [Google Scholar] [CrossRef] [Green Version]
- Jones, B.W.; Watt, C.B.; Frederick, J.M.; Baehr, W.; Chen, C.K.; Levine, E.M.; Milam, A.H.; Lavail, M.M.; Marc, R.E. Retinal remodeling triggered by photoreceptor degenerations. J. Comp. Neurol. 2003, 464, 1–16. [Google Scholar] [CrossRef]
- Marc, R.E.; Jones, B.W.; Watt, C.B.; Strettoi, E. Neural remodeling in retinal degeneration. Prog. Retin. Eye Res. 2003, 22, 607–655. [Google Scholar] [CrossRef]
- Yefimova, M.G.; Messaddeq, N.; Karam, A.; Jacquard, C.; Weber, C.; Jonet, L.; Wolfrum, U.; Jeanny, J.C.; Trottier, Y. Polyglutamine toxicity induces rod photoreceptor division, morphological transformation or death in Spinocerebellar ataxia 7 mouse retina. Neurobiol. Dis. 2010, 40, 311–324. [Google Scholar] [CrossRef]
- Niewiadomska-Cimicka, A.; Trottier, Y. Molecular Targets and Therapeutic Strategies in Spinocerebellar Ataxia Type 7. Neurotherapeutics 2019, 16, 1074–1096. [Google Scholar] [CrossRef]
- Yvert, G.; Lindenberg, K.S.; Picaud, S.; Landwehrmeyer, G.B.; Sahel, J.A.; Mandel, J.L. Expanded polyglutamines induce neurodegeneration and trans-neuronal alterations in cerebellum and retina of SCA7 transgenic mice. Hum. Mol. Genet. 2000, 9, 2491–2506. [Google Scholar] [CrossRef] [Green Version]
- Nagar, S.; Krishnamoorthy, V.; Cherukuri, P.; Jain, V.; Dhingra, N.K. Early remodeling in an inducible animal model of retinal degeneration. Neuroscience 2009, 160, 517–529. [Google Scholar] [CrossRef]
- Krishnamoorthy, V.; Cherukuri, P.; Poria, D.; Goel, M.; Dagar, S.; Dhingra, N.K. Retinal Remodeling: Concerns, Emerging Remedies and Future Prospects. Front. Cell Neurosci. 2016, 10, 38. [Google Scholar] [CrossRef] [Green Version]
- Denlinger, B.; Helft, Z.; Telias, M.; Lorach, H.; Palanker, D.; Kramer, R.H. Local photoreceptor degeneration causes local pathophysiological remodeling of retinal neurons. JCI Insight 2020, 5, e132114. [Google Scholar] [CrossRef]
- FitzGibbon, T.; Nestorovski, Z. Morphological consequences of myelination in the human retina. Exp. Eye Res. 1997, 65, 809–819. [Google Scholar] [CrossRef]
- Grignolo, A.; Orzalesi, N.; Calabria, G.A. Studies on the fine structure and the rhodopsin cycle of the rabbit retina in experimental degeneration induced by sodium iodate. Exp. Eye Res. 1966, 5, 86–97. [Google Scholar] [CrossRef]
- Matsusaka, T. Lamellar bodies in the synaptic cytoplasm of the accessory cone from the chick retina as revealed by electron microscopy. J. Ultrastruct. Res. 1967, 18, 55–70. [Google Scholar] [CrossRef]
- Fouquet, J.P.; Dang, D.C.; Meusy-Dessolle, N. Functional differentiation of Leydig cells in the testis of the fetal monkey (Macaca fascicularis). Ann. Biol. Anim. Biochim. Biophys. 1978, 18, 1205–1221. [Google Scholar] [CrossRef] [Green Version]
- Pack, R.J.; Al-Ugaily, L.H.; Morris, G.; Widdicombe, J.G. The distribution and structure of cells in the tracheal epithelium of the mouse. Cell Tissue Res. 1980, 208, 65–84. [Google Scholar] [CrossRef]
- Van Vorstenbosch, C.J.; Spek, E.; Colenbrander, B.; Wensing, C.J.G. The ultrastructure of normal fetal and neonatal pig testis germ cells and the influence of fetal decapitation on the germ cell development. Development 1987, 99, 553–563. [Google Scholar] [CrossRef]
- Miething, A. Morphological studies on prespermatogonia and pre-Sertoli cells in the testes of 6-to 11-day-old golden hamsters. Anat. Embryol. 1989, 179, 503–510. [Google Scholar] [CrossRef]
- Friedlander, M.; Rosenstrauch, A.; Bedrak, E. Leydig cell differentiation during the reproductive cycle of the seasonal breeder Camelus dromedarius: An ultrastructural analysis. Gen. Comp. Endocrinol. 1994, 55, 1–11. [Google Scholar] [CrossRef]
- Chung, E.Y.; Yang, Y.C.; Kang, H.W.; Choi, K.H.; Jun, J.C.; Lee, K.Y. Ultrastructure of Germ Cells and the Functions of Leydig Cells and Sertoli Cells Associated with Spermatogenesis in Pampus argenteus (Teleostei: Perciformes: Stromateidae). Zool. Stud. 2010, 49, 39–50. [Google Scholar] [CrossRef] [Green Version]
- Yan, X.; Ma, L.; Hovakimyan, M.; Lukas, J.; Wree, A.; Frank, M.; Guthoff, R.; Rolfs, A.; Witt, M.; Luo, J. Defects in the retina of Niemann-pick type C 1 mutant mice. BMC Neurosci. 2014, 15, 126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niewiadomska-Cimicka, A.; Doussau, F.; Perot, J.B.; Roux, M.J.; Keime, C.; Hache, A.; Piguet, F.; Novati, A.; Weber, C.; Yalcin, B.; et al. SCA7 Mouse Cerebellar Pathology Reveals Preferential Downregulation of Key Purkinje Cell-Identity Genes and Shared Disease Signature with SCA1 and SCA2. J. Neurosci. 2021, 41, 4910–4936. [Google Scholar] [CrossRef] [PubMed]
- Prince, J.S.; Kohen, C.; Kohen, E.; Jimenez, J.; Brada, Z. Direct connection between myelinosomes, endoplasmic reticulum and nuclear envelope in mouse hepatocytes grown with the amphiphilic drug, quinacrine. Tissue Cell 1993, 25, 103–110. [Google Scholar] [CrossRef]
- Kowal, J.; Arras, G.; Colombo, M.; Jouve, M.; Morath, J.P.; Primdal-Bengtson, B.; Dingli, F.; Loew, D.; Tkach, M.; Théry, C. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc. Natl. Acad. Sci. USA 2016, 113, E968–E977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith-Dijak, A.I.; Sepers, M.D.; Raymond, L.A. Alterations in synaptic function and plasticity in Huntington disease. J. Neurochem. 2019, 150, 346–365. [Google Scholar] [CrossRef] [PubMed]
- Morgans, C.W. Neurotransmitter release at ribbon synapses in the retina. Immunol. Cell Biol. 2000, 78, 442–446. [Google Scholar] [CrossRef]
- Haass, C.; Selkoe, D.J. Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer’s amyloid beta-peptide. Nat. Rev. Mol. Cell Biol. 2007, 8, 101–112. [Google Scholar] [CrossRef]
- Rieux, M.; Alpaugh, M.; Sciacca, G.; Saint-Pierre, M.; Masnata, M.; Denis, H.L.; Lévesque, S.A.; Herrmann, F.; Bazenet, C.; Garneau, A.P.; et al. Shedding a new light on Huntington’s disease: How blood can both propagate and ameliorate disease pathology. Mol. Psychiatry 2020, 26, 5441–5463. [Google Scholar] [CrossRef]
- Chen, J.; Marks, E.; Lai, B.; Zhang, Z.; Duce, J.A.; Lam, L.Q.; Volitakis, I.; Bush, A.I.; Hersch, S.; Fox, J.H. Iron accumulates in Huntington’s disease neurons: Protection by deferoxamine. PLoS ONE 2013, 8, e77023. [Google Scholar] [CrossRef]
- Yefimova, M.; Bere, E.; Neyroud, A.S.; Jegou, B.; Bourmeyster, N.; Ravel, C. Myelinosome-like vesicles in human seminal plasma: A cryo-electron microscopy study. Cryobiology 2020, 92, 15–20. [Google Scholar] [CrossRef]
- Reichenbach, A.; Bringmann, A. New Functions of Müller Cells. Glia 2013, 61, 651–678. [Google Scholar] [CrossRef]
- Wild, E.J.; Boggio, R.; Langbehn, D.; Robertson, N.; Haider, S.; Miller, J.R.; Zetterberg, H.; Leavitt, B.R.; Kuhn, R.; Tabrizi, S.J.; et al. Quantification of mutant huntingtin protein in cerebrospinal fluid from Huntington’s disease patients. J. Clin. Investig. 2015, 125, 1979–1986. [Google Scholar] [CrossRef]
- Silajdžić, E.; Björkqvist, M. A Critical Evaluation of Wet Biomarkers for Huntington’s Disease: Current Status and Ways Forward. J. Huntingtons Dis. 2018, 7, 109–135. [Google Scholar] [CrossRef] [Green Version]
- Newman, E.; Reichenbach, A. The Müller cell: A functional element of the retina. Trends Neurosci. 1996, 19, 307–312. [Google Scholar] [CrossRef]
- Pearce, M.M.P.; Spartz, E.J.; Hong, W.; Luo, L.; Kopito, R.R. Prion-like transmission of neuronal huntingtin aggregates to Phagocytic Glia in the Drosophila brain. Nat. Commun. 2015, 6, 6768. [Google Scholar] [CrossRef]
- Donnelly, K.M.; DeLorenzo, O.R.; Zaya, A.D.; Pisano, G.E.; Thu, W.M.; Luo, L.; Kopito, R.R.; Panning Pearce, M.M. Phagocytic glia are obligatory intermediates in transmission of mutant huntingtin aggregates across neuronal synapses. eLife 2020, 9, e58499. [Google Scholar] [CrossRef]
- Benraiss, A.; Wang, S.; Herrlinger, S.; Li, X.; Chandler-Militello, D.; Mauceri, J.; Burm, H.B.; Toner, M.; Osipovitch, M.; Xu, Q.J.; et al. Human glia can both induce and rescue aspects of disease phenotype in Huntington disease. Nat. Commun. 2016, 7, 11758. [Google Scholar] [CrossRef]
- Jeon, I.; Cicchetti, F.; Cisbani, G.; Lee, S.; Li, E.; Bae, J.; Lee, N.; Li, L.; Im, W.; Kim, M.; et al. Human-to-mouse prion-like propagation of mutant huntingtin protein. Acta Neuropathol. 2016, 132, 577–592. [Google Scholar] [CrossRef] [Green Version]
- Hong, Y.; Zhao, T.; Li, X.J.; Li, S. Mutant huntingtin inhibits αB-crystallin expression and impairs exosome secretion from astrocytes. J. Neurosci. 2017, 37, 9550–9563. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yefimova, M.G.; Béré, E.; Cantereau-Becq, A.; Meunier-Balandre, A.-C.; Merceron, B.; Burel, A.; Merienne, K.; Ravel, C.; Becq, F.; Bourmeyster, N. Myelinosome Organelles in the Retina of R6/1 Huntington Disease (HD) Mice: Ubiquitous Distribution and Possible Role in Disease Spreading. Int. J. Mol. Sci. 2021, 22, 12771. https://doi.org/10.3390/ijms222312771
Yefimova MG, Béré E, Cantereau-Becq A, Meunier-Balandre A-C, Merceron B, Burel A, Merienne K, Ravel C, Becq F, Bourmeyster N. Myelinosome Organelles in the Retina of R6/1 Huntington Disease (HD) Mice: Ubiquitous Distribution and Possible Role in Disease Spreading. International Journal of Molecular Sciences. 2021; 22(23):12771. https://doi.org/10.3390/ijms222312771
Chicago/Turabian StyleYefimova, Marina G., Emile Béré, Anne Cantereau-Becq, Annie-Claire Meunier-Balandre, Bruno Merceron, Agnès Burel, Karine Merienne, Célia Ravel, Frédéric Becq, and Nicolas Bourmeyster. 2021. "Myelinosome Organelles in the Retina of R6/1 Huntington Disease (HD) Mice: Ubiquitous Distribution and Possible Role in Disease Spreading" International Journal of Molecular Sciences 22, no. 23: 12771. https://doi.org/10.3390/ijms222312771