
BMP Receptor Inhibition Enhances Tissue Repair in Endoglin Heterozygous Mice

 , , ,
, , ,  and
and 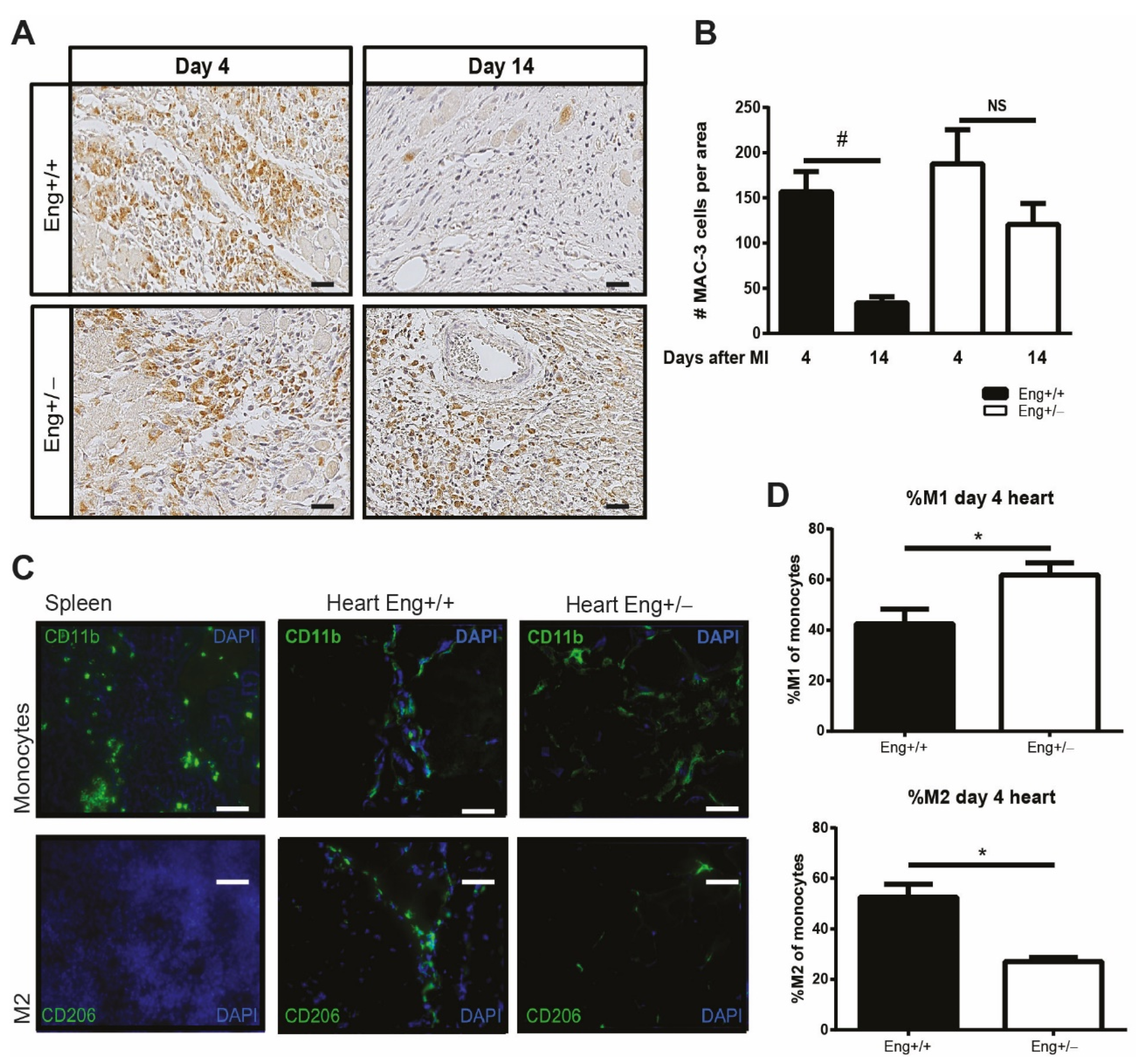{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Endoglin Deficiency Results in Prolonged Inflammation and Reduced M2 Macrophage Presence in the Heart after MI
2.2. Endoglin Deficiency Reduces In Vitro Differentiation of M2 Macrophages in Both HHT1 Mice and Patients
2.3. In Vitro Switch of Macrophage Differentiation by Adaptation of the TGFβ Signaling Response
2.4. TGFβ/BMP and Non-Smad Signaling in Eng+/− Macrophages Is Impaired
2.5. LDN Treatment Improves Cardiac Function after Experimentally Induced MI
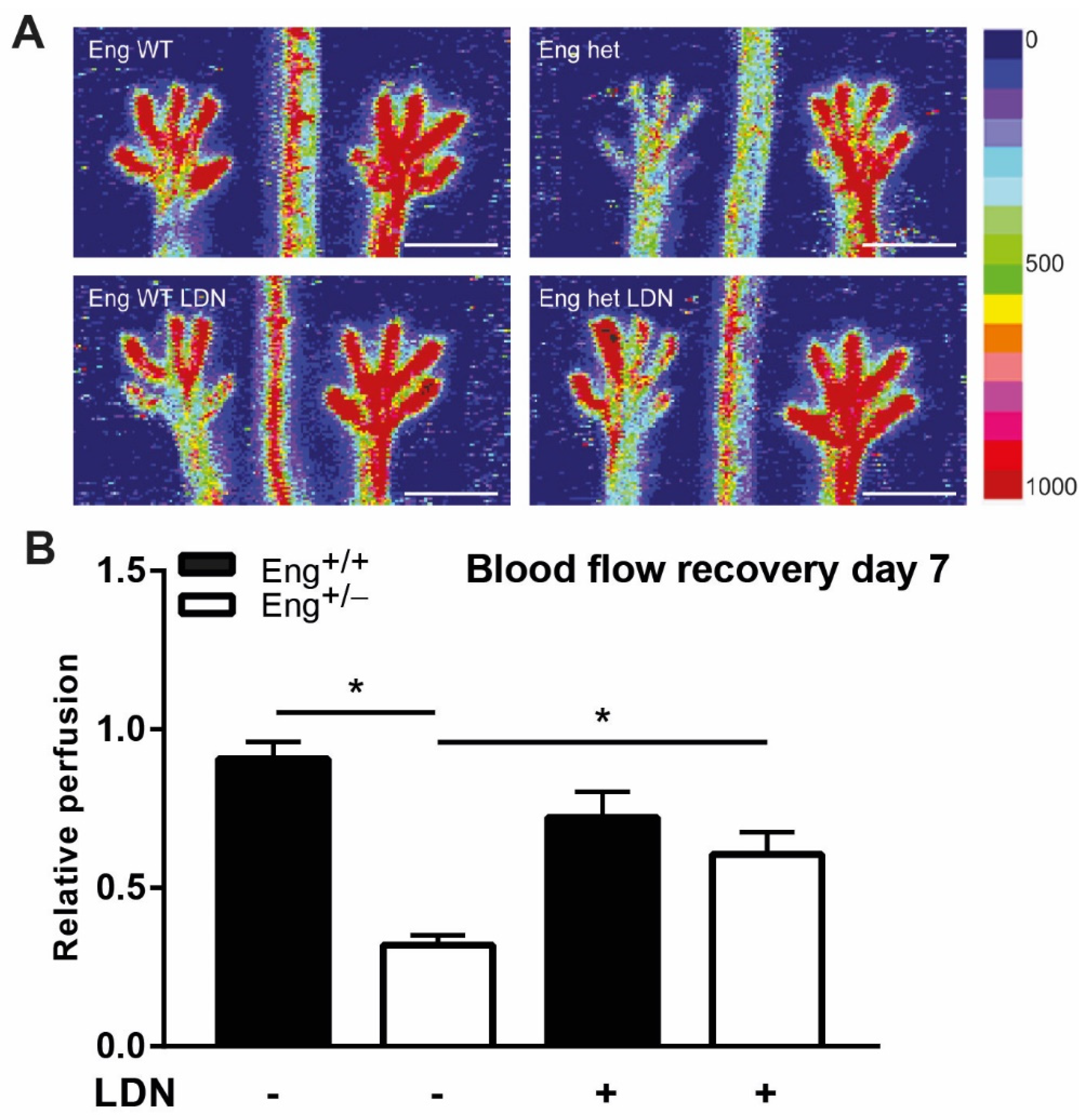
2.6. LDN Treatment Improves Perfusion Recovery after Hind Limb Ischemia
3. Discussion
4. Materials and Methods
4.1. Clinical Studies
4.2. Animals
4.3. Myocardial Infarction and BMPRI-Inhibitor Treatment
4.4. Cardiac Function Measurements
4.5. Hind Limb Ischemia and Perfusion Imaging
4.6. Immunohistochemistry
4.7. Isolation of Immune Cells from Murine Hearts
4.8. Flow Cytometry
4.9. Cultured Macrophages from Mouse Bone Marrow
4.10. Western Blot Analysis
4.11. Morphometry
4.12. Statistical Analysis
4.13. Data Availability
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Guilhem, A.; Malcus, C.; Clarivet, B.; Plauchu, H. Immunological abnormalities associated with hereditary haemorrhagic telangiec-tasia. J. Intern. Med. 2013, 351–362. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Gómez, E.; Jerkic, M.; Prieto, M.; Del Castillo, G.; Martín-Villar, E.; Letarte, M.; Bernabeu, C.; Pérez-Barriocanal, F.; Quintanilla, M.; López-Novoa, J.M. Impaired wound repair in adult endoglin heterozygous mice associated with lower NO bioavailability. J. Investig. Dermatol. 2014, 134, 247–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peter, M.R.; Jerkic, M.; Sotov, V.; Douda, D.N.; Ardelean, D.S.; Ghamami, N.; Lakschevitz, F.; Khan, M.A.; Robertson, S.J.; Glogauer, M.; et al. Impaired resolution of inflammation in the endoglin heterozygous mouse model of chronic colitis. Mediat. Inflamm. 2014, 2014, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Seghers, L.; De Vries, M.R.; Pardali, E.; Hoefer, I.E.; Hierck, B.P.; Dijke, P.T.T.; Goumans, M.J.; Quax, P.H.A. Shear induced collateral artery growth modulated by endoglin but not by ALK1. J. Cell. Mol. Med. 2012, 16, 2440–2450. [Google Scholar] [CrossRef] [PubMed]
- van Laake, L.W.; van den Driesche, S.; Post, S.; Feijen, A.; Jansen, M.A.; Driessens, M.H.; Mager, J.J.; Snijder, R.J.; Westermann, C.J.J.; Doevendans, P.A.; et al. Endoglin has a crucial role in blood cell-mediated vascular repair. Circulation 2006, 114, 2288–2297. [Google Scholar] [CrossRef] [Green Version]
- Torsney, E.; Charlton, R.; Parums, D.; Collis, M.; Arthur, H. Inducible expression of human endoglin during inflammation and wound healing in vivo. Inflamm. Res. Off. J. Eur. Histamine Res. Soc. 2002, 51, 464–470. [Google Scholar] [CrossRef] [PubMed]
- Rossi, E.; Sanz-Rodriguez, F.; Eleno, N.; Düwell, A.; Blanco, F.J.; Langa, C.; Botella, L.M.; Cabañas, C.; Lopez-Novoa, J.M.; Bernabéu, C. Endothelial endoglin is involved in inflammation: Role in leukocyte adhesion and transmigration. Blood 2013, 121, 403–415. [Google Scholar] [CrossRef] [Green Version]
- Rossi, E.; Lopez-Novoa, J.M.; Bernabéu, C. Endoglin involvement in integrin-mediated cell adhesion as a putative pathogenic mechanism in hereditary hemorrhagic telangiectasia type 1 (HHT1). Front. Genet. 2015, 5, 457. [Google Scholar] [CrossRef] [Green Version]
- Aristorena, M.; Blanco, F.J.; Casas-Engel, M.D.L.; Ojeda-Fernandez, L.; Gallardo-Vara, E.; Corbi, A.; Botella, L.M.; Bernabéu, C. Expression of endoglin isoforms in the myeloid lineage and their role during aging and macrophage polarization. J. Cell Sci. 2014, 127, 2723–2735. [Google Scholar] [CrossRef] [Green Version]
- Sanz-Rodríguez, F.; Fernández, L.A.; Zarrabeitia, R.; Perez-Molino, A.; Ramírez, J.R.; Coto, E.; Bernabéu, C.; Botella, L.M. Mutation analysis in spanish patients with hereditary hemorrhagic telangiectasia: Deficient endoglin up-regulation in activated monocytes. Clin. Chem. 2004, 50, 2003–2011. [Google Scholar] [CrossRef]
- Ojeda-Fernández, L.; Recio-Poveda, L.; Aristorena, M.; Lastres, P.; Blanco, F.J.; Sanz-Rodríguez, F.; Gallardo-Vara, E.; de las Casas-Engel, M.; Corbí, Á.; Arthur, H.M.; et al. Mice lacking endoglin in macrophages show an impaired immune response. PLoS Genet. 2016, 12, e1005935. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Han, Z.; Degos, V.; Shen, F.; Choi, E.-J.; Sun, Z.; Kang, S.; Wong, M.; Zhu, W.; Zhan, L.; et al. Persistent infiltration and pro-inflammatory differentiation of monocytes cause unresolved inflammation in brain arteriovenous malformation. Angiogenesis 2016, 19, 451–461. [Google Scholar] [CrossRef] [Green Version]
- Goumans, M.J.; ten Dijke, P. TGF-βsignaling in control of cardiovascular function. Cold Spring Harb. Perspect. Biol. 2018, 10, a022210. [Google Scholar] [CrossRef] [Green Version]
- Goumans, M.-J.; Zwijsen, A.; ten Dijke, P.; Bailly, S. Bone morphogenetic proteins in vascular homeostasis and disease. Cold Spring Harb. Perspect. Biol. 2018, 10, a031989. [Google Scholar] [CrossRef] [PubMed]
- Doetschman, T.; Barnett, J.V.; Runyan, R.B.; Camenisch, T.D.; Heimark, R.L.; Granzier, H.L.; Conway, S.J.; Azhar, M. Transforming growth factor beta signaling in adult cardiovascular diseases and repair. Cell Tissue Res. 2011, 347, 203–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishida, Y.; Kondo, T.; Takayasu, T.; Iwakura, Y.; Mukaida, N. The essential involvement of cross-talk between IFN-γ and TGF-beta in the skin wound-healing process. J. Immunol. 2004, 172, 1848–1855. [Google Scholar] [CrossRef] [Green Version]
- Kulkarni, A.B.; Huh, C.G.; Becker, D.; Geiser, A.; Lyght, M.; Flanders, K.C.; Roberts, A.B.; Sporn, M.B.; Ward, J.M.; Karlsson, S. Transforming growth factor β 1 null mutation in mice causes excessive inflammatory response and early death. Proc. Natl. Acad. Sci. USA 1993, 90, 770–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsson, J.; Goumans, M.; Sjöstrand, L.J.; Van Rooijen, M.A.; Ward, D.; Levéen, P.; Xu, X.; ten Dijke, P.; Mummery, C.L.; Karlsson, S. Abnormal angiogenesis but intact hematopoietic potential in TGF-beta type I receptor-deficient mice. EMBO J. 2001, 20, 1663–1673. [Google Scholar] [CrossRef] [Green Version]
- Russell, N.S.; Floot, B.; Van Werkhoven, E.; Schriemer, M.; De Jong-Korlaar, R.; Woerdeman, L.A.; Stewart, F.A.; Scharpfenecker, M. Blood and lymphatic microvessel damage in irradiated human skin: The role of TGF-β, endoglin and macrophages. Radiother. Oncol. 2015, 116, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Shull, M.M.; Ormsby, I.; Kier, A.B.; Pawlowski, S.; Diebold, R.J.; Yin, M.; Allen, R.; Sidman, C.; Proetzel, G.; Calvint, D.; et al. Targeted disruption of the mouse transforming growth factor-β1 gene results in multifocal inflammatory disease. Nat. Cell Biol. 1992, 359, 693–699. [Google Scholar] [CrossRef]
- Grainger, D.J.; Mosedale, D.E.; Metcalfe, J.C. TGF-β in blood: A complex problem. Cytokine Growth Factor Rev. 2000, 11, 133–145. [Google Scholar] [CrossRef]
- Wan, M.; Li, C.; Zhen, G.; Jiao, K.; He, W.; Jia, X.; Wang, W.; Shi, C.; Xing, Q.; Chen, Y.-F.; et al. Injury-Activated Transforming Growth Factor β Controls Mobilization of Mesenchymal Stem Cells for Tissue Remodeling. Stem Cells 2012, 30, 2498–2511. [Google Scholar] [CrossRef] [Green Version]
- de Sousa Lopes, S.M.C.; Feijen, A.; Korving, J.; Korchynskyi, O.; Larsson, J.; Karlsson, S.; ten Dijke, P.; Lyons, K.M.; Goldschmeding, R.; Doevendans, P.; et al. Connective tissue growth factor expression and Smad signaling during mouse heart development and myocardial infarction. Dev. Dyn. 2004, 231, 542–550. [Google Scholar] [CrossRef] [PubMed]
- Goumans, M.-J.; Van Zonneveld, A.J.; Dijke, P.T. Transforming growth factor β-induced endothelial-to-mesenchymal transition: A switch to cardiac fibrosis? Trends Cardiovasc. Med. 2008, 18, 293–298. [Google Scholar] [CrossRef]
- Lebrin, F.; Goumans, M.-J.; Jonker, L.; Carvalho, R.L.C.; Valdimarsdottir, G.; Thorikay, M.; Mummery, C.; Arthur, H.M.; ten Dijke, P. Endoglin promotes endothelial cell proliferation and TGF-β/ALK1 signal transduction. EMBO J. 2004, 23, 4018–4028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nahrendorf, M.; Swirski, F.K.; Aikawa, E.; Stangenberg, L.; Wurdinger, T.; Figueiredo, J.-L.; Libby, P.; Weissleder, R.; Pittet, M.J. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J. Exp. Med. 2007, 204, 3037–3047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mills, C.D.; Harris, R.A.; Ley, K. Macrophage polarization: Decisions that affect health. J. Clin. Cell. Immunol. 2015, 6. [Google Scholar] [CrossRef] [Green Version]
- Dingenouts, C.K.E.; Bakker, W.; Lodder, K.; Wiesmeijer, K.C.; Moerkamp, A.T.; Maring, J.A.; Arthur, H.; Smits, A.M.; Goumans, M.-J. Inhibiting DPP4 in a mouse model of HHT1 results in a shift towards regenerative macrophages and reduces fibrosis after myocardial infarction. PLoS ONE 2017, 12, e0189805. [Google Scholar] [CrossRef] [Green Version]
- Massagué, J. How cells read TGF-β signals. Nat. Rev. Mol. Cell Biol. 2000, 1, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Lan, H.Y.; Glushakova, O.; Zhu, H.J.; Kang, D.; Schreiner, G.F.; Böttinger, E.P.; Johnson, R.J.; Sautin, Y.Y. Role of ERK1/2 and p38 mitogen-activated protein kinases in the regulation of thrombospondin-1 by TGF-β1 in rat proximal tubular cells and mouse fibroblasts. J. Am. Soc. Nephrol. JASN 2005, 16, 899–904. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.E. Non-Smad Signaling Pathways of the TGF-β Family. Cold Spring Harb. Perspect. Biol. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. The inflammatory response in myocardial injury, repair, and remodelling. Nat. Rev. Cardiol. 2014, 11, 255–265. [Google Scholar] [CrossRef] [Green Version]
- Gombozhapova, A.; Rogovskaya, Y.; Shurupov, V.; Rebenkova, M.; Kzhyshkowska, J.; Popov, S.V.; Karpov, R.S.; Ryabov, V. Macrophage activation and polarization in post-infarction cardiac remodeling. J. Biomed. Sci. 2017, 24, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ten Dijke, P.; Goumans, M.-J.; Pardali, E. Endoglin in angiogenesis and vascular diseases. Angiogenesis 2008, 11, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Tual-Chalot, S.; Mahmoud, M.; Allinson, K.R.; Redgrave, R.E.; Zhai, Z.; Oh, S.P.; Fruttiger, M.; Arthur, H.M. Endothelial depletion of Acvrl1 in mice leads to arteriovenous malformations associated with reduced endoglin expression. PLoS ONE 2014, 9, e98646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Post, S.; Smits, A.M.; Broek, A.J.V.D.; Sluijter, J.P.; Hoefer, I.E.; Janssen, B.J.; Snijder, R.J.; Mager, J.J.; Pasterkamp, G.; Mummery, C.; et al. Impaired recruitment of HHT-1 mononuclear cells to the ischaemic heart is due to an altered CXCR4/CD26 balance. Cardiovasc. Res. 2009, 85, 494–502. [Google Scholar] [CrossRef] [PubMed]
- Dupuis-Girod, S.; Giraud, S.; Decullier, E.; Lesca, G.; Cottin, V.; Faure, F.; Merrot, O.; Saurin, J.; Cordier, J.; Plauchu, H. Hemorrhagic hereditary telangiectasia (Rendu-Osler disease) and infectious diseases: An underestimated association. Clin. Infect. Dis. 2007, 44, 841–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathis, S.; Dupuis-Girod, S.; Plauchu, H.; Giroud, M.; Barroso, B.; Ly, K.H.; Ingrand, P.; Gilbert, B.; Godeneche, G.; Neau, J.-P. Cerebral abscesses in hereditary haemorrhagic telangiectasia: A clinical and microbiological evaluation. Clin. Neurol. Neurosurg. 2012, 114, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Braga, T.T.; Agudelo, J.S.H.; Camara, N.O.S. Macrophages during the fibrotic process: M2 as friend and foe. Front. Immunol. 2015, 6, 602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vernon, M.A.; Mylonas, K.J.; Hughes, J. Macrophages and renal fibrosis. Semin. Nephrol. 2010, 30, 302–317. [Google Scholar] [CrossRef]
- Carvalho, R.L.; Jonker, L.; Goumans, M.; Larsson, J.; Bouwman, P.; Karlsson, S.; ten Dijke, P.; Arthur, H.M.; Mummery, C.L. Defective paracrine signalling by TGFβ in yolk sac vasculature of endoglin mutant mice: A paradigm for hereditary haemorrhagic telangiectasia. Development 2004, 131, 6237–6247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez-Gómez, E.; Eleno, N.; López-Novoa, J.M.; Ramirez, J.R.; Velasco, B.; Letarte, M.; Bernabéu, C.; Quintanilla, M. Characterization of murine S-endoglin isoform and its effects on tumor development. Oncogene 2005, 24, 4450–4461. [Google Scholar] [CrossRef] [Green Version]
- Velasco, S.; Alvarez-Muñoz, P.; Pericacho, M.; Dijke, P.T.; Bernabéu, C.; López-Novoa, J.M.; Rodríguez-Barbero, A. L- and S-endoglin differentially modulate TGFβ1 signaling mediated by ALK1 and ALK5 in L6E9 myoblasts. J. Cell Sci. 2008, 121, 913–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanco, F.J.; Grande, M.T.; Langa, C.; Oujo, B.; Velasco, S.; Rodriguez-Barbero, A.; Pérez-Gómez, E.; Quintanilla, M.; López-Novoa, J.M.; Bernabéu, C. S-Endoglin expression is induced in senescent endothelial cells and contributes to vascular pathology. Circ. Res. 2008, 103, 1383–1392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.; Lee, J.; Nam, S.K.; Hoon Jun, Y. S-endoglin expression is induced in hyperoxia and contributes to altered pulmonary angi-ogenesis in bronchopulmonary dysplasia development. Sci. Rep. 2020, 10, 3043. [Google Scholar] [CrossRef] [Green Version]
- Kebir, D.E.; Filep, J.G. Modulation of neutrophil apoptosis and the resolution of inflammation through β2 integrins. Front. Immunol. 2013, 4, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Pericacho, M.; Velasco, S.; Prieto, M.; Llano, E.; López-Novoa, J.M.; Rodríguez-Barbero, A. Endoglin haploinsufficiency promotes fibroblast accumulation during wound healing through Akt activation. PLoS ONE 2013, 8, e54687. [Google Scholar] [CrossRef] [Green Version]
- Park, S.; DiMaio, T.A.; Liu, W.; Wang, S.; Sorenson, C.M.; Sheibani, N. Endoglin regulates the activation and quiescence of endothelium by participating in canonical and non-canonical TGF-β signaling pathways. J. Cell Sci. 2013, 126, 1392–1405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monick, M.M.; Powers, L.S.; Barrett, C.W.; Hinde, S.; Ashare, A.; Groskreutz, D.J.; Nyunoya, T.; Coleman, M.; Spitz, D.R.; Hunninghake, G.W. Constitutive ERK MAPK activity regulates macrophage ATP production and mitochondrial integrity. J. Immunol. 2008, 180, 7485–7496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawatzky, D.A.; Willoughby, D.A.; Colville-Nash, P.R.; Rossi, A.G. The Involvement of the Apoptosis-Modulating Proteins ERK 1/2, Bcl-xL and Bax in the Resolution of Acute Inflammation in Vivo. Am. J. Pathol. 2006, 168, 33–41. [Google Scholar] [CrossRef] [Green Version]
- Olieslagers, S.; Pardali, E.; Tchaikovski, V.; ten Dijke, P.; Waltenberger, J. TGF-β1/ALK5-induced monocyte migration involves PI3K and p38 pathways and is not negatively affected by diabetes mellitus. Cardiovasc. Res. 2011, 91, 510–518. [Google Scholar] [CrossRef] [Green Version]
- Aguirre-Ghiso, J.A. Models, mechanisms and clinical evidence for cancer dormancy. Nat. Rev. Cancer 2007, 7, 834–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomes, E.; Rockwell, P. p38 MAPK as a negative regulator of VEGF/VEGFR2 signaling pathway in serum deprived human SK-N-SH neuroblastoma cells. Neurosci. Lett. 2008, 431, 95–100. [Google Scholar] [CrossRef] [Green Version]
- Abdalla, S.A. Hereditary haemorrhagic telangiectasia: Current views on genetics and mechanisms of disease. J. Med. Genet. 2005, 43, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Thalgott, J.; Dos-Santos-Luis, D.; Lebrin, F. Pericytes as targets in hereditary hemorrhagic telangiectasia. Front. Genet. 2015, 6, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Peet, C.; Ivetic, A.I.; Bromage, D.; Shah, A.M. Cardiac monocytes and macrophages after myocardial infarction. Cardiovasc. Res. 2020, 116, 1101–1112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panizzi, P.; Swirski, F.K.; Figueiredo, J.-L.; Waterman, P.; Sosnovik, D.E.; Aikawa, E.; Libby, P.; Pittet, M.; Weissleder, R.; Nahrendorf, M. Impaired Infarct Healing in Atherosclerotic Mice With Ly-6ChiMonocytosis. J. Am. Coll. Cardiol. 2010, 55, 1629–1638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maekawa, Y.; Anzai, T.; Yoshikawa, T.; Asakura, Y.; Takahashi, T.; Ishikawa, S.; Mitamura, H.; Ogawa, S. Prognostic significance of peripheral monocytosis after reperfused acute myocardial infarction:a possible role for left ventricular remodeling. J. Am. Coll. Cardiol. 2002, 39, 241–246. [Google Scholar] [CrossRef] [Green Version]
- Shintani, Y.; Shintani, Y.; Shintani, Y.; Ishida, H.; Saba, R.; Yamaguchi, A.; Adachi, H.; Yashiro, K.; Suzuki, K. Alternatively activated macrophages determine repair of the infarcted adult murine heart. J. Clin. Investig. 2016, 126, 2151–2166. [Google Scholar]
- Ma, Y.; Halade, G.V.; Zhang, J.; Ramirez, T.A.; Levin, D.; Voorhees, A.; Jin, Y.-F.; Han, H.-C.; Manicone, A.M.; Lindsey, M.L. Matrix Metalloproteinase-28 Deletion Exacerbates Cardiac Dysfunction and Rupture After Myocardial Infarction in Mice by Inhibiting M2 Macrophage Activation. Circ. Res. 2013, 112, 675–688. [Google Scholar] [CrossRef]
- De Vinuesa, A.G.; Bocci, M.; Pietras, K.; ten Dijke, P. Targeting tumour vasculature by inhibiting activin receptor-like kinase (ALK)1 function. Biochem. Soc. Trans. 2016, 44, 1142–1149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, P.B.; Deng, D.Y.; Lai, C.S.; Hong, C.C.; Cuny, G.D.; Bouxsein, M.L.; Hong, D.W.; McManus, P.M.; Katagiri, T.; Sachidanandan, C.; et al. BMP type I receptor inhibition reduces heterotopic ossification. Nat. Med. 2008, 14, 1363–1369. [Google Scholar] [CrossRef] [PubMed]
- Welten, S.M.; Bastiaansen, A.J.; De Jong, R.C.; De Vries, M.R.; Peters, E.A.; Boonstra, M.C.; Sheikh, S.P.; La Monica, N.; Kandimalla, E.R.; Quax, P.H.; et al. Inhibition of 14q32 MicroRNAs miR-329, miR-487b, miR-494, and miR-495 Increases Neovascularization and Blood Flow Recovery After Ischemia. Circ. Res. 2014, 115, 696–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duim, S.N.; Kurakula, K.; Goumans, M.-J.; Kruithof, B.P. Cardiac endothelial cells express Wilms’ tumor-1. Wt1 expression in the developing, adult and infarcted heart. J. Mol. Cell. Cardiol. 2015, 81, 127–135. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bakker, W.; Dingenouts, C.K.E.; Lodder, K.; Wiesmeijer, K.C.; de Jong, A.; Kurakula, K.; Mager, H.-J.J.; Smits, A.M.; de Vries, M.R.; Quax, P.H.A.; et al. BMP Receptor Inhibition Enhances Tissue Repair in Endoglin Heterozygous Mice. Int. J. Mol. Sci. 2021, 22, 2010. https://doi.org/10.3390/ijms22042010
Bakker W, Dingenouts CKE, Lodder K, Wiesmeijer KC, de Jong A, Kurakula K, Mager H-JJ, Smits AM, de Vries MR, Quax PHA, et al. BMP Receptor Inhibition Enhances Tissue Repair in Endoglin Heterozygous Mice. International Journal of Molecular Sciences. 2021; 22(4):2010. https://doi.org/10.3390/ijms22042010
Chicago/Turabian StyleBakker, Wineke, Calinda K. E. Dingenouts, Kirsten Lodder, Karien C. Wiesmeijer, Alwin de Jong, Kondababu Kurakula, Hans-Jurgen J. Mager, Anke M. Smits, Margreet R. de Vries, Paul H. A. Quax, and et al. 2021. "BMP Receptor Inhibition Enhances Tissue Repair in Endoglin Heterozygous Mice" International Journal of Molecular Sciences 22, no. 4: 2010. https://doi.org/10.3390/ijms22042010