
Proteolysis Targeting Chimeric Molecules: Tuning Molecular Strategies for a Clinically Sound Listening

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. The Essential: The Formation of the Ternary Complex E3 Ligase–PROTAC–POI
1.2. The Step Forward: Improvement of PROTACs Specificity and Pharmacodynamics
1.3. More Than a Bridge between Two Shoulders: The Pharmacokinetic Role of the Linker
1.4. Current Approaches for a Rational Design of PROTACs
2. Conclusions and Future Perspectives
- -
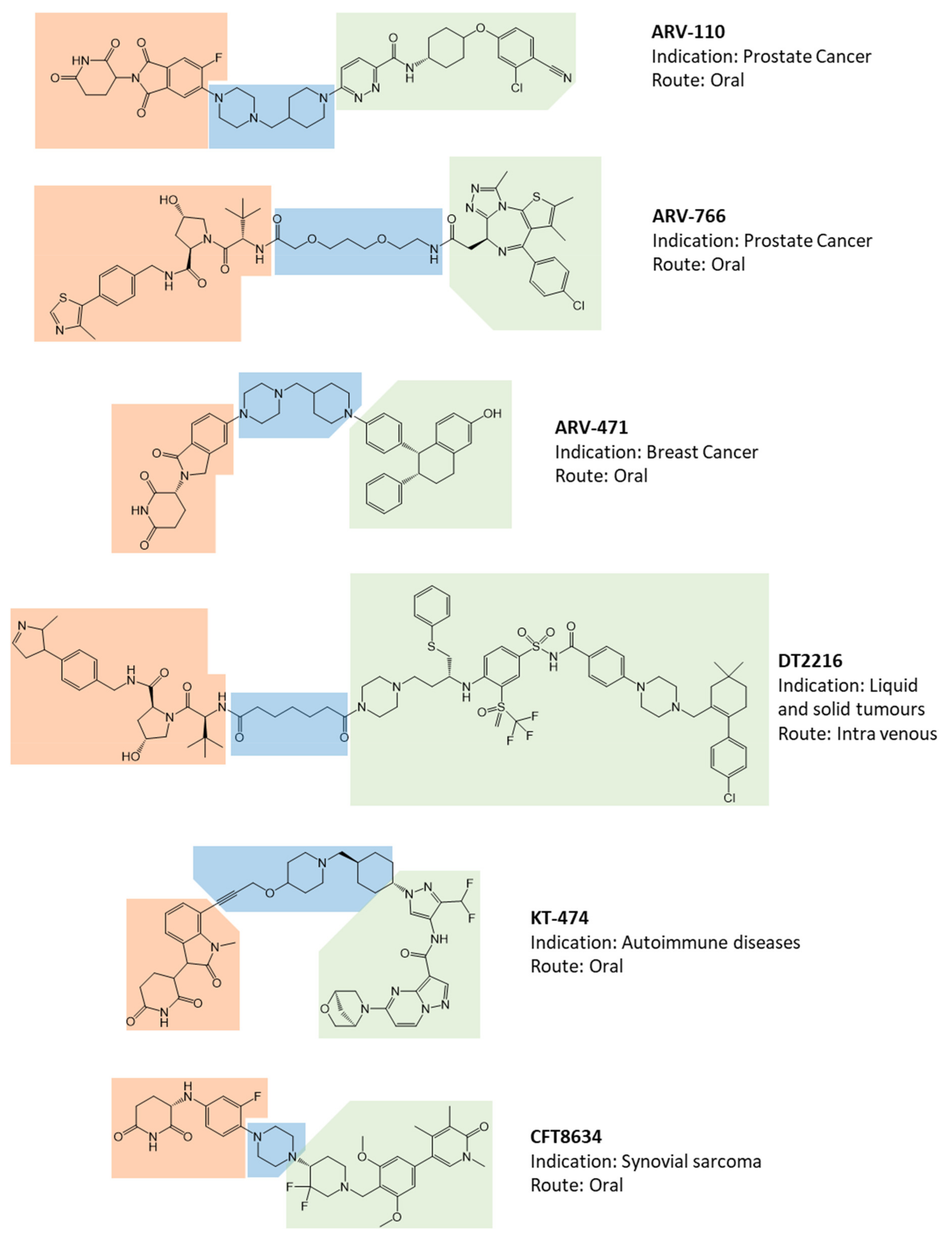
- The opportunity to obtain an oral drug essentially relies on the use of a CRBN-based degradation enhancer. In fact, the corresponding thalidomide-like moiety displays a favorable lipophilic/molecular weight ratio and consequent satisfactory gut absorption. Unless a deep design intervention on the linker, a VHL-based degradation enhancer, the only other alternative to the CRBN, is essentially suitable for parenteral administration;
- -
- According to available data, with the sole exception of BRD9 degradation enhancer CFT8634, all PROTAC’s warheads of clinical interest target the known POI’s binding sites through recognized ligands or structural derivatives. Data on the use of alternative binding sites of known target proteins are not available;
- -
- Despite the simple presumptive role of the linker portion, it represents a structural moiety of design interest. The use of saturated heterocycles or on available linkers with hydrogen bond acceptors and reduced conformational freedom is frequently associated with the development of oral drugs. Saturated linear hydrocarbon chains are more frequently found in parenteral administration.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sakamoto, K.M.; Kim, K.B.; Kumagai, A.; Mercurio, F.; Crews, C.M.; Deshaies, R.J. Protacs: Chimeric Molecules That Target Proteins to the Skp1-Cullin-F Box Complex for Ubiquitination and Degradation. Proc. Natl. Acad. Sci. USA 2001, 98, 8554–8559. [Google Scholar] [CrossRef] [Green Version]
- Hughes, S.J.; Testa, A.; Thompson, N.; Churcher, I. The Rise and Rise of Protein Degradation: Opportunities and Challenges Ahead. Drug Discov. Today 2021, 26, 2889–2897. [Google Scholar] [CrossRef]
- Mullard, A. First Targeted Protein Degrader Hits the Clinic. Nat. Rev. Drug Discov. 2019. [Google Scholar] [CrossRef]
- Békés, M.; Langley, D.R.; Crews, C.M. PROTAC Targeted Protein Degraders: The Past Is Prologue. Nat. Rev. Drug Discov. 2022, 21, 181–200. [Google Scholar] [CrossRef]
- Amm, I.; Sommer, T.; Wolf, D.H. Protein Quality Control and Elimination of Protein Waste: The Role of the Ubiquitin–Proteasome System. Biochim. Biophys. Acta Mol. Cell Res. 2014, 1843, 182–196. [Google Scholar] [CrossRef] [Green Version]
- Hershko, A.; Ciechanover, A. The Ubiquitin System. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef]
- Ito, T.; Ando, H.; Suzuki, T.; Ogura, T.; Hotta, K.; Imamura, Y.; Yamaguchi, Y.; Handa, H. Identification of a Primary Target of Thalidomide Teratogenicity. Science 2010, 327, 1345–1350. [Google Scholar] [CrossRef] [Green Version]
- Itoh, Y.; Ishikawa, M.; Naito, M.; Hashimoto, Y. Protein Knockdown Using Methyl Bestatin–Ligand Hybrid Molecules: Design and Synthesis of Inducers of Ubiquitination-Mediated Degradation of Cellular Retinoic Acid-Binding Proteins. J. Am. Chem. Soc. 2010, 132, 5820–5826. [Google Scholar] [CrossRef]
- Logan, I.R.; McNeill, H.V.; Cook, S.; Lu, X.; Lunec, J.; Robson, C.N. Analysis of the MDM2 Antagonist Nutlin-3 in Human Prostate Cancer Cells. Prostate 2007, 67, 900–906. [Google Scholar] [CrossRef]
- Zengerle, M.; Chan, K.-H.; Ciulli, A. Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4. ACS Chem. Biol. 2015, 10, 1770–1777. [Google Scholar] [CrossRef] [Green Version]
- Edmondson, S.D.; Yang, B.; Fallan, C. Proteolysis Targeting Chimeras (PROTACs)in ‘beyond Rule-of-Five’ Chemical Space: Recent Progress and Future Challenges. Bioorg. Med. Chem. Lett. 2019, 29, 1555–1564. [Google Scholar] [CrossRef]
- Pike, A.; Williamson, B.; Harlfinger, S.; Martin, S.; McGinnity, D.F. Optimising Proteolysis-Targeting Chimeras (PROTACs) for Oral Drug Delivery: A Drug Metabolism and Pharmacokinetics Perspective. Drug Discov. Today 2020, 25, 1793–1800. [Google Scholar] [CrossRef]
- Mori, T.; Ito, T.; Liu, S.; Ando, H.; Sakamoto, S.; Yamaguchi, Y.; Tokunaga, E.; Shibata, N.; Handa, H.; Hakoshima, T. Structural Basis of Thalidomide Enantiomer Binding to Cereblon. Sci. Rep. 2018, 8, 1294. [Google Scholar] [CrossRef]
- Hu, Z.; Crews, C.M. Recent Developments in PROTAC-Mediated Protein Degradation: From Bench to Clinic. ChemBioChem 2022, 23, e202100270. [Google Scholar] [CrossRef]
- Pettersson, M.; Crews, C.M. PROteolysis TArgeting Chimeras (PROTACs)—Past, Present and Future. Drug Discov. Today Technol. 2019, 31, 15–27. [Google Scholar] [CrossRef]
- Gadd, M.S.; Testa, A.; Lucas, X.; Chan, K.H.; Chen, W.; Lamont, D.J.; Zengerle, M.; Ciulli, A. Structural Basis of PROTAC Cooperative Recognition for Selective Protein Degradation. Nat. Chem. Biol. 2017, 13, 514–521. [Google Scholar] [CrossRef]
- Zhang, Y.; Loh, C.; Chen, J.; Mainolfi, N. Targeted Protein Degradation Mechanisms. Drug Discov. Today Technol. 2019, 31, 53–60. [Google Scholar] [CrossRef]
- Stathis, A.; Bertoni, F. BET Proteins as Targets for Anticancer Treatment. Cancer Discov. 2018, 8, 24–36. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Liu, Z.; Li, X. Recent Advances in Dual BRD4-Kinase Inhibitors Based on Polypharmacology. ChemMedChem 2022, 17, e202100731. [Google Scholar] [CrossRef]
- Testa, A.; Hughes, S.J.; Lucas, X.; Wright, J.E.; Ciulli, A. Structure-Based Design of a Macrocyclic PROTAC. Angew. Chem. 2020, 132, 1744–1751. [Google Scholar] [CrossRef] [Green Version]
- Roy, M.J.; Winkler, S.; Hughes, S.J.; Whitworth, C.; Galant, M.; Farnaby, W.; Rumpel, K.; Ciulli, A. SPR-Measured Dissociation Kinetics of PROTAC Ternary Complexes Influence Target Degradation Rate. ACS Chem. Biol. 2019, 14, 361–368. [Google Scholar] [CrossRef] [Green Version]
- Bondeson, D.P.; Smith, B.E.; Burslem, G.M.; Buhimschi, A.D.; Hines, J.; Jaime-Figueroa, S.; Wang, J.; Hamman, B.D.; Ishchenko, A.; Crews, C.M. Lessons in PROTAC Design from Selective Degradation with a Promiscuous Warhead. Cell Chem. Biol. 2018, 25, 78–87.e5. [Google Scholar] [CrossRef] [Green Version]
- Han, X.; Zhao, L.; Xiang, W.; Qin, C.; Miao, B.; Xu, T.; Wang, M.; Yang, C.Y.; Chinnaswamy, K.; Stuckey, J.; et al. Discovery of Highly Potent and Efficient PROTAC Degraders of Androgen Receptor (AR) by Employing Weak Binding Affinity VHL E3 Ligase Ligands. J. Med. Chem. 2019, 62, 11218–11231. [Google Scholar] [CrossRef]
- Pulliam, T.L.; Goli, P.; Awad, D.; Lin, C.; Wilkenfeld, S.R.; Frigo, D.E. Regulation and Role of CAMKK2 in Prostate Cancer. Nat. Rev. Urol. 2022, 19, 367–380. [Google Scholar] [CrossRef]
- Kargbo, R.B. PROTAC Compounds Targeting Androgen Receptor for Cancer Therapeutics: Prostate Cancer and Kennedy’s Disease. ACS Med. Chem. Lett. 2020, 11, 1092–1093. [Google Scholar] [CrossRef]
- Schlienger, N.; Lund, B.W.; Pawlas, J.; Badalassi, F.; Bertozzi, F.; Lewinsky, R.; Fejzic, A.; Thygesen, M.B.; Tabatabaei, A.; Bradley, S.R.; et al. Synthesis, Structure-Activity Relationships, and Characterization of Novel Nonsteroidal and Selective Androgen Receptor Modulators. J. Med. Chem. 2009, 52, 7186–7191. [Google Scholar] [CrossRef]
- Cattrini, C.; Caffo, O.; de Giorgi, U.; Mennitto, A.; Gennari, A.; Olmos, D.; Castro, E. Apalutamide, Darolutamide and Enzalutamide for Nonmetastatic Castration-Resistant Prostate Cancer (NmCRPC): A Critical Review. Cancers 2022, 14, 1792. [Google Scholar] [CrossRef]
- Nagata, N.; Miyakawa, M.; Amano, S.; Furuya, K.; Yamamoto, N.; Inoguchi, K. Design and Synthesis of Tricyclic Tetrahydroquinolines as a New Series of Nonsteroidal Selective Androgen Receptor Modulators (SARMs). Bioorg. Med. Chem. Lett. 2011, 21, 1744–1747. [Google Scholar] [CrossRef]
- Vajdos, F.F.; Hoth, L.R.; Geoghegan, K.F.; Simons, S.P.; LeMotte, P.K.; Danley, D.E.; Ammirati, M.J.; Pandit, J. The 2.0 Å Crystal Structure of the ERα Ligand-Binding Domain Complexed with Lasofoxifene. Protein Sci. 2007, 16, 897–905. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Ashkenazi, A.; Fairbrother, W.J.; Leverson, J.D.; Souers, A.J. From Basic Apoptosis Discoveries to Advanced Selective BCL-2 Family Inhibitors. Nat. Rev. Drug Discov. 2017, 16, 273–284. [Google Scholar] [CrossRef]
- Kaefer, A.; Yang, J.; Noertersheuser, P.; Mensing, S.; Humerickhouse, R.; Awni, W.; Xiong, H. Mechanism-Based Pharmacokinetic/Pharmacodynamic Meta-Analysis of Navitoclax (ABT-263) Induced Thrombocytopenia. Cancer Chemother. Pharmacol. 2014, 74, 593–602. [Google Scholar] [CrossRef]
- Schoenwaelder, S.M.; Jarman, K.E.; Gardiner, E.E.; Hua, M.; Qiao, J.; White, M.J.; Josefsson, E.C.; Alwis, I.; Ono, A.; Willcox, A.; et al. Bcl-XL-Inhibitory BH3 Mimetics Can Induce a Transient Thrombocytopathy That Undermines the Hemostatic Function of Platelets. Blood 2011, 118, 1663–1674. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.; Zhang, X.; Lv, D.; Zhang, Q.; He, Y.; Zhang, P.; Liu, X.; Thummuri, D.; Yuan, Y.; Wiegand, J.S.; et al. A Selective BCL-XL PROTAC Degrader Achieves Safe and Potent Antitumor Activity. Nat. Med. 2019, 25, 1938–1947. [Google Scholar] [CrossRef]
- Lv, D.; Pal, P.; Liu, X.; Jia, Y.; Thummuri, D.; Zhang, P.; Hu, W.; Pei, J.; Zhang, Q.; Zhou, S.; et al. Development of a BCL-XL and BCL-2 Dual Degrader with Improved Anti-Leukemic Activity. Nat. Commun. 2021, 12, 6896. [Google Scholar] [CrossRef]
- De Nardo, D.; Balka, K.R.; Gloria, Y.C.; Rao, V.R.; Latz, E.; Masters, S.L. Interleukin-1 Receptor-Associated Kinase 4 (IRAK4) Plays a Dual Role in Myddosome Formation and Toll-like Receptor Signaling. J. Biol. Chem. 2018, 293, 15195–15207. [Google Scholar] [CrossRef] [Green Version]
- Somani, V.K.; Zhang, D.; Dodhiawala, P.B.; Lander, V.E.; Liu, X.; Kang, L.-I.; Chen, H.-P.; Knolhoff, B.L.; Li, L.; Grierson, P.M.; et al. IRAK4 Signaling Drives Resistance to Checkpoint Immunotherapy in Pancreatic Ductal Adenocarcinoma. Gastroenterology 2022, 162, 2047–2062. [Google Scholar] [CrossRef]
- Chen, Y.; Ning, Y.; Bai, G.; Tong, L.; Zhang, T.; Zhou, J.; Zhang, H.; Xie, H.; Ding, J.; Duan, W. Design, Synthesis, and Biological Evaluation of IRAK4-Targeting PROTACs. ACS Med. Chem. Lett. 2021, 12, 82–87. [Google Scholar] [CrossRef]
- De Novellis, D.; Cacace, F.; Caprioli, V.; Wierda, W.G.; Mahadeo, K.M.; Tambaro, F.P. The Tki Era in Chronic Leukemias. Pharmaceutics 2021, 13, 2201. [Google Scholar] [CrossRef]
- Zullow, H.J.; Sankar, A.; Ingram, D.R.; Samé Guerra, D.D.; D’Avino, A.R.; Collings, C.K.; Lazcano, R.; Wang, W.-L.; Liang, Y.; Qi, J.; et al. The FUS::DDIT3 Fusion Oncoprotein Inhibits BAF Complex Targeting and Activity in Myxoid Liposarcoma. Mol. Cell 2022, 82, 1737–1750.e8. [Google Scholar] [CrossRef]
- Michel, B.C.; D’Avino, A.R.; Cassel, S.H.; Mashtalir, N.; McKenzie, Z.M.; McBride, M.J.; Valencia, A.M.; Zhou, Q.; Bocker, M.; Soares, L.M.M.; et al. A Non-Canonical SWI/SNF Complex Is a Synthetic Lethal Target in Cancers Driven by BAF Complex Perturbation. Nat. Cell Biol. 2018, 20, 1410–1420. [Google Scholar] [CrossRef]
- Bechter, O.; Schöffski, P. Make Your Best BET: The Emerging Role of BET Inhibitor Treatment in Malignant Tumors. Pharmacol. Ther. 2020, 208, 107479. [Google Scholar] [CrossRef]
- Sabnis, R.W. BRD9 Bifunctional Degraders for Treating Cancer. ACS Med. Chem. Lett. 2021, 12, 1879–1880. [Google Scholar] [CrossRef]
- Liu, J.; Peng, Y.; Wei, W. Light-Controllable PROTACs for Temporospatial Control of Protein Degradation. Front. Cell Dev. Biol. 2021, 9, 678077. [Google Scholar] [CrossRef]
- Xue, G.; Wang, K.; Zhou, D.; Zhong, H.; Pan, Z. Light-Induced Protein Degradation with Photocaged PROTACs. J. Am. Chem. Soc. 2019, 141, 18370–18374. [Google Scholar] [CrossRef]
- Reynders, M.; Matsuura, B.S.; Bérouti, M.; Simoneschi, D.; Marzio, A.; Pagano, M.; Trauner, D. PHOTACs enable optical control of protein degradation. Sci. Adv. 2020, 6, eaay5064. [Google Scholar]
- Jin, Y.-H.; Lu, M.-C.; Wang, Y.; Shan, W.-X.; Wang, X.-Y.; You, Q.-D.; Jiang, Z.-Y. Azo-PROTAC: Novel Light-Controlled Small-Molecule Tool for Protein Knockdown. J. Med. Chem. 2020, 63, 4654. [Google Scholar] [CrossRef]
- Pfaff, P.; Kusal, T.; Samarasinghe, T.G.; Crews, C.M.; Carreira, E.M. Reversible Spatiotemporal Control of Induced Protein Degradation by Bistable PhotoPROTACs. ACS Cent. Sci. 2019, 5, 1682–1690. [Google Scholar] [CrossRef] [Green Version]
- Kounde, C.S.; Shchepinova, M.M.; Saunders, C.N.; Muelbaier, M.; Rackham, M.D.; Harling, J.D.; Tate, E.W. A Caged E3 Ligase Ligand for PROTAC-Mediated Protein Degradation with Light. Chem. Commun. 2020, 56, 5532–5535. [Google Scholar] [CrossRef]
- Seung Lee, J.; Kim, J.; Ye, Y.; Kim, T. Materials and Device Design for Advanced Phototherapy Systems. Adv. Drug Deliv. Rev. 2022, 186, 114339. [Google Scholar] [CrossRef]
- Van den Brand, J.; de Kok, M.; Koetse, M.; Cauwe, M.; Verplancke, R.; Bossuyt, F.; Jablonski, M.; Vanfleteren, J. Flexible and Stretchable Electronics for Wearable Health Devices. Solid-State Electron. 2015, 113, 116–120. [Google Scholar] [CrossRef]
- Lu, L.; Gutruf, P.; Xia, L.; Bhatti, D.L.; Wang, X.; Vazquez-Guardado, A.; Ning, X.; Shen, X.; Sang, T.; Ma, R.; et al. Wireless Optoelectronic Photometers for Monitoring Neuronal Dynamics in the Deep Brain. Proc. Natl. Acad. Sci. USA 2018, 115, E1374–E1383. [Google Scholar] [CrossRef] [Green Version]
- Lebraud, H.; Wright, D.J.; Johnson, C.N.; Heightman, T.D. Protein Degradation by In-Cell Self-Assembly of Proteolysis Targeting Chimeras. ACS Cent. Sci. 2016, 2, 927–934. [Google Scholar] [CrossRef] [Green Version]
- Maneiro, M.; Forte, N.; Shchepinova, M.M.; Kounde, C.S.; Chudasama, V.; Baker, J.R.; Tate, E.W. Antibody–PROTAC Conjugates Enable HER2-Dependent Targeted Protein Degradation of BRD4. ACS Chem. Biol. 2020, 15, 1306–1312. [Google Scholar] [CrossRef]
- Zagidullin, A.; Milyukov, V.; Rizvanov, A.; Bulatov, E. Novel Approaches for the Rational Design of PROTAC Linkers. Explor. Target. AntiTumor Ther. 2020, 1, 381–390. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Maple, H.J.; Clayden, N.; Baron, A.; Stacey, C.; Felix, R. Developing Degraders: Principles and Perspectives on Design and Chemical Space. MedChemComm 2019, 10, 1755–1764. [Google Scholar] [CrossRef]
- Cyrus, K.; Wehenkel, M.; Choi, E.Y.; Han, H.J.; Lee, H.; Swanson, H.; Kim, K.B. Impact of Linker Length on the Activity of PROTACs. Mol. BioSyst. 2011, 7, 359–364. [Google Scholar] [CrossRef] [Green Version]
- Negi, A.; Voisin-Chiret, A.S. Strategies to Reduce the On-Target Platelet Toxicity of Bcl-XL Inhibitors: PROTACs, SNIPERs and Prodrug-Based Approaches. ChemBioChem 2022, e202100689. [Google Scholar] [CrossRef]
- Troup, R.I.; Fallan, C.; Baud, M.G.J. Current Strategies for the Design of PROTAC Linkers: A Critical Review. Explor. Target. AntiTumor Ther. 2020, 1, 273–312. [Google Scholar] [CrossRef]
- Atilaw, Y.; Poongavanam, V.; Svensson Nilsson, C.; Nguyen, D.; Giese, A.; Meibom, D.; Erdelyi, M.; Kihlberg, J. Solution Conformations Shed Light on PROTAC Cell Permeability. ACS Med. Chem. Lett. 2021, 12, 107–114. [Google Scholar] [CrossRef]
- Pereira, D.M.; Rodrigues, C.M.P. Targeted Avenues for Cancer Treatment: The MEK5–ERK5 Signaling Pathway. Trends Mol. Med. 2020, 26, 394–407. [Google Scholar] [CrossRef]
- Danelius, E.; Poongavanam, V.; Peintner, S.; Wieske, L.H.E.; Erdélyi, M.; Kihlberg, J. Solution Conformations Explain the Chameleonic Behaviour of Macrocyclic Drugs. Chem. Eur. J. 2020, 26, 5231–5244. [Google Scholar] [CrossRef]
- Cicero, D.O.; Barbato, G.; Bazzo, R. NMR Analysis of Molecular Flexibility in Solution: A New Method for the Study of Complex Distributions of Rapidly Exchanging Conformations. Application to a 13-Residue Peptide with an 8-Residue Loop. J. Am. Chem. Soc. 1995, 117, 1027–1033. [Google Scholar] [CrossRef]
- Gilson, M.K.; Zhou, H.X. Calculation of Protein-Ligand Binding Affinities. Annu. Rev. Biophys. Biomol. Struct. 2007, 36, 21–42. [Google Scholar] [CrossRef]
- Han, B. A Suite of Mathematical Solutions to Describe Ternary Complex Formation and Their Application to Targeted Protein Degradation by Heterobifunctional Ligands. J. Biol. Chem. 2020, 295, 15280–15291. [Google Scholar] [CrossRef]
- Guo, W.H.; Qi, X.; Yu, X.; Liu, Y.; Chung, C.I.; Bai, F.; Lin, X.; Lu, D.; Wang, L.; Chen, J.; et al. Enhancing Intracellular Accumulation and Target Engagement of PROTACs with Reversible Covalent Chemistry. Nat. Commun. 2020, 11, 4268. [Google Scholar] [CrossRef]
- Drummond, M.L.; Williams, C.I. In Silico Modeling of PROTAC-Mediated Ternary Complexes: Validation and Application. J. Chem. Inf. Model. 2019, 59, 1634–1644. [Google Scholar] [CrossRef] [Green Version]
- Molecular Operating Environment (MOE). 2020.09 Chemical Computing Group ULC, 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7. 2022. Available online: https://www.chemcomp.com/Products.htm (accessed on 1 February 2022).
- Zaidman, D.; Prilusky, J.; London, N. ProsetTac: Rosetta Based Modeling of PROTAC Mediated Ternary Complexes. J. Chem. Inf. Model. 2020, 60, 4894–4903. [Google Scholar] [CrossRef]
- Bai, N.; Miller, S.A.; Andrianov, G.V.; Yates, M.; Kirubakaran, P.; Karanicolas, J. Rationalizing PROTAC-Mediated Ternary Complex Formation Using Rosetta. J. Chem. Inf. Model. 2021, 61, 1368–1382. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pedrucci, F.; Pappalardo, C.; Marzaro, G.; Ferri, N.; Ferlin, A.; De Toni, L. Proteolysis Targeting Chimeric Molecules: Tuning Molecular Strategies for a Clinically Sound Listening. Int. J. Mol. Sci. 2022, 23, 6630. https://doi.org/10.3390/ijms23126630
Pedrucci F, Pappalardo C, Marzaro G, Ferri N, Ferlin A, De Toni L. Proteolysis Targeting Chimeric Molecules: Tuning Molecular Strategies for a Clinically Sound Listening. International Journal of Molecular Sciences. 2022; 23(12):6630. https://doi.org/10.3390/ijms23126630
Chicago/Turabian StylePedrucci, Federica, Claudia Pappalardo, Giovanni Marzaro, Nicola Ferri, Alberto Ferlin, and Luca De Toni. 2022. "Proteolysis Targeting Chimeric Molecules: Tuning Molecular Strategies for a Clinically Sound Listening" International Journal of Molecular Sciences 23, no. 12: 6630. https://doi.org/10.3390/ijms23126630