
Genome-Wide Identification and Characterization of RNA/DNA Differences Associated with Fusarium graminearum Infection in Wheat

 ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
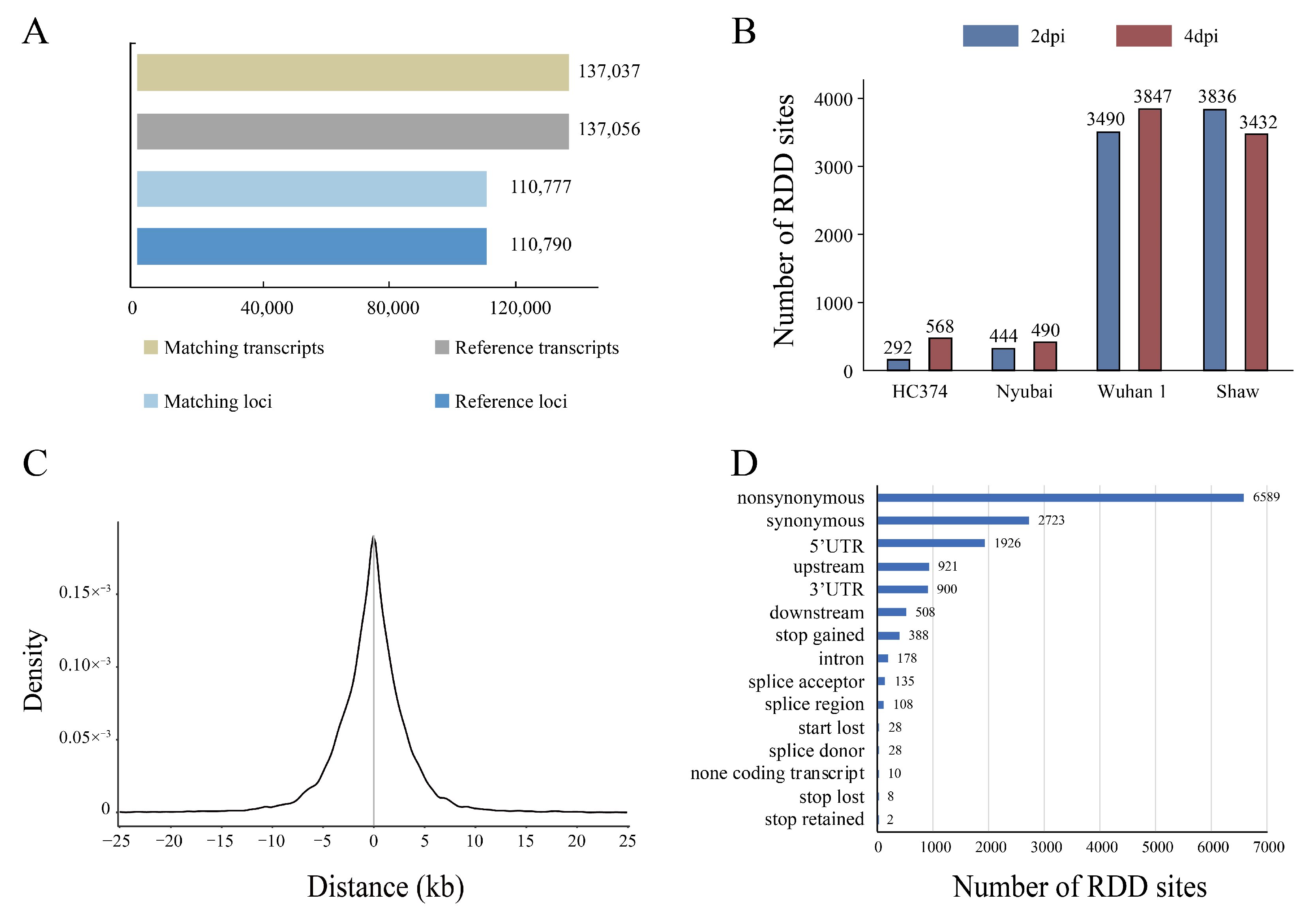
2.1. Identification of RDDs Associated with FHB Using RNA-Seq Data
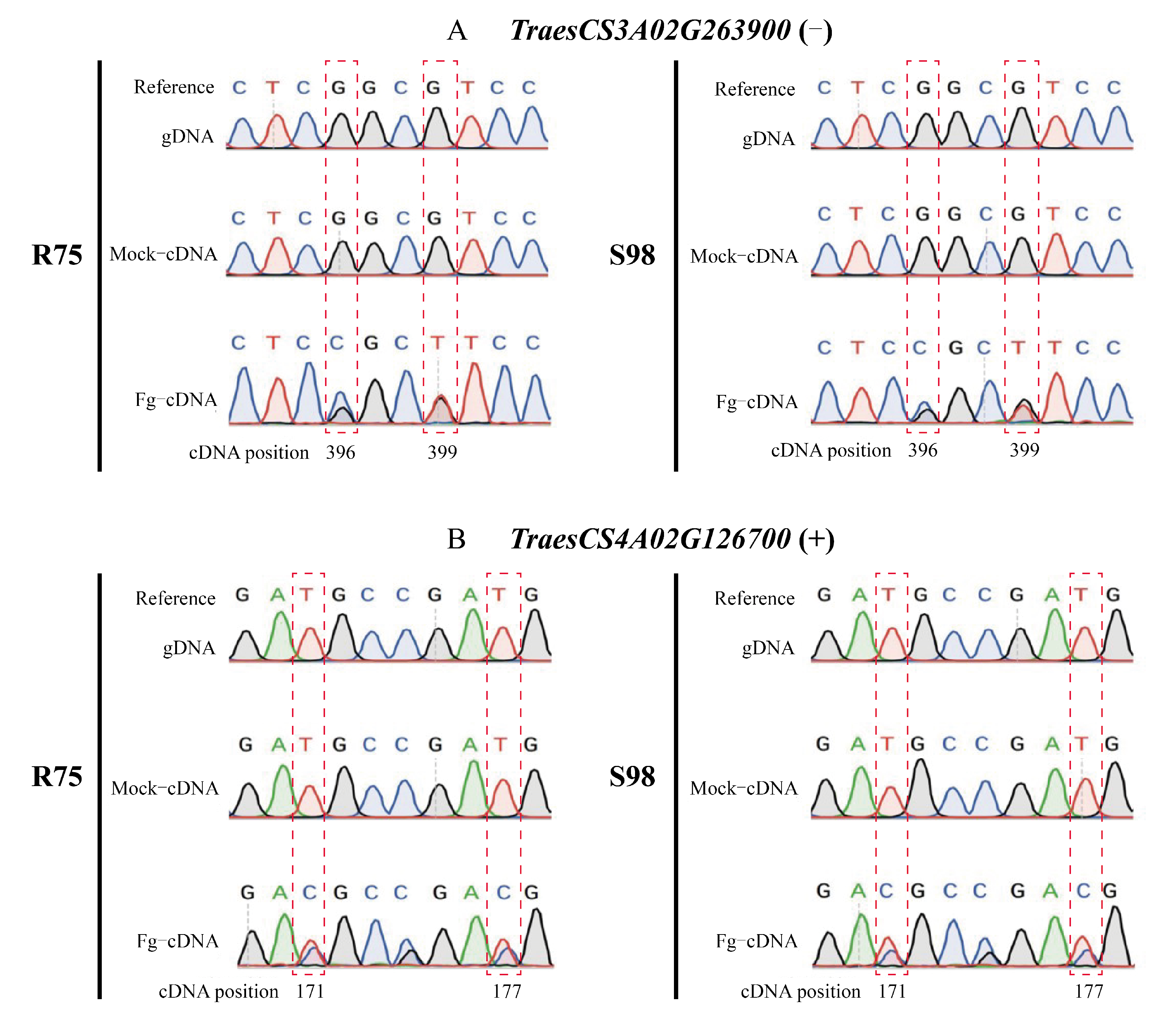
2.2. Identification of Putative High-Confidence RDDs Associated with FHB
2.3. Expression and Enrichment Analysis of RDD Genes
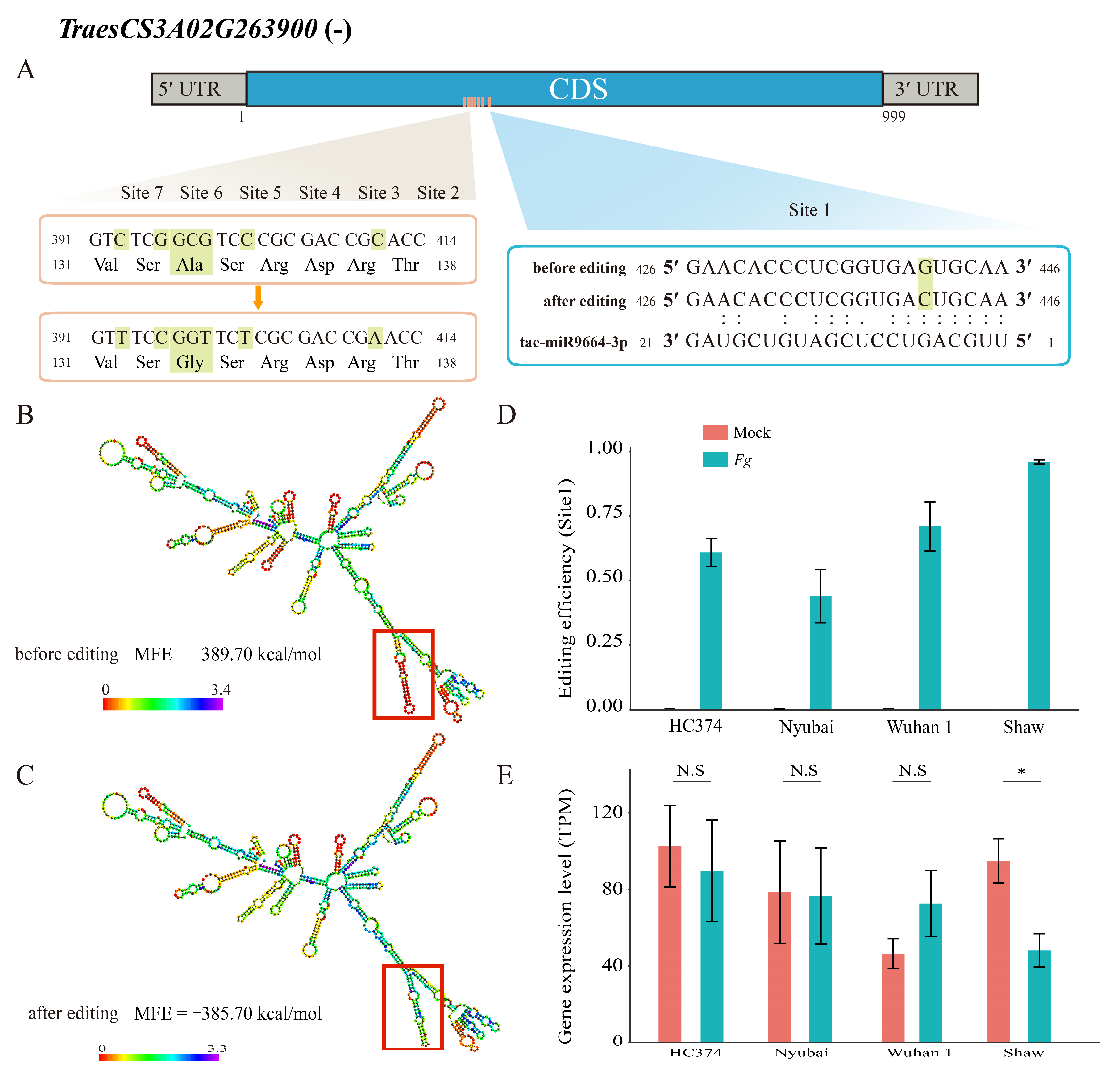
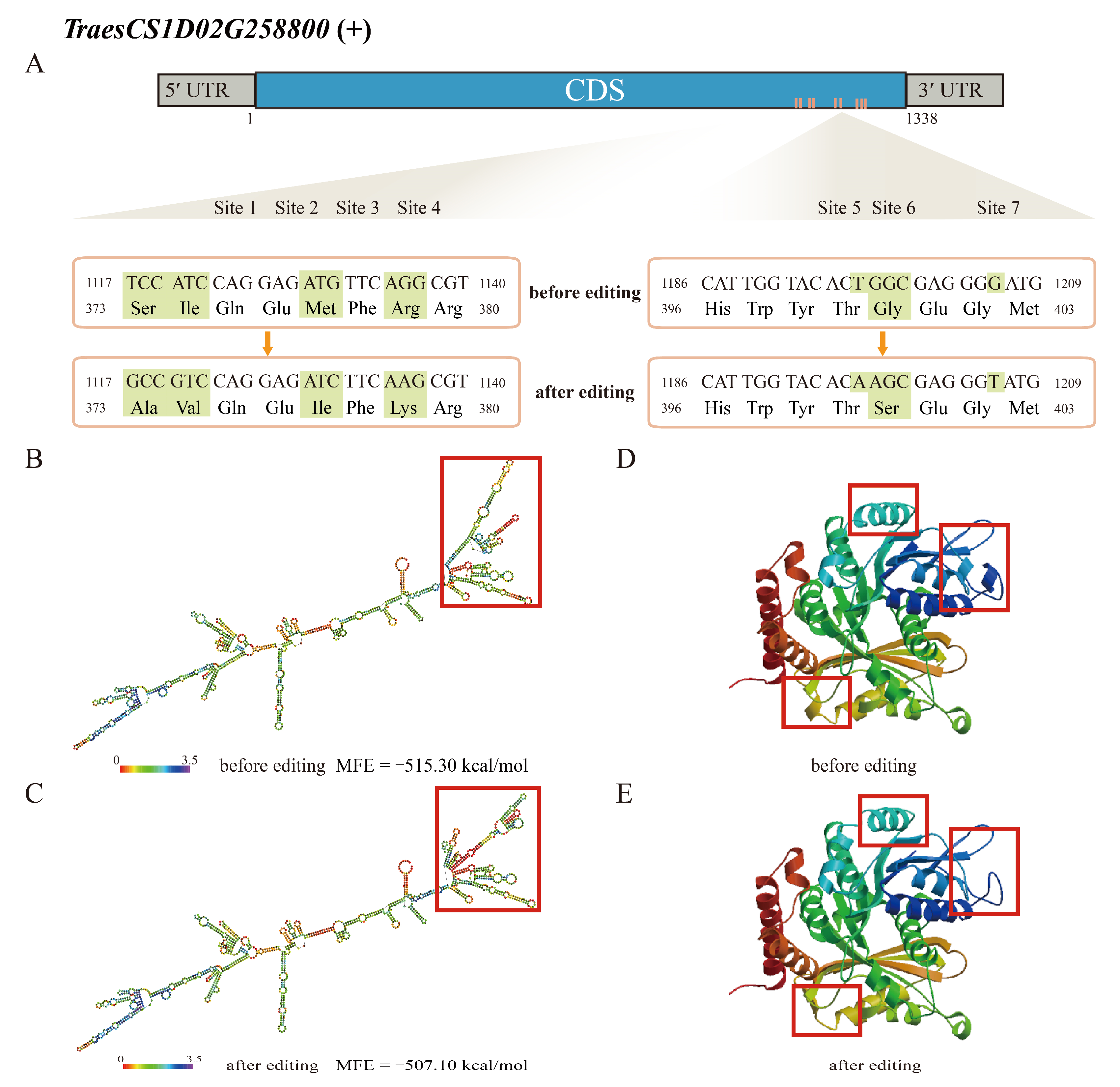
2.4. The Effect of RDDs on RNA Structure
2.5. The Effect of RDDs on Binding Ability and Protein Structure
3. Discussion
4. Materials and Methods
4.1. RNA-Seq Data and Read Mapping
4.2. Identification of RNA/DNA Difference Sites Related to F. graminearum Infection
- (1)
- Systematic errors of the sequencing platform and software were corrected by the GATK VariantFiltration tool, with the initial filter parameter -filter “FS > 30.0”, -filter “QD < 2.0”;
- (2)
- Only RDDs identified in all three biological replicates and mapped by more than 10 reads in each sample with reference read > 2 and edited read > 3 were kept;
- (3)
- RDDs detected in both the F. graminearum-treated samples and their mock control samples within a genotype were removed to exclude genotype-specific genomic polymorphisms;
- (4)
- Approximately 103.8 M single-nucleotide polymorphisms (SNP) from the resequencing data of 414 wheat lines [59] were used to map the obtained RDDs from the above analysis and filter out the putative SNP sites with the same genomic physical position, further removing genomic SNP loci;
- (5)
- Finally, the RDDs shared across all four genotypes, or among the three FHB-resistant genotypes (Nyubai, Wuhan 1 and HC374), were retained and the sequence-mapped information for each RDD was further verified by confirming the alignment information of reads with the Integrative Genomics Viewer (IGV) tool.
4.3. Correlation Analysis between RDD Genes and Traits
4.4. RNA Structure Analysis
4.5. MicroRNA Target Analysis
4.6. Protein Domain and Structure Analysis
4.7. Orthologous Gene Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shewry, P.R. Wheat. J. Exp. Bot. 2009, 60, 1537–1553. [Google Scholar] [CrossRef] [PubMed]
- Appels, R.; Eversole, K.; Feuillet, C.; Keller, B.; Rogers, J.; Stein, N.; Pozniak, C.J.; Stein, N.; Choulet, F.; Distelfeld, A.; et al. Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science 2018, 361, eaar7191. [Google Scholar]
- Gill, B.S.; Appels, R.; Botha-Oberholster, A.-M.; Buell, C.R.; Bennetzen, J.L.; Chalhoub, B.; Chumley, F.; Dvořák, J.; Iwanaga, M.; Keller, B.; et al. A workshop report on wheat genome sequencing: International Genome Research on Wheat Consortium. Genetics 2004, 168, 1087–1096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miransari, M.; Smith, D. Sustainable wheat (Triticum aestivum L.) production in saline fields: A review. Crit. Rev. Biotechnol. 2019, 39, 999–1014. [Google Scholar] [CrossRef]
- Guihua, B.; Shaner, G. Scab of wheat: Prospects for control. Plant Dis. 1994, 78, 760–766. [Google Scholar]
- Dexter, J.E.; Clear, R.M.; Preston, K.R. Fusarium head blight: Effect on the milling and baking of some Canadian wheats. Cereal Chem. 1996, 73, 695–701. [Google Scholar]
- Zhu, Z.; Hao, Y.; Mergoum, M.; Bai, G.; Humphreys, G.; Cloutier, S.; Xia, X.; He, Z. Breeding wheat for resistance to Fusarium head blight in the Global North: China, USA, and Canada. Crop J. 2019, 7, 730–738. [Google Scholar]
- Buerstmayr, H.; Ban, T.; Anderson, J. QTL mapping and marker-assisted selection for Fusarium head blight resistance in wheat: A review. Plant Breed. 2009, 128, 1–26. [Google Scholar] [CrossRef]
- Rawat, N.; Pumphrey, M.O.; Liu, S.; Zhang, X.; Tiwari, V.K.; Ando, K.; Trick, H.N.; Bockus, W.W.; Akhunov, E.; Anderson, J.A.; et al. Wheat Fhb1 encodes a chimeric lectin with agglutinin domains and a pore-forming toxin-like domain conferring resistance to Fusarium head blight. Nat. Genet. 2016, 48, 1576–1580. [Google Scholar] [CrossRef]
- Jia, H.; Zhou, J.; Xue, S.; Li, G.; Yan, H.; Ran, C.; Zhang, Y.; Shi, J.; Jia, L.; Wang, X.; et al. A journey to understand wheat Fusarium head blight resistance in the Chinese wheat landrace Wangshuibai. Crop J. 2018, 6, 48–59. [Google Scholar] [CrossRef]
- Li, G.; Zhou, J.; Jia, H.; Gao, Z.; Fan, M.; Luo, Y.; Zhao, P.; Xue, S.; Li, N.; Yuan, Y.; et al. Mutation of a histidine-rich calcium-binding-protein gene in wheat confers resistance to Fusarium head blight. Nat. Genet. 2019, 51, 1106–1112. [Google Scholar] [CrossRef]
- Su, Z.; Bernardo, A.; Tian, B.; Chen, H.; Wang, S.; Ma, H.; Cai, S.; Liu, D.; Zhang, D.; Li, T.; et al. A deletion mutation in TaHRC confers Fhb1 resistance to Fusarium head blight in wheat. Nat. Genet. 2019, 51, 1099–1105. [Google Scholar] [CrossRef]
- Wang, H.; Sun, S.; Ge, W.; Zhao, L.; Hou, B.; Wang, K.; Lyu, Z.; Chen, L.; Xu, S.; Guo, J.; et al. Horizontal gene transfer of Fhb7 from fungus underlies Fusarium head blight resistance in wheat. Science 2020, 368, eaba5435. [Google Scholar] [CrossRef]
- Pan, Y.; Liu, Z.; Rocheleau, H.; Fauteux, F.; Wang, Y.; McCartney, C.; Ouellet, T. Transcriptome dynamics associated with resistance and susceptibility against fusarium head blight in four wheat genotypes. BMC Genom. 2018, 19, 642. [Google Scholar] [CrossRef]
- Hofstad, A.N.; Nussbaumer, T.; Akhunov, E.; Shin, S.; Kugler, K.G.; Kistler, H.C.; Mayer, K.F.; Muehlbauer, G.J. Examining the Transcriptional Response in Wheat Near-Isogenic Lines to Infection and Deoxynivalenol Treatment. Plant Genome 2016, 9, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Keller, W.; Wolf, J.; Gerber, A. Editing of messenger RNA precursors and of tRNAs by adenosine to inosine conversion. FEBS Lett. 1999, 452, 71–76. [Google Scholar] [CrossRef] [Green Version]
- Stern, D.B.; Goldschmidt-Clermont, M.; Hanson, M.R. Chloroplast RNA metabolism. Annu. Rev. Plant Biol. 2010, 61, 125–155. [Google Scholar] [CrossRef]
- Wang, C.; Xu, J.R.; Liu, H. A-to-I RNA editing independent of ADARs in filamentous fungi. RNA Biol. 2016, 13, 940–945. [Google Scholar] [CrossRef] [Green Version]
- Takenaka, M.; Zehrmann, A.; Verbitskiy, D.; Hartel, B.; Brennicke, A. RNA editing in plants and its evolution. Annu. Rev. Genet. 2013, 47, 335–352. [Google Scholar] [CrossRef]
- Wakasugi, T.; Hirose, T.; Horihata, M.; Tsudzuki, T.; Kössel, H.; Sugiura, M. Creation of a novel protein-coding region at the RNA level in black pine chloroplasts: The pattern of RNA editing in the gymnosperm chloroplast is different from that in angiosperms. Proc. Natl. Acad. Sci. USA 1996, 93, 8766–8770. [Google Scholar] [CrossRef] [Green Version]
- Bock, R.; Kössel, H.; Maliga, P. Introduction of a heterologous editing site into the tobacco plastid genome: The lack of RNA editing leads to a mutant phenotype. EMBO J. 1994, 13, 4623–4628. [Google Scholar] [CrossRef]
- Drescher, A.; Hupfer, H.; Nickel, C.; Albertazzi, F.; Hohmann, U.; Herrmann, R.G.; Maier, R.M. C-to-U conversion in the intercistronic ndhI/ ndhG RNA of plastids from monocot plants: Conventional editing in an unconventional small reading frame? Mol. Genet. Genom. 2002, 267, 262–269. [Google Scholar] [CrossRef]
- Liu, H.; Wang, Q.; He, Y.; Chen, L.; Hao, C.; Jiang, C.; Li, Y.; Dai, Y.; Kang, Z.; Xu, J.R. Genome-wide A-to-I RNA editing in fungi independent of ADAR enzymes. Genome Res. 2016, 26, 499–509. [Google Scholar] [CrossRef] [Green Version]
- Savva, Y.A.; Rieder, L.E.; Reenan, R.A. The ADAR protein family. Genome Biol. 2012, 13, 252. [Google Scholar] [CrossRef]
- Liu, H.; Li, Y.; Chen, D.; Qi, Z.; Wang, Q.; Wang, J.; Jiang, C.; Xu, J.R. A-to-I RNA editing is developmentally regulated and generally adaptive for sexual reproduction in Neurospora crassa. Proc. Natl. Acad. Sci. USA 2017, 114, E7756–E7765. [Google Scholar] [CrossRef] [Green Version]
- Bian, Z.; Ni, Y.; Xu, J.R.; Liu, H. A-to-I mRNA editing in fungi: Occurrence, function, and evolution. Cell. Mol. Life Sci. 2019, 76, 329–340. [Google Scholar] [CrossRef]
- Shikanai, T. RNA editing in plant organelles: Machinery, physiological function and evolution. Cell. Mol. Life Sci. 2006, 63, 698–708. [Google Scholar] [CrossRef]
- Peng, Z.; Cheng, Y.; Tan, B.C.; Kang, L.; Tian, Z.; Zhu, Y.; Zhang, W.; Liang, Y.; Hu, X.; Tan, X.; et al. Comprehensive analysis of RNA-Seq data reveals extensive RNA editing in a human transcriptome. Nat. Biotechnol. 2012, 30, 253–260. [Google Scholar] [CrossRef]
- Meng, Y.; Chen, D.; Jin, Y.; Mao, C.; Wu, P.; Chen, M. RNA editing of nuclear transcripts in Arabidopsis thaliana. BMC Genom. 2010, 11 (Suppl. 4), S12. [Google Scholar] [CrossRef] [Green Version]
- Pachter, L. A closer look at RNA editing. Nat. Biotechnol. 2012, 30, 246–247. [Google Scholar] [CrossRef]
- Wan, Y.; Kertesz, M.; Spitale, R.C.; Segal, E.; Chang, H.Y. Understanding the transcriptome through RNA structure. Nat. Rev. Genet. 2011, 12, 641–655. [Google Scholar] [CrossRef] [Green Version]
- Dethoff, E.A.; Chugh, J.; Mustoe, A.M.; Al-Hashimi, H.M. Functional complexity and regulation through RNA dynamics. Nature 2012, 482, 322–330. [Google Scholar] [CrossRef] [Green Version]
- Mao, L.; Ling, H.-Q.; Wan, J. Wheat functional genomics research in China: A decade of development. Crop J. 2018, 6, 1–6. [Google Scholar] [CrossRef]
- Steiner, B.; Buerstmayr, M.; Michel, S.; Schweiger, W.; Lemmens, M.; Buerstmayr, H. Breeding strategies and advances in line selection for Fusarium head blight resistance in wheat. Trop. Plant Pathol. 2017, 42, 165–174. [Google Scholar] [CrossRef] [Green Version]
- Buerstmayr, M.; Steiner, B.; Buerstmayr, H.; Léon, J. Breeding for Fusarium head blight resistance in wheat—progress and challenges. Plant Breed. 2019, 139, 429–454. [Google Scholar] [CrossRef]
- Gong, X.; He, X.; Zhang, Y.; Li, L.; Sun, Z.; Bai, G.; Singh, P.K.; Li, T. Development of an Evaluation System for Fusarium Resistance in Wheat Grains and Its Application in Assessment of the Corresponding Effects of Fhb1. Plant Dis. 2020, 104, 2210–2216. [Google Scholar] [CrossRef]
- Qulsum, U.; Azad, M.T.A.; Tsukahara, T. Analysis of Tissue-specific RNA Editing Events of Genes Involved in RNA Editing in Arabidopsis thaliana. J. Plant Biol. 2019, 62, 351–358. [Google Scholar] [CrossRef]
- Zhang, A.; Jiang, X.; Zhang, F.; Wang, T.; Zhang, X. Dynamic response of RNA editing to temperature in Grape by RNA deep-sequencing. Funct. Integr. Genom. 2019, 20, 421–432. [Google Scholar] [CrossRef] [Green Version]
- Solomon, O.; Segni, A.D.; Cesarkas, K.; Porath, H.T.; Marcu-Malina, V.; Mizrahi, O.; Stern-Ginossar, N.; Kol, N.; Farage-Barhom, S.; Glick-Saar, E.; et al. RNA editing by ADAR1 leads to context-dependent transcriptome-wide changes in RNA secondary structure. Nat. Commun. 2017, 8, 1440. [Google Scholar] [CrossRef]
- Warf, M.B.; Berglund, J.A. Role of RNA structure in regulating pre-mRNA splicing. Trends Biochem. Sci. 2010, 35, 169–178. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Ye, J.; Yu, C.; Xie, Q.; Wang, X.; Yu, H.; Song, J.; Li, C.; Cui, L.; Han, H.; et al. Endogenous RNA editing of a nuclear gene BOSS triggers flowering in tomato. BioRxiv 2021. [Google Scholar] [CrossRef]
- Dutcher, S.K. The tubulin fraternity: Alpha to eta. Curr. Opin. Cell Biol. 2001, 13, 49–54. [Google Scholar] [CrossRef]
- Yoshikawa, M.; Yang, G.X.; Kawaguchi, K.; Komatsu, S. Expression analyses of β-tubulin isotype genes in rice. Plant Cell Physiol. 2003, 44, 1202–1207. [Google Scholar] [CrossRef] [Green Version]
- Lefèvre, J.; Chernov, K.G.; Joshi, V.; Delga, S.; Toma, F.; Pastré, D.; Curmi, D.; Savarin, P. The C terminus of tubulin, a versatile partner for cationic molecules—Binding of Tau, polyamines and calcium. J. Biol. Chem. 2011, 286, 3065–3078. [Google Scholar] [CrossRef] [Green Version]
- Livanos, P.; Galatis, B.; Apostolakos, P. The interplay between ROS and tubulin cytoskeleton in plants. Plant Signal. Behav. 2014, 9, e28069. [Google Scholar] [CrossRef]
- Waszczak, C.; Carmody, M.; Kangasjärvi, J. Reactive Oxygen Species in Plant Signaling. Annu. Rev. Plant Biol. 2018, 69, 209–236. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Castillo-González, C.; Yu, B.; Zhang, X. The functions of plant small RNAs in development and in stress responses. Plant J. 2017, 90, 654–670. [Google Scholar] [CrossRef] [Green Version]
- Thornton, C.; Tang, K.C.; Phamluong, K.; Luong, K.; Vagts, A.; Nikanjam, D.; Yaka, R.; Ron, D. Spatial and temporal regulation of RACK1 function and N-methyl-D-aspartate receptor activity through WD40 motif-mediated dimerization. J. Biol. Chem. 2004, 279, 31357–31364. [Google Scholar] [CrossRef] [Green Version]
- Nakashima, A.; Chen, L.; Thao, N.P.; Fujiwara, M.; Wong, H.L.; Kuwano, M.; Umemura, K.; Shirasu, K.; Kawasaki, T.; Shimamoto, K. Rack1 functions in rice innate immunity by interacting with the rac1 immune complex. Plant Cell 2008, 20, 2265–2279. [Google Scholar] [CrossRef] [Green Version]
- Ono, E.; Wong, H.L.; Kawasaki, T.; Hasegawa, M.; Kodama, O.; Shimamoto, K. Essential role of the small GTPase Rac in disease resistance of rice. Proc. Natl. Acad. Sci. USA 2001, 98, 759–764. [Google Scholar] [CrossRef]
- Li, J.; Zhang, H.; Yang, R.; Zeng, Q.; Han, G.; Du, Y.; Yang, J.; Yang, G.; Luo, Q. Identification of miRNAs Contributing to the Broad-Spectrum and Durable Blast Resistance in the Yunnan Local Rice Germplasm. Front. Plant Sci. 2021, 12, 749919. [Google Scholar] [CrossRef]
- Bukau, B.; Horwich, A.L. The Hsp70 and Hsp60 chaperone machines. Cell 1998, 92, 351–366. [Google Scholar] [CrossRef] [Green Version]
- Berka, M.; Kopecká, R.; Berková, V.; Brzobohatý, B.; Cerný, M. Regulation of heat shock proteins 70 and their role in plant immunity. J. Exp. Bot. 2022, 73, 1894–1909. [Google Scholar] [CrossRef]
- Payá-Milans, M.; Olmstead, J.W.; Nunez, G.; Rinehart, T.A.; Staton, M. Comprehensive evaluation of RNA-seq analysis pipelines in diploid and polyploid species. Gigascience 2018, 7, giy132. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Ramaswami, G.; Zhang, R.; Piskol, R.; Keegan, L.P.; Deng, P.; O’Connell, M.A.; Li, J.B. Identifying RNA editing sites using RNA sequencing data alone. Nat. Methods 2013, 10, 128–132. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhao, X.; Li, Y.; Xu, J.; Bi, A.; Kang, L.; Xu, D.; Chen, H.; Wang, Y.; Wang, Y.; et al. Triticum population sequencing provides insights into wheat adaptation. Nat. Genet. 2020, 52, 1412–1422. [Google Scholar] [CrossRef]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [Green Version]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.Y.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gruber, A.R.; Lorenz, R.; Bernhart, S.H.; Neubock, R.; Hofacker, I.L. The Vienna RNA websuite. Nucleic Acids Res. 2008, 36, W70–W74. [Google Scholar] [CrossRef] [Green Version]
- Mao, Y.; Li, Q.; Wang, W.; Liang, P.; Tao, S. Number variation of high stability regions is correlated with gene functions. Genome Biol. Evol. 2013, 5, 484–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, X.; Zhao, P.X. psRNATarget: A plant small RNA target analysis server. Nucleic Acids Res. 2011, 39, W155–W159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011, 39, W29–W37. [Google Scholar] [CrossRef] [Green Version]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, G.; Pan, Y.; Zhao, Q.; Huang, J.; Pan, W.; Cui, L.; Song, W.; Ouellet, T.; Pan, Y.; Nie, X. Genome-Wide Identification and Characterization of RNA/DNA Differences Associated with Fusarium graminearum Infection in Wheat. Int. J. Mol. Sci. 2022, 23, 7982. https://doi.org/10.3390/ijms23147982
Yang G, Pan Y, Zhao Q, Huang J, Pan W, Cui L, Song W, Ouellet T, Pan Y, Nie X. Genome-Wide Identification and Characterization of RNA/DNA Differences Associated with Fusarium graminearum Infection in Wheat. International Journal of Molecular Sciences. 2022; 23(14):7982. https://doi.org/10.3390/ijms23147982
Chicago/Turabian StyleYang, Guang, Yan Pan, Qinlong Zhao, Jiaqian Huang, Wenqiu Pan, Licao Cui, Weining Song, Therese Ouellet, Youlian Pan, and Xiaojun Nie. 2022. "Genome-Wide Identification and Characterization of RNA/DNA Differences Associated with Fusarium graminearum Infection in Wheat" International Journal of Molecular Sciences 23, no. 14: 7982. https://doi.org/10.3390/ijms23147982
APA StyleYang, G., Pan, Y., Zhao, Q., Huang, J., Pan, W., Cui, L., Song, W., Ouellet, T., Pan, Y., & Nie, X. (2022). Genome-Wide Identification and Characterization of RNA/DNA Differences Associated with Fusarium graminearum Infection in Wheat. International Journal of Molecular Sciences, 23(14), 7982. https://doi.org/10.3390/ijms23147982