
The Role of Histology-Agnostic Drugs in the Treatment of Metastatic Castration-Resistant Prostate Cancer

 , , , , and
, , , , and 
Abstract
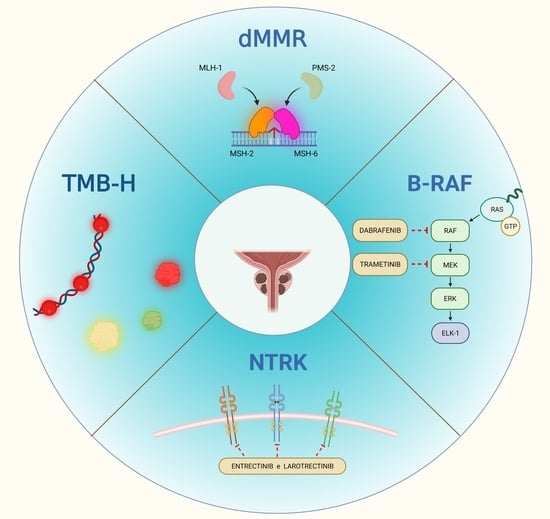
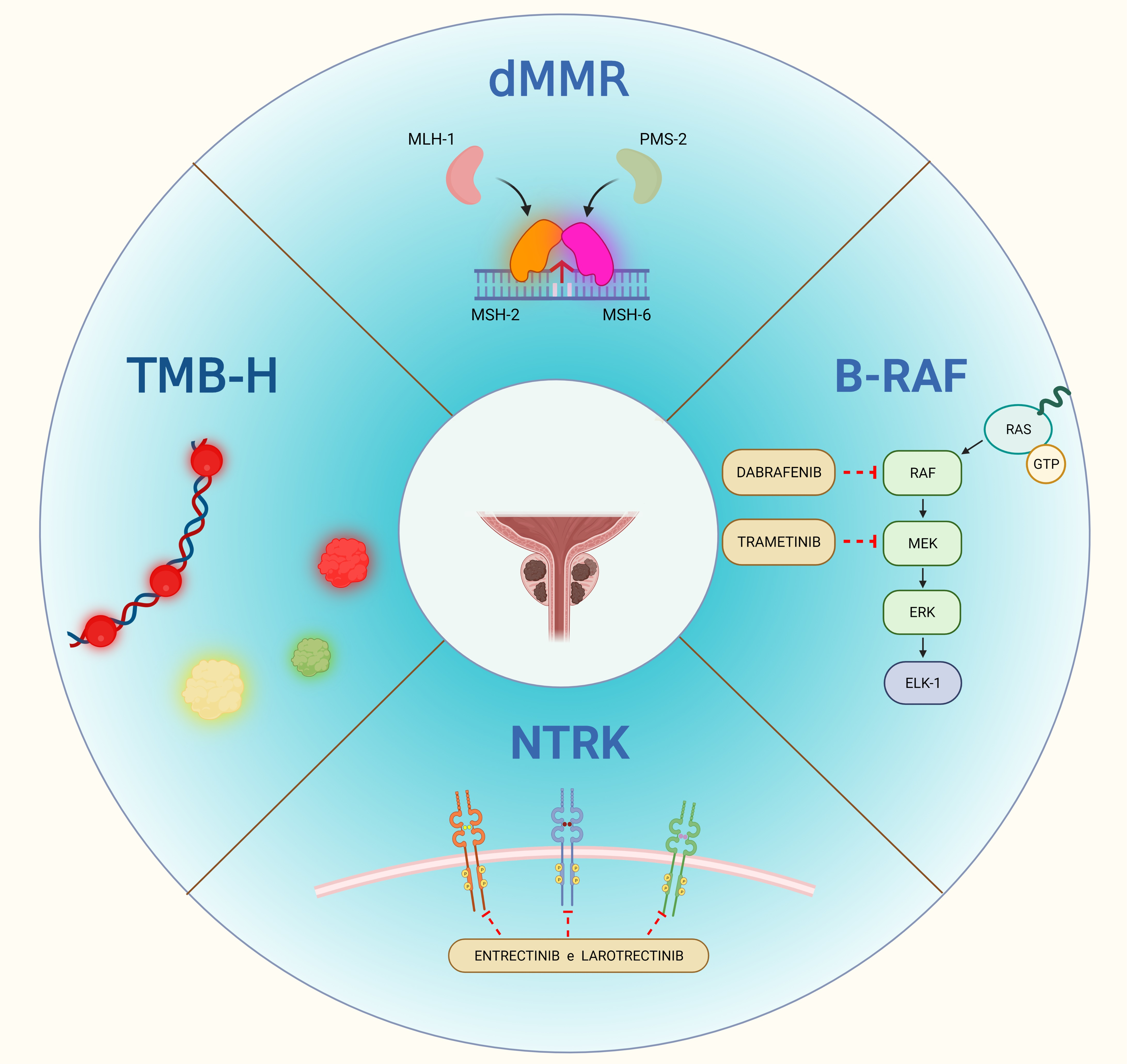
:
1. Introduction
Objectives
2. Methods
3. Results
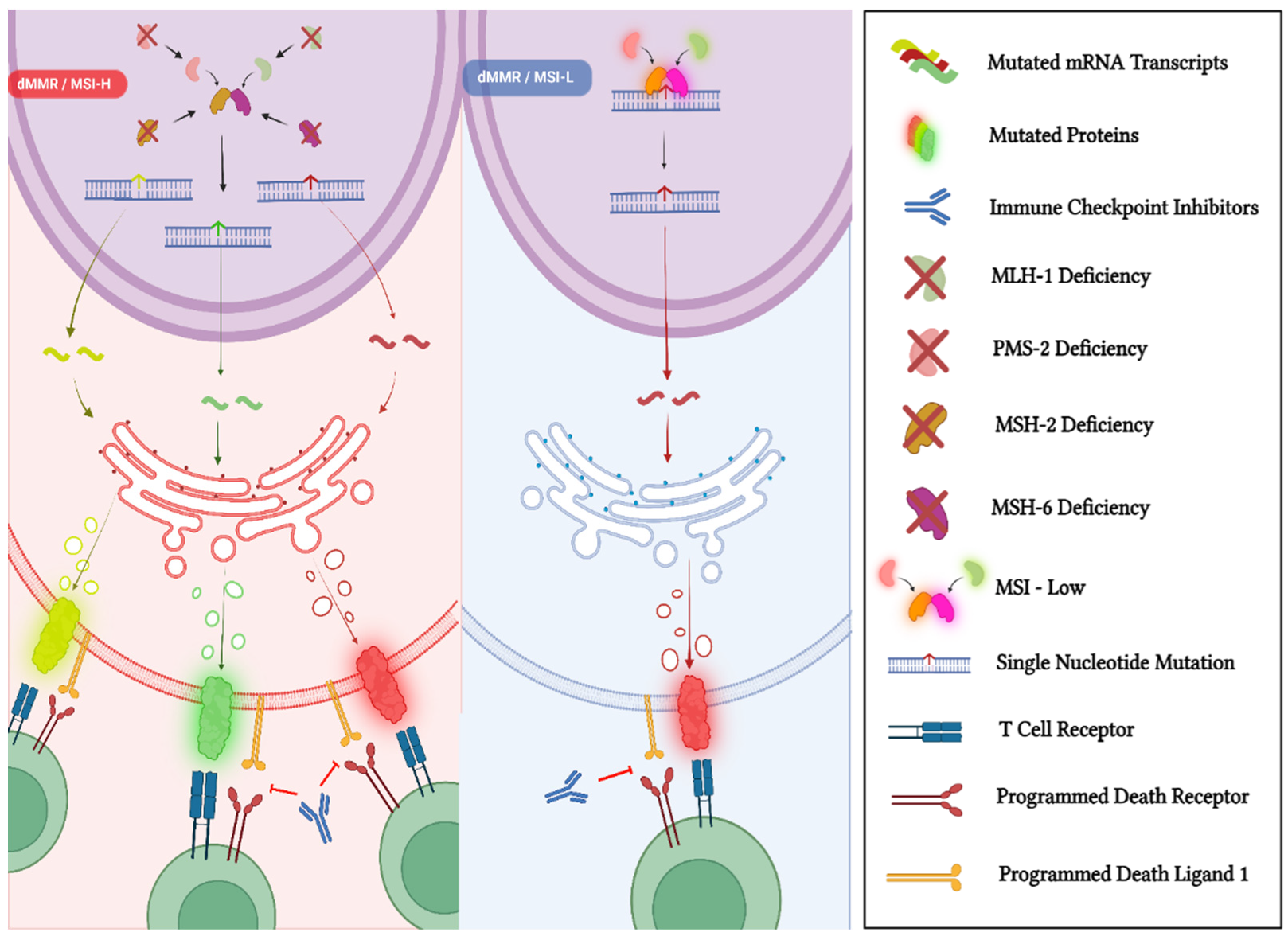
3.1. Mismatch-Repair Deficiency and High Microsatellite Instability
3.1.1. dMMR/MSI-H Status in PCa
3.1.2. Approval of Pembrolizumab and Dostarlimab for MSI-H/dMMR Solid Tumors
3.1.3. Pembrolizumab and Dostarlimab in MSI-H/dMMR mCRPC
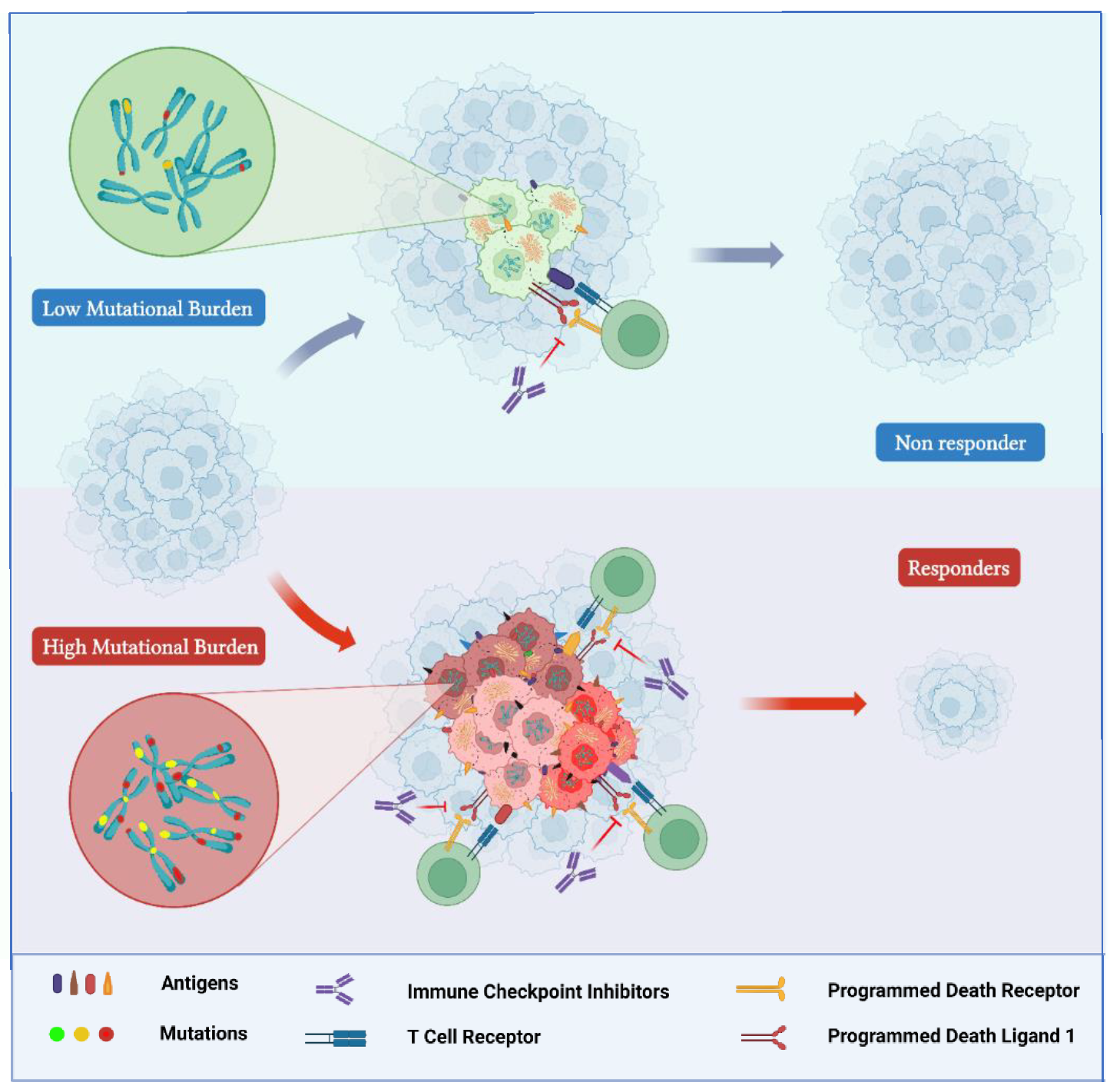
3.2. Tumor Mutational Burden
3.2.1. TMB-H Status in PCa
3.2.2. Approval of Pembrolizumab for TMB-H Solid Tumors
3.2.3. Pembrolizumab in TMB-H mCRPC
3.3. Neurotrophic Tropomyosin Receptor Kinase Gene Fusions
3.3.1. The NTRK Gene Fusions in PCa
3.3.2. Larotrectinib and Entrectinib in Tumors Harboring NTRK Fusion Genes
3.3.3. Larotrectinib and Entrectinib in mCRPC
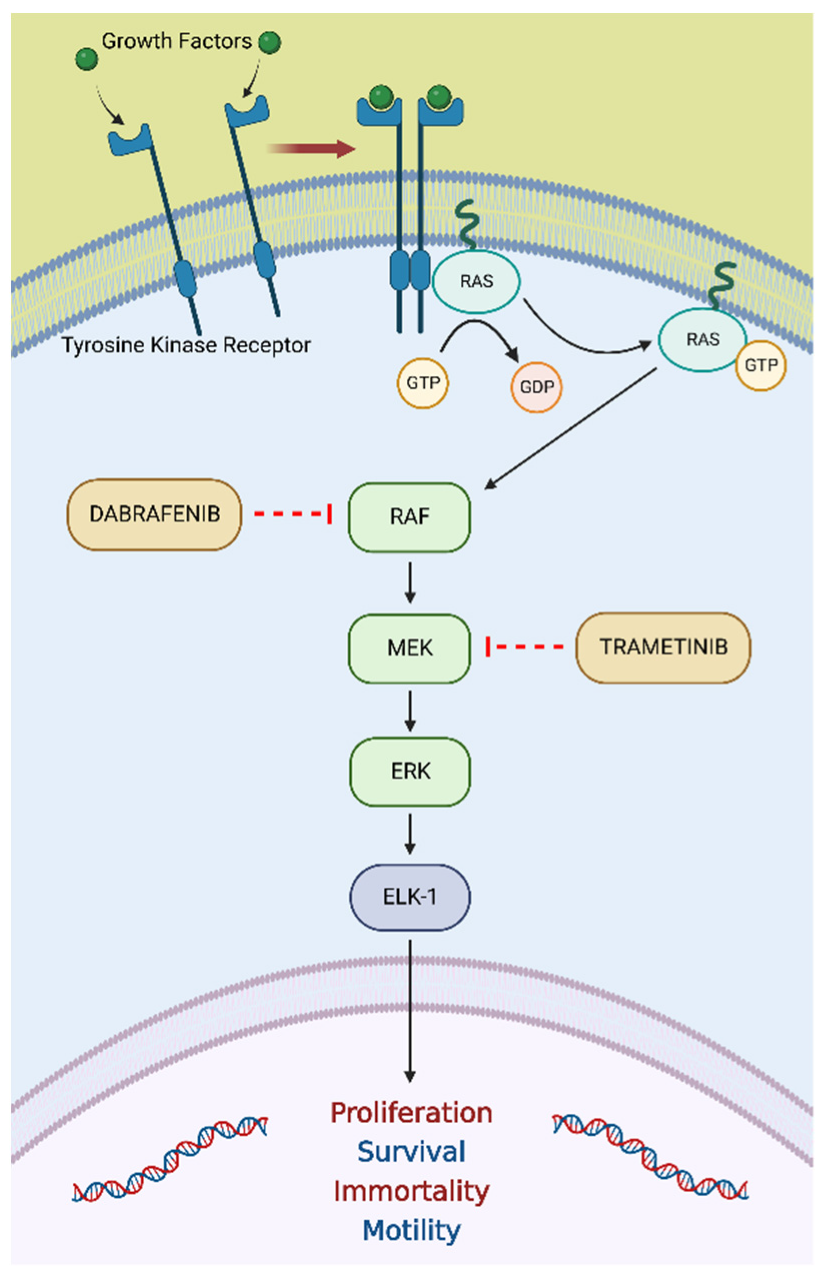
3.4. BRAF V600E Mutation
3.4.1. BRAF V600E Mutation in PCa
3.4.2. Dabrafenib plus Trametinib in Tumors Harboring a BRAF V600E Mutation
3.4.3. Dabrafenib plus Trametinib in mCRPC Harboring a BRAF V600E Mutation
4. Discussion
4.1. Future Perspectives
4.2. Limitations
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Cattrini, C.; España, R.; Mennitto, A.; Bersanelli, M.; Castro, E.; Olmos, D.; Lorente, D.; Gennari, A. Optimal Sequencing and Predictive Biomarkers in Patients with Advanced Prostate Cancer. Cancers 2021, 13, 4522. [Google Scholar] [CrossRef] [PubMed]
- Sandhu, S.; Moore, C.M.; Chiong, E.; Beltran, H.; Bristow, R.G.; Williams, S.G. Prostate Cancer. Lancet 2021, 398, 1075–1090. [Google Scholar] [CrossRef]
- Tannock, I.F.; de Wit, R.; Berry, W.R.; Horti, J.; Pluzanska, A.; Chi, K.N.; Oudard, S.; Théodore, C.; James, N.D.; Turesson, I.; et al. Docetaxel plus Prednisone or Mitoxantrone plus Prednisone for Advanced Prostate Cancer. N. Engl. J. Med. 2004, 351, 1502–1512. [Google Scholar] [CrossRef] [Green Version]
- De Bono, J.S.; Oudard, S.; Ozguroglu, M.; Hansen, S.; Machiels, J.-P.; Kocak, I.; Gravis, G.; Bodrogi, I.; Mackenzie, M.J.; Shen, L.; et al. Prednisone plus Cabazitaxel or Mitoxantrone for Metastatic Castration-Resistant Prostate Cancer Progressing after Docetaxel Treatment: A Randomised Open-Label Trial. Lancet 2010, 376, 1147–1154. [Google Scholar] [CrossRef]
- de Bono, J.S.; Logothetis, C.J.; Molina, A.; Fizazi, K.; North, S.; Chu, L.; Chi, K.N.; Jones, R.J.; Goodman, O.B.; Saad, F.; et al. Abiraterone and Increased Survival in Metastatic Prostate Cancer. N. Engl. J. Med. 2011, 364, 1995–2005. [Google Scholar] [CrossRef]
- Scher, H.I.; Fizazi, K.; Saad, F.; Taplin, M.-E.; Sternberg, C.N.; Miller, K.; de Wit, R.; Mulders, P.; Chi, K.N.; Shore, N.D.; et al. Increased Survival with Enzalutamide in Prostate Cancer after Chemotherapy. N. Engl. J. Med. 2012, 367, 1187–1197. [Google Scholar] [CrossRef] [Green Version]
- De Bono, J.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Olaparib for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2020, 382, 2091–2102. [Google Scholar] [CrossRef]
- Parker, C.; Nilsson, S.; Heinrich, D.; Helle, S.I.; O’Sullivan, J.M.; Fosså, S.D.; Chodacki, A.; Wiechno, P.; Logue, J.; Seke, M.; et al. Alpha Emitter Radium-223 and Survival in Metastatic Prostate Cancer. N. Engl. J. Med. 2013, 369, 213–223. [Google Scholar] [CrossRef] [Green Version]
- Sartor, O.; de Bono, J.; Chi, K.N.; Fizazi, K.; Herrmann, K.; Rahbar, K.; Tagawa, S.T.; Nordquist, L.T.; Vaishampayan, N.; El-Haddad, G.; et al. Lutetium-177-PSMA-617 for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2021, 385, 1091–1103. [Google Scholar] [CrossRef]
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. Sipuleucel-T Immunotherapy for Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2010, 363, 411–422. [Google Scholar] [CrossRef] [Green Version]
- Rizzo, A.; Mollica, V.; Cimadamore, A.; Santoni, M.; Scarpelli, M.; Giunchi, F.; Cheng, L.; Lopez-Beltran, A.; Fiorentino, M.; Montironi, R.; et al. Is There a Role for Immunotherapy in Prostate Cancer? Cells 2020, 9, 2051. [Google Scholar] [CrossRef]
- Turco, F.; Tucci, M.; Angusti, T.; Parente, A.; Di Stefano, R.F.; Urban, S.; Pisano, C.; Samuelly, A.; Audisio, A.; Audisio, M.; et al. Role of Radium-223 Discontinuation Due to Adverse Events in Castration-Resistant Prostate Cancer Patients. A Retrospective Monocentric Analysis. Tumori 2022, 3008916221077144. [Google Scholar] [CrossRef]
- Pestana, R.C.; Sen, S.; Hobbs, B.P.; Hong, D.S. Histology-Agnostic Drug Development—Considering Issues beyond the Tissue. Nat. Rev. Clin. Oncol 2020, 17, 555–568. [Google Scholar] [CrossRef]
- Marshall, J.L.; Peshkin, B.N.; Yoshino, T.; Vowinckel, J.; Danielsen, H.E.; Melino, G.; Tsamardinos, I.; Haudenschild, C.; Kerr, D.J.; Sampaio, C.; et al. The Essentials of Multiomics. Oncologist 2022, 27, 272–284. [Google Scholar] [CrossRef]
- Tarantino, P.; Mazzarella, L.; Marra, A.; Trapani, D.; Curigliano, G. The Evolving Paradigm of Biomarker Actionability: Histology-Agnosticism as a Spectrum, Rather than a Binary Quality. Cancer Treat. Rev. 2021, 94, 102169. [Google Scholar] [CrossRef]
- Park, J.J.H.; Siden, E.; Zoratti, M.J.; Dron, L.; Harari, O.; Singer, J.; Lester, R.T.; Thorlund, K.; Mills, E.J. Systematic Review of Basket Trials, Umbrella Trials, and Platform Trials: A Landscape Analysis of Master Protocols. Trials 2019, 20, 572. [Google Scholar] [CrossRef] [Green Version]
- FDA Grants Accelerated Approval to Pembrolizumab for First Tissue/Site Agnostic Indication. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-pembrolizumab-first-tissuesite-agnostic-indication (accessed on 24 June 2022).
- Marcus, L.; Lemery, S.J.; Keegan, P.; Pazdur, R. FDA Approval Summary: Pembrolizumab for the Treatment of Microsatellite Instability-High Solid Tumors. Clin. Cancer Res. 2019, 25, 3753–3758. [Google Scholar] [CrossRef] [Green Version]
- FDA Approves Larotrectinib for Solid Tumors with NTRK Gene Fusions. Available online: https://www.fda.gov/drugs/fda-approves-larotrectinib-solid-tumors-ntrk-gene-fusions (accessed on 24 June 2022).
- FDA Approves Entrectinib for NTRK Solid Tumors and ROS-1 NSCLC. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-entrectinib-ntrk-solid-tumors-and-ros-1-nsclc (accessed on 24 June 2022).
- Marcus, L.; Donoghue, M.; Aungst, S.; Myers, C.E.; Helms, W.S.; Shen, G.; Zhao, H.; Stephens, O.; Keegan, P.; Pazdur, R. FDA Approval Summary: Entrectinib for the Treatment of NTRK Gene Fusion Solid Tumors. Clin. Cancer Res. 2021, 27, 928–932. [Google Scholar] [CrossRef]
- FDA Approves Pembrolizumab for Adults and Children with TMB-H Solid Tumors. Available online: https://www.fda.gov/drugs/drug-approvals-and-databases/fda-approves-pembrolizumab-adults-and-children-tmb-h-solid-tumors (accessed on 24 June 2022).
- Marcus, L.; Fashoyin-Aje, L.A.; Donoghue, M.; Yuan, M.; Rodriguez, L.; Gallagher, P.S.; Philip, R.; Ghosh, S.; Theoret, M.R.; Beaver, J.A.; et al. FDA Approval Summary: Pembrolizumab for the Treatment of Tumor Mutational Burden-High Solid Tumors. Clin. Cancer Res. 2021, 27, 4685–4689. [Google Scholar] [CrossRef]
- FDA Grants Accelerated Approval to Dostarlimab-Gxly for dMMR Advanced Solid Tumors. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-dostarlimab-gxly-dmmr-advanced-solid-tumors (accessed on 24 June 2022).
- FDA Grants Accelerated Approval to Dabrafenib in Combination with Trametinib for Unresectable or Metastatic Solid Tumors with BRAF V600E Mutation. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-dabrafenib-combination-trametinib-unresectable-or-metastatic-solid (accessed on 24 June 2022).
- Li, G.-M. Mechanisms and Functions of DNA Mismatch Repair. Cell Res. 2008, 18, 85–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiricny, J. The Multifaceted Mismatch-Repair System. Nat. Rev. Mol. Cell Biol. 2006, 7, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Graham, L.S.; Schweizer, M.T. Mismatch Repair Deficiency and Clinical Implications in Prostate Cancer. Prostate 2022, 82 (Suppl. S1), S37–S44. [Google Scholar] [CrossRef] [PubMed]
- Pećina-Šlaus, N.; Kafka, A.; Salamon, I.; Bukovac, A. Mismatch Repair Pathway, Genome Stability and Cancer. Front. Mol. Biosci. 2020, 7, 122. [Google Scholar] [CrossRef]
- Li, K.; Luo, H.; Huang, L.; Luo, H.; Zhu, X. Microsatellite Instability: A Review of What the Oncologist Should Know. Cancer Cell Int. 2020, 20, 16. [Google Scholar] [CrossRef] [Green Version]
- Kok, M.; Chalabi, M.; Haanen, J. How I Treat MSI Cancers with Advanced Disease. ESMO Open 2019, 4, e000511. [Google Scholar] [CrossRef] [Green Version]
- EAU Prostate Cancer Guidelines. Available online: https://uroweb.org/guidelines/prostate-cancer (accessed on 24 June 2022).
- NCCN Prostate Cancer Guidelines Version 4.2022. Available online: https://www.nccn.org/guidelines/guidelines-detail?category=1&id=1459 (accessed on 24 June 2022).
- Robinson, D.; Van Allen, E.M.; Wu, Y.-M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.-M.; Montgomery, B.; Taplin, M.-E.; Pritchard, C.C.; Attard, G.; et al. Integrative Clinical Genomics of Advanced Prostate Cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef] [Green Version]
- Pritchard, C.C.; Mateo, J.; Walsh, M.F.; De Sarkar, N.; Abida, W.; Beltran, H.; Garofalo, A.; Gulati, R.; Carreira, S.; Eeles, R.; et al. Inherited DNA-Repair Gene Mutations in Men with Metastatic Prostate Cancer. N. Engl. J. Med. 2016, 375, 443–453. [Google Scholar] [CrossRef]
- Guedes, L.B.; Antonarakis, E.S.; Schweizer, M.T.; Mirkheshti, N.; Almutairi, F.; Park, J.C.; Glavaris, S.; Hicks, J.; Eisenberger, M.A.; De Marzo, A.M.; et al. MSH2 Loss in Primary Prostate Cancer. Clin. Cancer Res. 2017, 23, 6863–6874. [Google Scholar] [CrossRef] [Green Version]
- Abida, W.; Cheng, M.L.; Armenia, J.; Middha, S.; Autio, K.A.; Vargas, H.A.; Rathkopf, D.; Morris, M.J.; Danila, D.C.; Slovin, S.F.; et al. Analysis of the Prevalence of Microsatellite Instability in Prostate Cancer and Response to Immune Checkpoint Blockade. JAMA Oncol. 2019, 5, 471–478. [Google Scholar] [CrossRef]
- Latham, A.; Srinivasan, P.; Kemel, Y.; Shia, J.; Bandlamudi, C.; Mandelker, D.; Middha, S.; Hechtman, J.; Zehir, A.; Dubard-Gault, M.; et al. Microsatellite Instability Is Associated with the Presence of Lynch Syndrome Pan-Cancer. J. Clin. Oncol. 2019, 37, 286–295. [Google Scholar] [CrossRef]
- Nicolosi, P.; Ledet, E.; Yang, S.; Michalski, S.; Freschi, B.; O’Leary, E.; Esplin, E.D.; Nussbaum, R.L.; Sartor, O. Prevalence of Germline Variants in Prostate Cancer and Implications for Current Genetic Testing Guidelines. JAMA Oncol. 2019, 5, 523–528. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Wei, Y.; Pan, J.; Jin, S.; Gu, W.; Gan, H.; Zhu, Y.; Ye, D.-W. Prevalence of Comprehensive DNA Damage Repair Gene Germline Mutations in Chinese Prostate Cancer Patients. Int. J. Cancer 2021, 148, 673–681. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Network. Comprehensive Molecular Characterization of Human Colon and Rectal Cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [Green Version]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch Repair Deficiency Predicts Response of Solid Tumors to PD-1 Blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [Green Version]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [Green Version]
- Marabelle, A.; Le, D.T.; Ascierto, P.A.; Di Giacomo, A.M.; De Jesus-Acosta, A.; Delord, J.-P.; Geva, R.; Gottfried, M.; Penel, N.; Hansen, A.R.; et al. Efficacy of Pembrolizumab in Patients with Noncolorectal High Microsatellite Instability/Mismatch Repair-Deficient Cancer: Results From the Phase II KEYNOTE-158 Study. J. Clin. Oncol. 2020, 38, 1–10. [Google Scholar] [CrossRef]
- Safety and Efficacy of anti–PD-1 Antibody Dostarlimab in Patients (pts) with Mismatch Repair-Deficient (dMMR) Solid Cancers: Results from GARNET Study. Available online: https://ascopubs.org/doi/abs/10.1200/JCO.2021.39.3_suppl.9 (accessed on 24 June 2022).
- Antonarakis, E.S.; Shaukat, F.; Isaacsson Velho, P.; Kaur, H.; Shenderov, E.; Pardoll, D.M.; Lotan, T.L. Clinical Features and Therapeutic Outcomes in Men with Advanced Prostate Cancer and DNA Mismatch Repair Gene Mutations. Eur. Urol. 2019, 75, 378–382. [Google Scholar] [CrossRef]
- Graham, L.S.; Montgomery, B.; Cheng, H.H.; Yu, E.Y.; Nelson, P.S.; Pritchard, C.; Erickson, S.; Alva, A.; Schweizer, M.T. Mismatch Repair Deficiency in Metastatic Prostate Cancer: Response to PD-1 Blockade and Standard Therapies. PLoS ONE 2020, 15, e0233260. [Google Scholar] [CrossRef]
- Barata, P.; Agarwal, N.; Nussenzveig, R.; Gerendash, B.; Jaeger, E.; Hatton, W.; Ledet, E.; Lewis, B.; Layton, J.; Babiker, H.; et al. Clinical Activity of Pembrolizumab in Metastatic Prostate Cancer with Microsatellite Instability High (MSI-H) Detected by Circulating Tumor DNA. J. Immunother. Cancer 2020, 8, e001065. [Google Scholar] [CrossRef]
- Sena, L.A.; Fountain, J.; Isaacsson Velho, P.; Lim, S.J.; Wang, H.; Nizialek, E.; Rathi, N.; Nussenzveig, R.; Maughan, B.L.; Velez, M.G.; et al. Tumor Frameshift Mutation Proportion Predicts Response to Immunotherapy in Mismatch Repair-Deficient Prostate Cancer. Oncologist 2021, 26, e270–e278. [Google Scholar] [CrossRef]
- Shimizu, K.; Sano, T.; Mizuno, K.; Sunada, T.; Makita, N.; Hagimoto, H.; Goto, T.; Sawada, A.; Fujimoto, M.; Ichioka, K.; et al. A Case of Microsatellite Instability-High Clinically Advanced Castration-Resistant Prostate Cancer Showing a Remarkable Response to Pembrolizumab Sustained over at Least 18 Months. Cold Spring Harb. Mol. Case Stud. 2022, 8, a006194. [Google Scholar] [CrossRef]
- Ravindranathan, D.; Russler, G.A.; Yantorni, L.; Drusbosky, L.M.; Bilen, M.A. Detection of Microsatellite Instability via Circulating Tumor DNA and Response to Immunotherapy in Metastatic Castration-Resistant Prostate Cancer: A Case Series. Case Rep. Oncol. 2021, 14, 190–196. [Google Scholar] [CrossRef]
- Sena, L.A.; Salles, D.C.; Engle, E.L.; Zhu, Q.; Tukachinsky, H.; Lotan, T.L.; Antonarakis, E.S. Mismatch Repair-Deficient Prostate Cancer with Parenchymal Brain Metastases Treated with Immune Checkpoint Blockade. Cold Spring Harb. Mol. Case Stud. 2021, 7, a006094. [Google Scholar] [CrossRef]
- Fujiwara, M.; Komai, Y.; Yuasa, T.; Numao, N.; Yamamoto, S.; Fukui, I.; Yonese, J. Pembrolizumab for a Patient with Metastatic Castration-Resistant Prostate Cancer with Microsatellite Instability-High. IJU Case Rep. 2020, 3, 62–64. [Google Scholar] [CrossRef] [Green Version]
- Han, H.J.; Li, Y.R.; Roach, M.; Aggarwal, R. Dramatic Response to Combination Pembrolizumab and Radiation in Metastatic Castration Resistant Prostate Cancer. Adv. Med. Oncol. 2020, 12, 1758835920936084. [Google Scholar] [CrossRef]
- Manogue, C.; Cotogno, P.; Ledet, E.; Lewis, B.; Wyatt, A.W.; Sartor, O. Biomarkers for Programmed Death-1 Inhibition in Prostate Cancer. Oncologist 2019, 24, 444–448. [Google Scholar] [CrossRef] [Green Version]
- Costa, L.B.; Queiroz, M.A.; de Barbosa, F.G.; Nunes, R.F.; Marin, J.F.G.; Dzik, C.; Buchpiguel, C.A. Pseudoprogression on PSMA PET Imaging of a MCRPC Patient under Anti-PD1 Treatment. Eur J. Nucl. Med. Mol. Imaging 2019, 46, 1576–1577. [Google Scholar] [CrossRef] [PubMed]
- Strickler, J.H.; Hanks, B.A.; Khasraw, M. Tumor Mutational Burden as a Predictor of Immunotherapy Response: Is More Always Better? Clin. Cancer Res. 2021, 27, 1236–1241. [Google Scholar] [CrossRef] [PubMed]
- Fancello, L.; Gandini, S.; Pelicci, P.G.; Mazzarella, L. Tumor Mutational Burden Quantification from Targeted Gene Panels: Major Advancements and Challenges. J. Immunother. Cancer 2019, 7, 183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chalmers, Z.R.; Connelly, C.F.; Fabrizio, D.; Gay, L.; Ali, S.M.; Ennis, R.; Schrock, A.; Campbell, B.; Shlien, A.; Chmielecki, J.; et al. Analysis of 100,000 Human Cancer Genomes Reveals the Landscape of Tumor Mutational Burden. Genome Med. 2017, 9, 34. [Google Scholar] [CrossRef] [Green Version]
- Huang, T.; Chen, X.; Zhang, H.; Liang, Y.; Li, L.; Wei, H.; Sun, W.; Wang, Y. Prognostic Role of Tumor Mutational Burden in Cancer Patients Treated with Immune Checkpoint Inhibitors: A Systematic Review and Meta-Analysis. Front. Oncol. 2021, 11, 706652. [Google Scholar] [CrossRef]
- Weis, L.N.; Tolaney, S.M.; Barrios, C.H.; Barroso-Sousa, R. Tissue-Agnostic Drug Approvals: How Does This Apply to Patients with Breast Cancer? NPJ Breast Cancer 2021, 7, 120. [Google Scholar] [CrossRef]
- Ryan, M.J.; Bose, R. Genomic Alteration Burden in Advanced Prostate Cancer and Therapeutic Implications. Front. Oncol. 2019, 9, 1287. [Google Scholar] [CrossRef] [Green Version]
- Marabelle, A.; Fakih, M.; Lopez, J.; Shah, M.; Shapira-Frommer, R.; Nakagawa, K.; Chung, H.C.; Kindler, H.L.; Lopez-Martin, J.A.; Miller, W.H.; et al. Association of Tumour Mutational Burden with Outcomes in Patients with Advanced Solid Tumours Treated with Pembrolizumab: Prospective Biomarker Analysis of the Multicohort, Open-Label, Phase 2 KEYNOTE-158 Study. Lancet Oncol. 2020, 21, 1353–1365. [Google Scholar] [CrossRef]
- Graf, R.P.; Fisher, V.; Weberpals, J.; Gjoerup, O.; Tierno, M.B.; Huang, R.S.P.; Sayegh, N.; Lin, D.I.; Raskina, K.; Schrock, A.B.; et al. Comparative Effectiveness of Immune Checkpoint Inhibitors vs Chemotherapy by Tumor Mutational Burden in Metastatic Castration-Resistant Prostate Cancer. JAMA Netw. Open 2022, 5, e225394. [Google Scholar] [CrossRef]
- Rosen, E.Y.; Goldman, D.A.; Hechtman, J.F.; Benayed, R.; Schram, A.M.; Cocco, E.; Shifman, S.; Gong, Y.; Kundra, R.; Solomon, J.P.; et al. TRK Fusions Are Enriched in Cancers with Uncommon Histologies and the Absence of Canonical Driver Mutations. Clin. Cancer Res. 2020, 26, 1624–1632. [Google Scholar] [CrossRef] [Green Version]
- Amatu, A.; Sartore-Bianchi, A.; Siena, S. NTRK Gene Fusions as Novel Targets of Cancer Therapy across Multiple Tumour Types. ESMO Open 2016, 1, e000023. [Google Scholar] [CrossRef] [Green Version]
- Chao, M.V. Neurotrophins and Their Receptors: A Convergence Point for Many Signalling Pathways. Nat. Rev. Neurosci. 2003, 4, 299–309. [Google Scholar] [CrossRef]
- Cocco, E.; Scaltriti, M.; Drilon, A. NTRK Fusion-Positive Cancers and TRK Inhibitor Therapy. Nat. Rev. Clin. Oncol. 2018, 15, 731–747. [Google Scholar] [CrossRef]
- Marchiò, C.; Scaltriti, M.; Ladanyi, M.; Iafrate, A.J.; Bibeau, F.; Dietel, M.; Hechtman, J.F.; Troiani, T.; López-Rios, F.; Douillard, J.-Y.; et al. ESMO Recommendations on the Standard Methods to Detect NTRK Fusions in Daily Practice and Clinical Research. Ann. Oncol. 2019, 30, 1417–1427. [Google Scholar] [CrossRef] [Green Version]
- Ling, Q.; Li, B.; Wu, X.; Wang, H.; Shen, Y.; Xiao, M.; Yang, Z.; Ma, R.; Chen, D.; Chen, H.; et al. The Landscape of NTRK Fusions in Chinese Patients with Solid Tumor. Ann. Oncol. 2018, 29, viii22–viii23. [Google Scholar] [CrossRef]
- Forsythe, A.; Zhang, W.; Phillip Strauss, U.; Fellous, M.; Korei, M.; Keating, K. A Systematic Review and Meta-Analysis of Neurotrophic Tyrosine Receptor Kinase Gene Fusion Frequencies in Solid Tumors. Adv. Med. Oncol. 2020, 12, 1758835920975613. [Google Scholar] [CrossRef]
- Westphalen, C.B.; Krebs, M.G.; Le Tourneau, C.; Sokol, E.S.; Maund, S.L.; Wilson, T.R.; Jin, D.X.; Newberg, J.Y.; Fabrizio, D.; Veronese, L.; et al. Genomic Context of NTRK1/2/3 Fusion-Positive Tumours from a Large Real-World Population. NPJ Precis. Oncol. 2021, 5, 69. [Google Scholar] [CrossRef]
- Yeh, Y.A.; Yang, S.; Constantinescu, M.; Chaudoir, C.; Tanner, A.; Henry, M.; Anderson, S.; Saldivar, J.-S.; Serkin, F.; Fazili, T.; et al. Prostatic Adenocarcinoma with Novel NTRK3 Gene Fusion: A Case Report. Am. J. Clin. Exp. Urol. 2019, 7, 341–345. [Google Scholar]
- Lassen, U. How I Treat NTRK Gene Fusion-Positive Cancers. ESMO Open 2019, 4, e000612. [Google Scholar] [CrossRef] [Green Version]
- Drilon, A.; Laetsch, T.W.; Kummar, S.; DuBois, S.G.; Lassen, U.N.; Demetri, G.D.; Nathenson, M.; Doebele, R.C.; Farago, A.F.; Pappo, A.S.; et al. Efficacy of Larotrectinib in TRK Fusion-Positive Cancers in Adults and Children. N. Engl. J. Med. 2018, 378, 731–739. [Google Scholar] [CrossRef]
- Hong, D.S.; DuBois, S.G.; Kummar, S.; Farago, A.F.; Albert, C.M.; Rohrberg, K.S.; van Tilburg, C.M.; Nagasubramanian, R.; Berlin, J.D.; Federman, N.; et al. Larotrectinib in Patients with TRK Fusion-Positive Solid Tumours: A Pooled Analysis of Three Phase 1/2 Clinical Trials. Lancet Oncol. 2020, 21, 531–540. [Google Scholar] [CrossRef]
- Doebele, R.C.; Drilon, A.; Paz-Ares, L.; Siena, S.; Shaw, A.T.; Farago, A.F.; Blakely, C.M.; Seto, T.; Cho, B.C.; Tosi, D.; et al. Entrectinib in Patients with Advanced or Metastatic NTRK Fusion-Positive Solid Tumours: Integrated Analysis of Three Phase 1–2 Trials. Lancet Oncol. 2020, 21, 271–282. [Google Scholar] [CrossRef]
- Holderfield, M.; Deuker, M.M.; McCormick, F.; McMahon, M. Targeting RAF Kinases for Cancer Therapy: BRAF-Mutated Melanoma and Beyond. Nat. Rev. Cancer 2014, 14, 455–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaman, A.; Wu, W.; Bivona, T.G. Targeting Oncogenic BRAF: Past, Present, and Future. Cancers 2019, 11, 1197. [Google Scholar] [CrossRef] [Green Version]
- Sholl, L.M. A Narrative Review of BRAF Alterations in Human Tumors: Diagnostic and Predictive Implications. Precis. Cancer Med. 2020, 3, 26. [Google Scholar] [CrossRef]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF Gene in Human Cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef]
- Vanni, I.; Tanda, E.T.; Spagnolo, F.; Andreotti, V.; Bruno, W.; Ghiorzo, P. The Current State of Molecular Testing in the BRAF-Mutated Melanoma Landscape. Front. Mol. Biosci. 2020, 7, 113. [Google Scholar] [CrossRef]
- Sullivan, R.J.; Flaherty, K.T. BRAF in Melanoma: Pathogenesis, Diagnosis, Inhibition, and Resistance. J. Skin Cancer 2011, 2011, 423239. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Willmore-Payne, C.; Layfield, L.J.; Holden, J.A. Lack of BRAF Activating Mutations in Prostate Adenocarcinoma: A Study of 93 Cases. Appl. Immunohistochem. Mol. Morphol. 2009, 17, 121–125. [Google Scholar] [CrossRef]
- Shen, Y.; Lu, Y.; Yin, X.; Zhu, G.; Zhu, J. KRAS and BRAF Mutations in Prostate Carcinomas of Chinese Patients. Cancer Genet. Cytogenet. 2010, 198, 35–39. [Google Scholar] [CrossRef]
- Jafarian, A.H.; Mirshekar Nasirabadi, K.; Etemad, S.; Jafaripour, M.; Darijani, M.; Sheikhi, M.; Ayatollahi, H.; Shakeri, S.; Shams, S.F.; Davari, S. Molecular Status of BRAF Mutation in Prostate Adenocarcinoma: The Analysis of 100 Cases in North-East of IRAN. Iran. J. Pathol. 2018, 13, 415–421. [Google Scholar]
- Cyrta, J.; Prandi, D.; Arora, A.; Hovelson, D.H.; Sboner, A.; Rodriguez, A.; Fedrizzi, T.; Beltran, H.; Robinson, D.R.; Gopalan, A.; et al. Comparative Genomics of Primary Prostate Cancer and Paired Metastases: Insights from 12 Molecular Case Studies. J. Pathol. 2022, 257, 274–284. [Google Scholar] [CrossRef]
- Alhamar, M.; Tudor Vladislav, I.; Smith, S.C.; Gao, Y.; Cheng, L.; Favazza, L.A.; Alani, A.M.; Ittmann, M.M.; Riddle, N.D.; Whiteley, L.J.; et al. Gene Fusion Characterisation of Rare Aggressive Prostate Cancer Variants-Adenosquamous Carcinoma, Pleomorphic Giant-Cell Carcinoma, and Sarcomatoid Carcinoma: An Analysis of 19 Cases. Histopathology 2020, 77, 890–899. [Google Scholar] [CrossRef]
- Kasajima, R.; Yamaguchi, R.; Shimizu, E.; Tamada, Y.; Niida, A.; Tremmel, G.; Kishida, T.; Aoki, I.; Imoto, S.; Miyano, S.; et al. Variant Analysis of Prostate Cancer in Japanese Patients and a New Attempt to Predict Related Biological Pathways. Oncol. Rep. 2020, 43, 943–952. [Google Scholar] [CrossRef]
- Suh, J.; Jeong, C.W.; Choi, S.; Ku, J.H.; Kim, H.H.; Kim, K.S.; Kwak, C. Targeted Next-Generation Sequencing for Locally Advanced Prostate Cancer in the Korean Population. Investig. Clin. Urol. 2020, 61, 127–135. [Google Scholar] [CrossRef]
- Ikeda, S.; Elkin, S.K.; Tomson, B.N.; Carter, J.L.; Kurzrock, R. Next-Generation Sequencing of Prostate Cancer: Genomic and Pathway Alterations, Potential Actionability Patterns, and Relative Rate of Use of Clinical-Grade Testing. Cancer Biol. 2019, 20, 219–226. [Google Scholar] [CrossRef]
- Barata, P.C.; Mendiratta, P.; Heald, B.; Klek, S.; Grivas, P.; Sohal, D.P.S.; Garcia, J.A. Targeted Next-Generation Sequencing in Men with Metastatic Prostate Cancer: A Pilot Study. Target. Oncol. 2018, 13, 495–500. [Google Scholar] [CrossRef]
- Ateeq, B.; Kunju, L.P.; Carskadon, S.L.; Pandey, S.K.; Singh, G.; Pradeep, I.; Tandon, V.; Singhai, A.; Goel, A.; Amit, S.; et al. Molecular Profiling of ETS and Non-ETS Aberrations in Prostate Cancer Patients from Northern India. Prostate 2015, 75, 1051–1062. [Google Scholar] [CrossRef] [Green Version]
- Efficacy and Safety of the Combination Therapy of Dabrafenib and Trametinib in Subjects with BRAF V600E-Mutated Rare Cancers. Available online: https://clinicaltrials.gov/ct2/show/NCT02034110 (accessed on 24 June 2022).
- Targeted Therapy Directed by Genetic Testing in Treating Patients with Advanced Refractory Solid Tumors, Lymphomas, or Multiple Myeloma (The MATCH Screening Trial). Available online: https://clinicaltrials.gov/ct2/show/NCT02465060 (accessed on 24 June 2022).
- Subbiah, V.; Kreitman, R.J.; Wainberg, Z.A.; Cho, J.Y.; Schellens, J.H.M.; Soria, J.C.; Wen, P.Y.; Zielinski, C.; Cabanillas, M.E.; Urbanowitz, G.; et al. Dabrafenib and Trametinib Treatment in Patients with Locally Advanced or Metastatic BRAF V600-Mutant Anaplastic Thyroid Cancer. J. Clin. Oncol. 2018, 36, 7–13. [Google Scholar] [CrossRef] [Green Version]
- Subbiah, V.; Kreitman, R.J.; Wainberg, Z.A.; Cho, J.Y.; Schellens, J.H.M.; Soria, J.C.; Wen, P.Y.; Zielinski, C.C.; Cabanillas, M.E.; Boran, A.; et al. Dabrafenib plus Trametinib in Patients with BRAF V600E-Mutant Anaplastic Thyroid Cancer: Updated Analysis from the Phase II ROAR Basket Study. Ann. Oncol. 2022, 33, 406–415. [Google Scholar] [CrossRef] [PubMed]
- Subbiah, V.; Lassen, U.; Élez, E.; Italiano, A.; Curigliano, G.; Javle, M.; de Braud, F.; Prager, G.W.; Greil, R.; Stein, A.; et al. Dabrafenib plus Trametinib in Patients with BRAFV600E-Mutated Biliary Tract Cancer (ROAR): A Phase 2, Open-Label, Single-Arm, Multicentre Basket Trial. Lancet Oncol. 2020, 21, 1234–1243. [Google Scholar] [CrossRef]
- Wen, P.Y.; Stein, A.; van den Bent, M.; De Greve, J.; Wick, A.; de Vos, F.Y.F.L.; von Bubnoff, N.; van Linde, M.E.; Lai, A.; Prager, G.W.; et al. Dabrafenib plus Trametinib in Patients with BRAFV600E-Mutant Low-Grade and High-Grade Glioma (ROAR): A Multicentre, Open-Label, Single-Arm, Phase 2, Basket Trial. Lancet Oncol. 2022, 23, 53–64. [Google Scholar] [CrossRef]
- Murciano-Goroff, Y.R.; Drilon, A.; Stadler, Z.K. The NCI-MATCH: A National, Collaborative Precision Oncology Trial for Diverse Tumor Histologies. Cancer Cell 2021, 39, 22–24. [Google Scholar] [CrossRef]
- Salama, A.K.S.; Li, S.; Macrae, E.R.; Park, J.-I.; Mitchell, E.P.; Zwiebel, J.A.; Chen, H.X.; Gray, R.J.; McShane, L.M.; Rubinstein, L.V.; et al. Dabrafenib and Trametinib in Patients with Tumors with BRAFV600E Mutations: Results of the NCI-MATCH Trial Subprotocol H. J. Clin. Oncol. 2020, 38, 3895–3904. [Google Scholar] [CrossRef]
- Study to Investigate Safety, Pharmacokinetic (PK), Pharmacodynamic (PD) and Clinical Activity of Trametinib in Subjects with Cancer or Plexiform Neurofibromas and Trametinib in Combination with Dabrafenib in Subjects with Cancers Harboring V600 Mutations. Available online: https://clinicaltrials.gov/ct2/show/NCT02124772 (accessed on 24 June 2022).
- Robert, C.; Karaszewska, B.; Schachter, J.; Rutkowski, P.; Mackiewicz, A.; Stroiakovski, D.; Lichinitser, M.; Dummer, R.; Grange, F.; Mortier, L.; et al. Improved Overall Survival in Melanoma with Combined Dabrafenib and Trametinib. N. Engl. J. Med. 2015, 372, 30–39. [Google Scholar] [CrossRef] [Green Version]
- Robert, C.; Grob, J.J.; Stroyakovskiy, D.; Karaszewska, B.; Hauschild, A.; Levchenko, E.; Chiarion Sileni, V.; Schachter, J.; Garbe, C.; Bondarenko, I.; et al. Five-Year Outcomes with Dabrafenib plus Trametinib in Metastatic Melanoma. N. Engl. J. Med. 2019, 381, 626–636. [Google Scholar] [CrossRef]
- Planchard, D.; Smit, E.F.; Groen, H.J.M.; Mazieres, J.; Besse, B.; Helland, Å.; Giannone, V.; D’Amelio, A.M.; Zhang, P.; Mookerjee, B.; et al. Dabrafenib plus Trametinib in Patients with Previously Untreated BRAFV600E-Mutant Metastatic Non-Small-Cell Lung Cancer: An Open-Label, Phase 2 Trial. Lancet Oncol. 2017, 18, 1307–1316. [Google Scholar] [CrossRef]
- Deininger, M.; Buchdunger, E.; Druker, B.J. The Development of Imatinib as a Therapeutic Agent for Chronic Myeloid Leukemia. Blood 2005, 105, 2640–2653. [Google Scholar] [CrossRef] [Green Version]
- Carr, T.H.; McEwen, R.; Dougherty, B.; Johnson, J.H.; Dry, J.R.; Lai, Z.; Ghazoui, Z.; Laing, N.M.; Hodgson, D.R.; Cruzalegui, F.; et al. Defining Actionable Mutations for Oncology Therapeutic Development. Nat. Rev. Cancer 2016, 16, 319–329. [Google Scholar] [CrossRef]
- EMA Recommends Extension of Indications for Pembrolizumab to MSI-H or dMMR Cancers and to Metastatic Cervical Cancer with PD-L1 CPS ≥1. Available online: https://www.esmo.org/oncology-news/ema-recommends-extension-of-indications-for-pembrolizumab-to-msi-h-or-dmmr-cancers-and-to-metastatic-cervical-cancer-with-pd-l1-cps-1 (accessed on 24 June 2022).
- Trullas, A.; Delgado, J.; Genazzani, A.; Mueller-Berghaus, J.; Migali, C.; Müller-Egert, S.; Zander, H.; Enzmann, H.; Pignatti, F. The EMA Assessment of Pembrolizumab as Monotherapy for the First-Line Treatment of Adult Patients with Metastatic Microsatellite Instability-High or Mismatch Repair Deficient Colorectal Cancer. ESMO Open 2021, 6, 100145. [Google Scholar] [CrossRef]
- Pharmaceutical Evaluation Division, Pharmaceutical Safety and Environmental Health Bureau Ministry of Health, Labour and Welfare. Pembrolizumab. Available online: https://www.pmda.go.jp/files/000231921.pdf (accessed on 24 June 2022).
- Vitrakvi (Larotrectinib). Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/vitrakvi (accessed on 24 June 2022).
- Rozlytrek (Entrectinib). Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/rozlytrek (accessed on 24 June 2022).
- Ardini, E.; Siena, S. Entrectinib Approval by EMA Reinforces Options for ROS1 and Tumour Agnostic NTRK Targeted Cancer Therapies. ESMO Open 2020, 5, e000867. [Google Scholar] [CrossRef]
- Pharmaceutical Evaluation Division, Pharmaceutical Safety and Environmental Health Bureau Ministry of Health, Labour and Welfare. Entrectinib. Available online: https://www.pmda.go.jp/files/000232794.pdf (accessed on 24 June 2022).
- Pederzoli, F.; Bandini, M.; Marandino, L.; Ali, S.M.; Madison, R.; Chung, J.; Ross, J.S.; Necchi, A. Targetable Gene Fusions and Aberrations in Genitourinary Oncology. Nat. Rev. Urol. 2020, 17, 613–625. [Google Scholar] [CrossRef]
- Iannantuono, G.M.; Riondino, S.; Sganga, S.; Roselli, M.; Torino, F. Activity of ALK Inhibitors in Renal Cancer with ALK Alterations: A Systematic Review. Int. J. Mol. Sci. 2022, 23, 3995. [Google Scholar] [CrossRef]
- Sayegh, N.; Swami, U.; Agarwal, N. Recent Advances in the Management of Metastatic Prostate Cancer. JCO Oncol. Pract. 2022, 18, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Cereda, V.; Formica, V.; Massimiani, G.; Tosetto, L.; Roselli, M. Targeting Metastatic Castration-Resistant Prostate Cancer: Mechanisms of Progression and Novel Early Therapeutic Approaches. Expert Opin. Investig. Drugs 2014, 23, 469–487. [Google Scholar] [CrossRef] [PubMed]
- Ioannidou, E.; Moschetta, M.; Shah, S.; Parker, J.S.; Ozturk, M.A.; Pappas-Gogos, G.; Sheriff, M.; Rassy, E.; Boussios, S. Angiogenesis and Anti-Angiogenic Treatment in Prostate Cancer: Mechanisms of Action and Molecular Targets. Int. J. Mol. Sci. 2021, 22, 9926. [Google Scholar] [CrossRef] [PubMed]
- Cereda, V.; Formica, V.; Roselli, M. Issues and Promises of Bevacizumab in Prostate Cancer Treatment. Expert Opin. Biol. 2018, 18, 707–717. [Google Scholar] [CrossRef] [PubMed]
- Devlies, W.; Eckstein, M.; Cimadamore, A.; Devos, G.; Moris, L.; Van den Broeck, T.; Montironi, R.; Joniau, S.; Claessens, F.; Gevaert, T. Clinical Actionability of the Genomic Landscape of Metastatic Castration Resistant Prostate Cancer. Cells 2020, 9, 2494. [Google Scholar] [CrossRef] [PubMed]
- Ku, S.-Y.; Gleave, M.E.; Beltran, H. Towards Precision Oncology in Advanced Prostate Cancer. Nat. Rev. Urol. 2019, 16, 645–654. [Google Scholar] [CrossRef]
- Abida, W.; Cyrta, J.; Heller, G.; Prandi, D.; Armenia, J.; Coleman, I.; Cieslik, M.; Benelli, M.; Robinson, D.; Van Allen, E.M.; et al. Genomic Correlates of Clinical Outcome in Advanced Prostate Cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 11428–11436. [Google Scholar] [CrossRef] [Green Version]
- Bagherabadi, A.; Hooshmand, A.; Shekari, N.; Singh, P.; Zolghadri, S.; Stanek, A.; Dohare, R. Correlation of NTRK1 Downregulation with Low Levels of Tumor-Infiltrating Immune Cells and Poor Prognosis of Prostate Cancer Revealed by Gene Network Analysis. Genes 2022, 13, 840. [Google Scholar] [CrossRef]
- Steinwald, P.; Ledet, E.; Sartor, O. Eradication of BRAF K601E Mutation in Metastatic Castrate-Resistant Prostate Cancer Treated with Cabazitaxel and Carboplatin: A Case Report. Clin. Genitourin. Cancer 2020, 18, e312–e314. [Google Scholar] [CrossRef]
- Su, P.-L.; Lin, C.-Y.; Chen, Y.-L.; Chen, W.-L.; Lin, C.-C.; Su, W.-C. Durable Response to Combined Dabrafenib and Trametinib in a Patient with BRAF K601E Mutation-Positive Lung Adenocarcinoma: A Case Report. JTO Clin. Res. Rep. 2021, 2, 100202. [Google Scholar] [CrossRef]
- Rogiers, A.; Thomas, D.; Vander Borght, S.; van den Oord, J.J.; Bechter, O.; Dewaele, M.; Rambow, F.; Marine, J.-C.; Wolter, P. Dabrafenib plus Trametinib in BRAF K601E-Mutant Melanoma. Br. J. Derm. 2019, 180, 421–422. [Google Scholar] [CrossRef]
- Venkatachalam, S.; McFarland, T.R.; Agarwal, N.; Swami, U. Immune Checkpoint Inhibitors in Prostate Cancer. Cancers 2021, 13, 2187. [Google Scholar] [CrossRef]
- Rebuzzi, S.E.; Rescigno, P.; Catalano, F.; Mollica, V.; Vogl, U.M.; Marandino, L.; Massari, F.; Pereira Mestre, R.; Zanardi, E.; Signori, A.; et al. Immune Checkpoint Inhibitors in Advanced Prostate Cancer: Current Data and Future Perspectives. Cancers 2022, 14, 1245. [Google Scholar] [CrossRef]
- Fizazi, K.; González Mella, P.; Castellano, D.; Minatta, J.N.; Rezazadeh Kalebasty, A.; Shaffer, D.; Vázquez Limón, J.C.; Sánchez López, H.M.; Armstrong, A.J.; Horvath, L.; et al. Nivolumab plus Docetaxel in Patients with Chemotherapy-Naïve Metastatic Castration-Resistant Prostate Cancer: Results from the Phase II CheckMate 9KD Trial. Eur. J. Cancer 2022, 160, 61–71. [Google Scholar] [CrossRef]
- Yu, E.Y.; Kolinsky, M.P.; Berry, W.R.; Retz, M.; Mourey, L.; Piulats, J.M.; Appleman, L.J.; Romano, E.; Gravis, G.; Gurney, H.; et al. Pembrolizumab Plus Docetaxel and Prednisone in Patients with Metastatic Castration-Resistant Prostate Cancer: Long-Term Results from the Phase 1b/2 KEYNOTE-365 Cohort B Study. Eur. Urol. 2022, 82, 22–30. [Google Scholar] [CrossRef]
- Drake, C.G.; Saad, F.; Clark, W.R.; Ciprotti, M.; Sharkey, B.; Subudhi, S.K.; Fizazi, K. 690TiP A Phase III, Randomized, Double-Blind Trial of Nivolumab or Placebo Combined with Docetaxel for Metastatic Castration-Resistant Prostate Cancer (MCRPC; CheckMate 7DX). Ann. Oncol. 2020, 31, S546. [Google Scholar] [CrossRef]
- Petrylak, D.P.; Ratta, R.; Gafanov, R.; Facchini, G.; Piulats, J.M.; Kramer, G.; Flaig, T.W.; Chandana, S.R.; Li, B.; Burgents, J.; et al. KEYNOTE-921: Phase III Study of Pembrolizumab plus Docetaxel for Metastatic Castration-Resistant Prostate Cancer. Future Oncol. 2021, 17, 3291–3299. [Google Scholar] [CrossRef]
- Wolff, L.; Kiesewetter, B. Applicability of ESMO-MCBS and ESCAT for Molecular Tumor Boards. memo Mag. Eur. Med. Oncol. 2022. [Google Scholar] [CrossRef]
- Mateo, J.; Chakravarty, D.; Dienstmann, R.; Jezdic, S.; Gonzalez-Perez, A.; Lopez-Bigas, N.; Ng, C.K.Y.; Bedard, P.L.; Tortora, G.; Douillard, J.-Y.; et al. A Framework to Rank Genomic Alterations as Targets for Cancer Precision Medicine: The ESMO Scale for Clinical Actionability of Molecular Targets (ESCAT). Ann. Oncol. 2018, 29, 1895–1902. [Google Scholar] [CrossRef]
- Adashek, J.J.; Subbiah, V.; Kurzrock, R. From Tissue-Agnostic to N-of-One Therapies: (R)Evolution of the Precision Paradigm. Trends Cancer 2021, 7, 15–28. [Google Scholar] [CrossRef]
- Mateo, J.; Steuten, L.; Aftimos, P.; André, F.; Davies, M.; Garralda, E.; Geissler, J.; Husereau, D.; Martinez-Lopez, I.; Normanno, N.; et al. Delivering Precision Oncology to Patients with Cancer. Nat. Med. 2022, 28, 658–665. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| First Author | Year of Publication | Type of PCa | Number of Patients | Staging | Somatic dMMR | Germline dMMR | ||
|---|---|---|---|---|---|---|---|---|
| Frequency (n) | Gene(s) Involved | Frequency (n) | Gene(s) Involved | |||||
| Robinson et al. [35] | 2015 | CRPC | 150 | Advanced | 2.7% (3) | MLH1-MSH2 | NA | NA |
| Pritchard et al. [36] | 2016 | NA | 692 | Advanced | NA | NA | 0.6% (4) | MSH2-MSH6-PMS2 |
| Guedes et al. [37] | 2017 | Mixed | 1176 | Localized/Advanced | 1.2% (14) | MSH2-MSH6 | 0.3% (4) | MSH2 |
| Abida et al. [38] | 2018 | CRPC | 1033 | Localized/Advanced | 3.1% (32) | MLH1-MSH2-MSH6-PMS2 | 0.8% (8) | MSH2-MSH6-PMS2 |
| Latham et al. [39] | 2019 | NA | 1048 | Localized/Advanced | 5.6% (54) | NA | 0.3% (3) | MSH2-PMS2 |
| Nicolosi et al. [40] | 2019 | Mixed | 3350 | Localized/Advanced | 1.7% (58) | MLH1-MSH2-MSH6-PMS2 | NA | NA |
| Wu et al. [41] | 2021 | Mixed | 246 | Localized/Advanced | NA | NA | 2.4% (6) | MSH2 |
| Publication | Patient Characteristics | Treatments before Pembrolizumab | Treatment with Pembrolizumab | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| First Author | Year | Age | Histotype of PCa (Gleason Score) | Staging at Diagnosis | MSI-H/dMMR | TMB | Treatments for Localized Disease * | Treatment for Metastatic Disease * | PSA before the First Cycle | Number of Cycles | Best PSA Response | Best Radiological Response | Outcome |
| Shimizu et al. [51] | 2022 | 67 | NA (5 + 4) | Localized | MSH2-MSH6 (IHC-Tissue) | 61 mut/Mb (Tissue) | ADT | AA-RT-Doce-Caba | 35.67 ng/mL | NA | Undetectable | PR | AwD |
| Ravindranathan et al. [52] | 2021 | 51 | ADC (5 + 5) | Advanced | MSI-H (NGS-ctDNA) | - | - | Doce-AA-Enza-Carbo/VP-16 | 39.9 ng/dL | 12 ** | Undetectable | CR | AwD |
| 81 | ADC (4 + 4) | Advanced | MSI-H (NGS-ctDNA) | - | - | ADT + RT-Enza-AA-SipT-Doce-R/223 | 86.8 ng/mL | 5 ** | 0.11 ng/mL | PR | AwD | ||
| Sena et al. [53] | 2021 | 60 | ADC (4 + 5) | Localized | MLH1 and PMS2 (IHC-Tissue) | 25 mut/Mb (Tissue) | RT + ADT | SipT-AA-RT-Enza-Pembro-Doce | NA | NA | Reduction of 79% from baseline | PR | DoD |
| Fujiwara et al. [54] | 2020 | 52 | NA (4 + 4) | Localized | MSI-H (NA) | - | RPr-RT | AA-Enza-Doce-Caba | 16.6 ng/mL | 5 ** | 6.1 ng/mL | PR | AwD |
| Han et al. [55] | 2019 | 75 | ADC (5 + 5) | Advanced | MSI-H (NGS-ctDNA) | - | - | AA-Carbo/Doce-RT-Carbo/Caba | NA | 6 ** | Undetectable | PR | AwD |
| Manogue et al. [56] | 2019 | 64 | NA (4 + 5) | Advanced | MSH2 (PCR-Tissue) | 40.9 mut/Mb (ctDNA) | - | ADT-AA-Doce | 15 ng/dL | 12 | <0.01 ng/mL | CR | AwD |
| Costa et al. [57] | 2019 | 85 | NA (5 + 5) | Localized | MSI-H (NA -Tissue) | - | RT-RPr | ADT-AA | 16 ng/mL | 8 | Undetectable | PR | NA |
| First Author | Year of Publication | Type of PCa | Number of Patients Enrolled | Staging | BRAF Alteration | ||
|---|---|---|---|---|---|---|---|
| Mutation (n) | Fusion (n) | Amplification (n) | |||||
| Cyrta et al. [88] | 2022 | CRPC | 12 | Advanced | K601E (1) | - | - |
| Alhamar et al. [89] | 2020 | Mixed | 19 | Mixed | - | FAM131A-BRAF (1) | - |
| - | SND1-BRAF (1) | - | |||||
| Kasajima et al. [90] | 2020 | HSPC | 21 | Localized | K601E (1) | - | - |
| Suh et al. [91] | 2020 | HSPC | 20 | Locally advanced | K601E (3) | - | - |
| Ikeda et al. [92] | 2019 | NA | 67 | NA | NA (1) | - | NA (2) |
| Barata et al. [93] | 2018 | Mixed | 66 | Advanced | - | - | NA (4) |
| Ateeq et al. [94] | 2015 | NA | 121 | NA | - | NA (1) | NA (2) |
| Anticancer Agent Class | Drug | Trade Name (Pharma. Industry) | Target | FDA Approval Date | Indication | Other Agencies’ Approvals (Date) | References |
|---|---|---|---|---|---|---|---|
| Immune Checkpoint Inhibitors (Monoclonal Antibodies) | Pembrolizumab | Keytruda (Merck & Co.) | PD-1 | 23 May 2017 | Adult and pediatric patients affected by unresectable or metastatic MSI-H/dMMR solid tumors that progressed on prior treatments and have no satisfactory alternative treatment options or by MSI-H/dMMR CRC that progressed following therapy with fluoropyrimidine, oxaliplatin, and irinotecan. | PMDA (30 November 2018) EMA * (24 March 2022) ** | [18,19,109,110,111] |
| 16 June 2020 | Adult and pediatric patients affected by unresectable or metastatic TMB-H *** solid tumors, as determined by an FDA-approved test, that progressed on prior treatments and have no satisfactory alternative treatment options. | - | [23,24] | ||||
| Dostarlimab | Jemperli (GlaxoSmithKline) | PD-1 | 17 August 2021 | Adult patients affected by dMMR recurrent or advanced solid tumors (as determined by an FDA-approved test) that have progressed on prior treatments and have no satisfactory alternative therapeutic options. | - | [25] | |
| Targeted Therapies (Small Molecules/Kinase Inhibitors) | Larotrectinib | Vitrakvi (Loxo Oncology Inc. and Bayer) | NTRK | 26 November 2018 | Adult and pediatric patients with solid tumors harboring an NTRK gene fusion, without a known acquired resistance mutation, that are either metastatic or where surgical resection is likely to result in severe morbidity, and who have no satisfactory alternative treatments or whose cancer has progressed following treatment. | EMA (19 July 2019) | [20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102,103,104,105,106,107,108,109,110,111,112] |
| Entrectinib | Rozlytrek (Genentech Inc.) | NTRK | 15 August 2019 | Adult and pediatric patients 12 years of age and older with solid tumors that have an NTRK gene fusion, without a known acquired resistance mutation, that are metastatic or where surgical resection is likely to result in severe morbidity, and who have progressed following treatment or have no satisfactory alternative therapy. | PMDA (3 June 2019) EMA (31 July 2020) | [21,22,113,114,115] | |
| Dabrafenib– Trametinib | Tafinlar–Mekinist (Novartis) | BRAF and MEK 1–2 | 22 June 2022 | Adult and pediatric patients (older than six years of age) with unresectable or metastatic solid tumors harboring a BRAF V600E mutation who both progressed on prior treatment and have no satisfactory alternative therapeutic options, excluding CRC patients. | - | [26] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iannantuono, G.M.; Torino, F.; Rosenfeld, R.; Guerriero, S.; Carlucci, M.; Sganga, S.; Capotondi, B.; Riondino, S.; Roselli, M. The Role of Histology-Agnostic Drugs in the Treatment of Metastatic Castration-Resistant Prostate Cancer. Int. J. Mol. Sci. 2022, 23, 8535. https://doi.org/10.3390/ijms23158535
Iannantuono GM, Torino F, Rosenfeld R, Guerriero S, Carlucci M, Sganga S, Capotondi B, Riondino S, Roselli M. The Role of Histology-Agnostic Drugs in the Treatment of Metastatic Castration-Resistant Prostate Cancer. International Journal of Molecular Sciences. 2022; 23(15):8535. https://doi.org/10.3390/ijms23158535
Chicago/Turabian StyleIannantuono, Giovanni Maria, Francesco Torino, Roberto Rosenfeld, Simona Guerriero, Manuela Carlucci, Stefano Sganga, Barbara Capotondi, Silvia Riondino, and Mario Roselli. 2022. "The Role of Histology-Agnostic Drugs in the Treatment of Metastatic Castration-Resistant Prostate Cancer" International Journal of Molecular Sciences 23, no. 15: 8535. https://doi.org/10.3390/ijms23158535