
Improved Trimethylangelicin Analogs for Cystic Fibrosis: Design, Synthesis and Preliminary Screening

 , , , ,
, , , ,  ,
,  , , , and
, , , and
Abstract
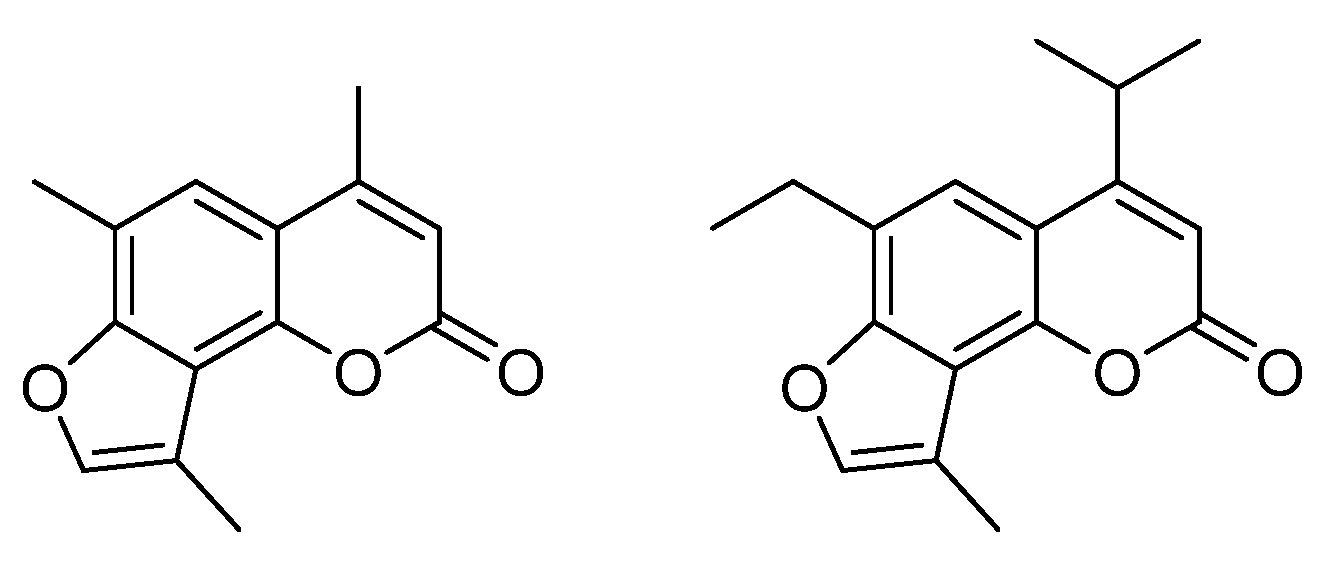
:1. Introduction
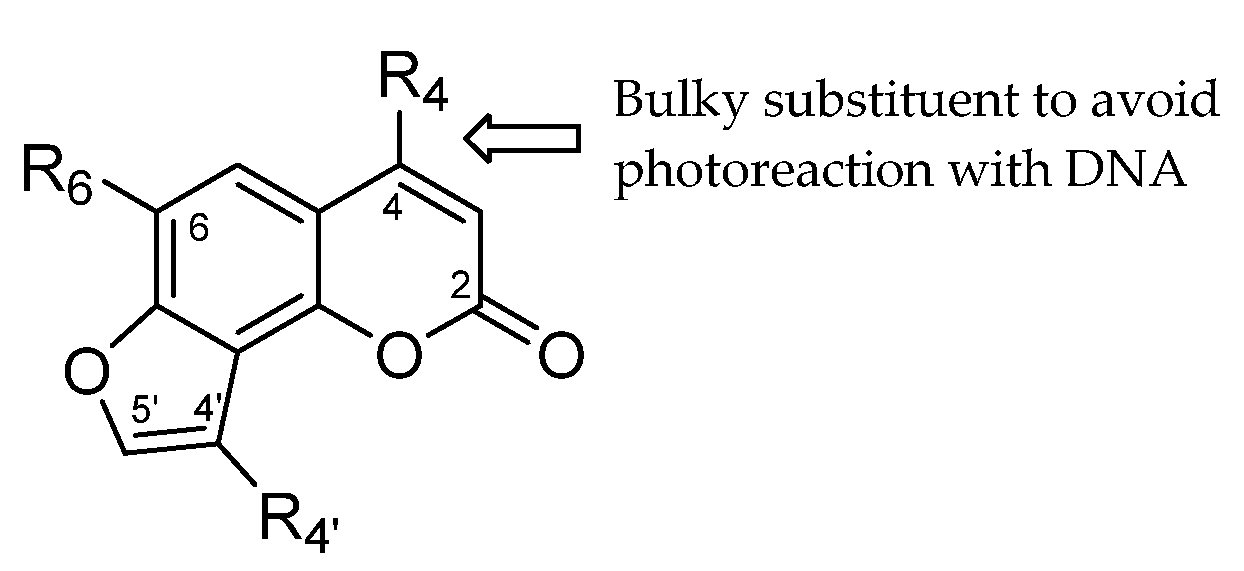
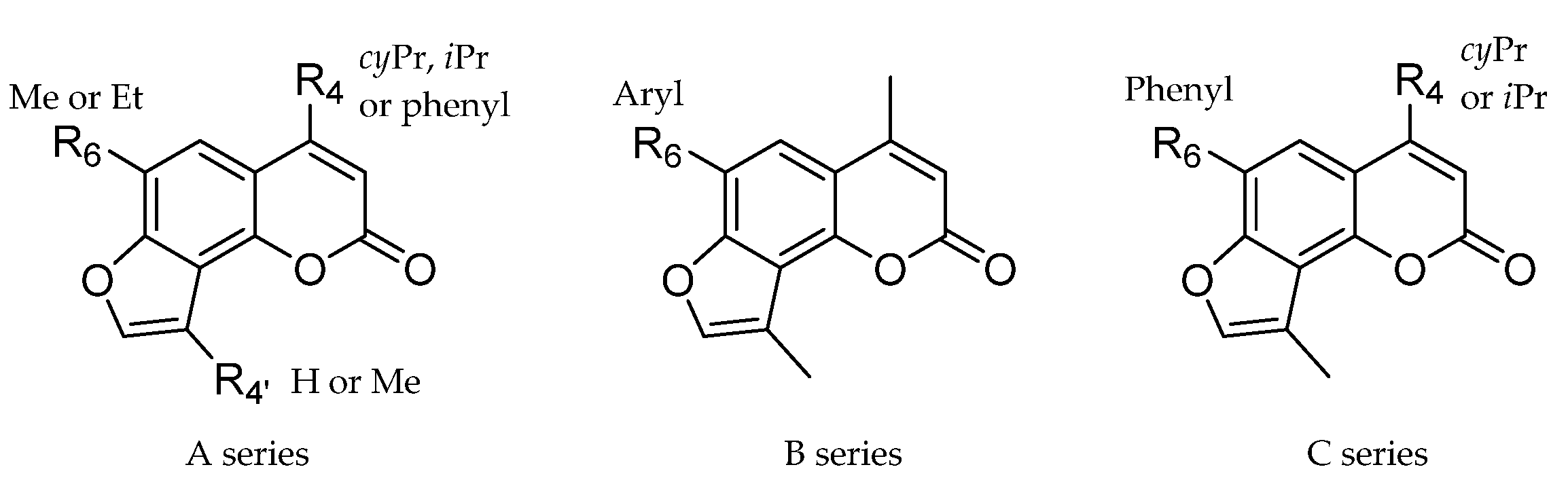
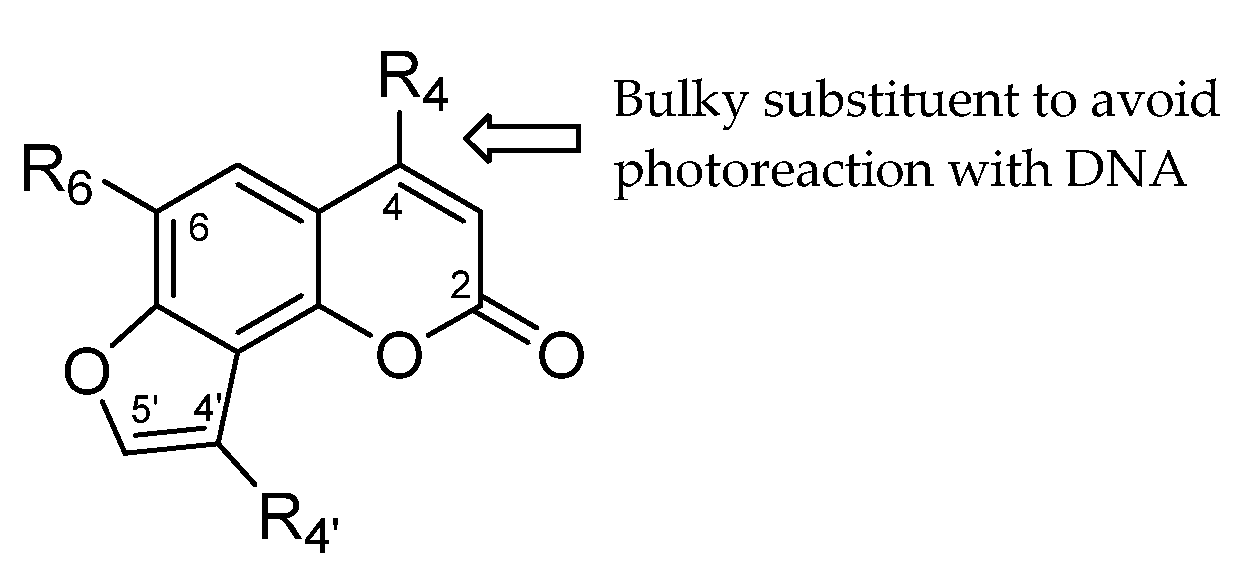
2. Results and Discussion
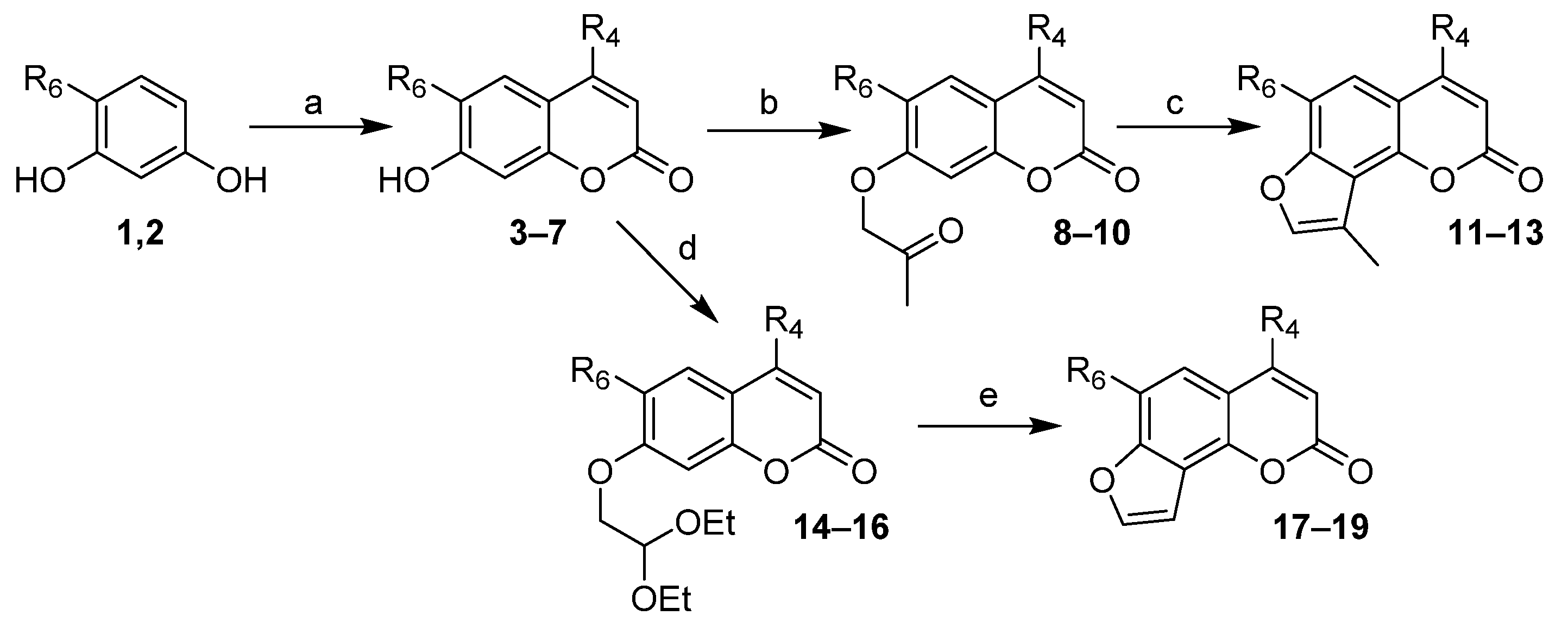
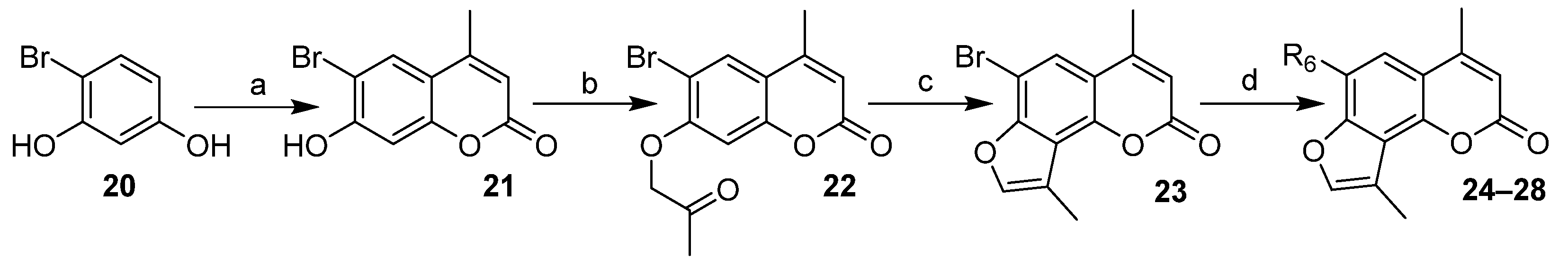
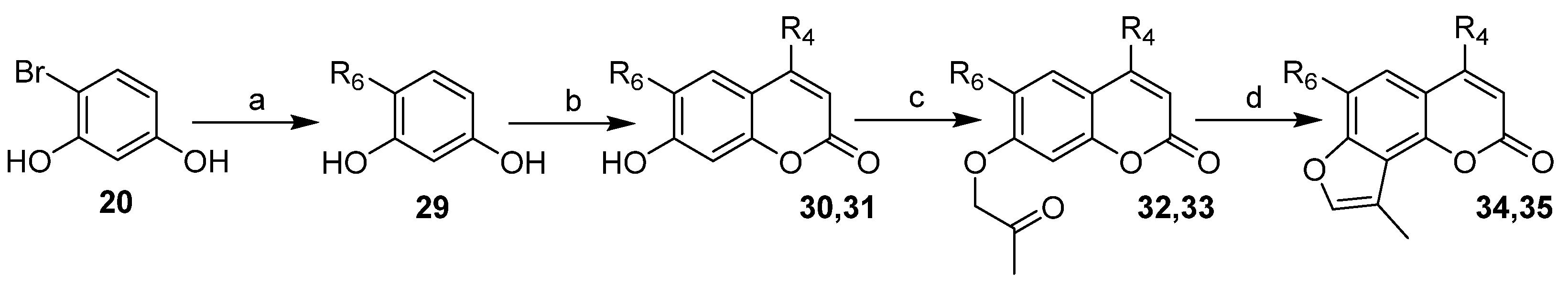
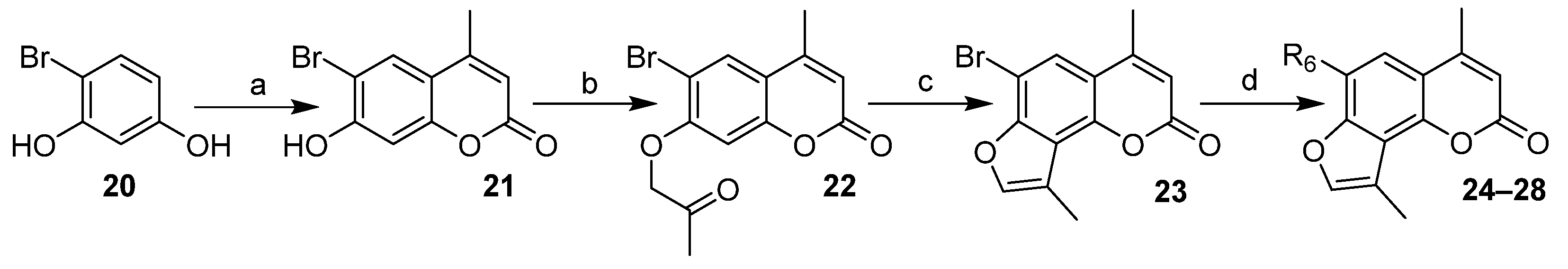
2.1. Chemistry
2.2. Biological Activity
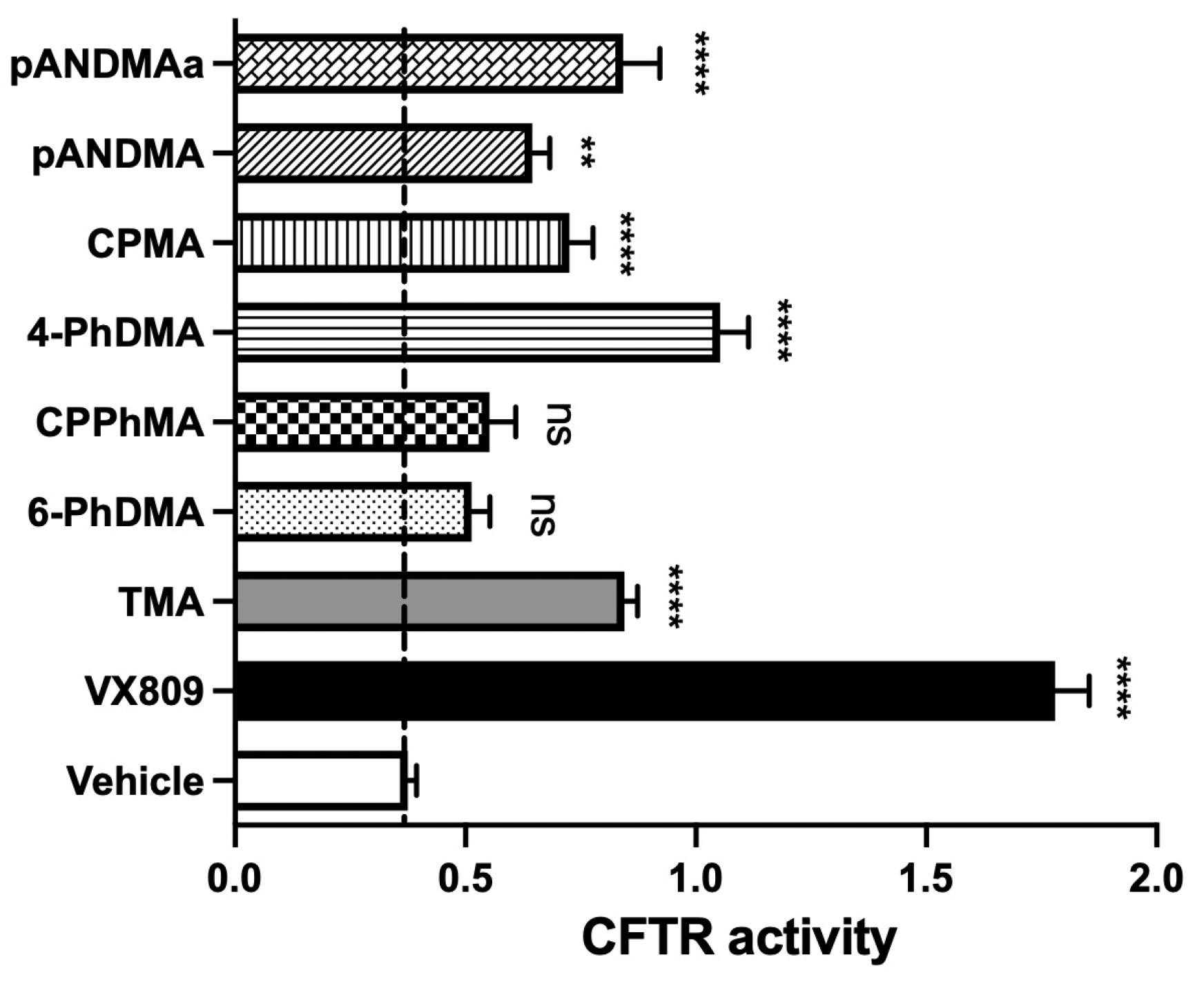
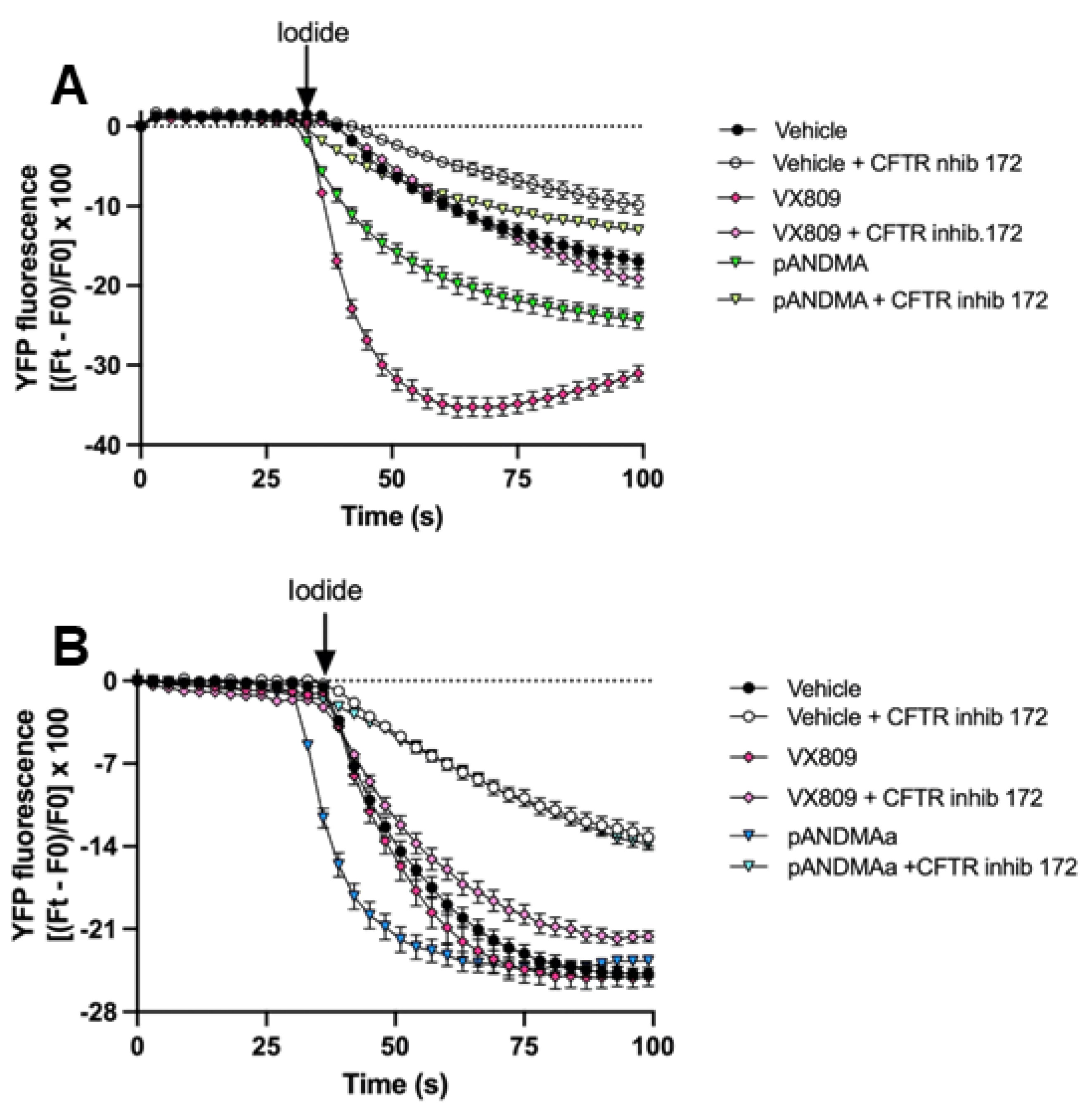
2.2.1. Functional Rescue of Mutant F508del CFTR Protein in CF Airway Cells
2.2.2. In Vitro Toxicity Studies: Photochemical Behavior
2.2.3. In Vitro Toxicity Studies: Evaluation of Mutagenic Effects (Ames Test)
2.2.4. In Vitro Toxicity Studies: Evaluation of the Cytotoxic Activity toward HepG2 Cells
2.3. Pharmacokinetic Profile
2.3.1. Formulation of Lipophilic TMA Analogs
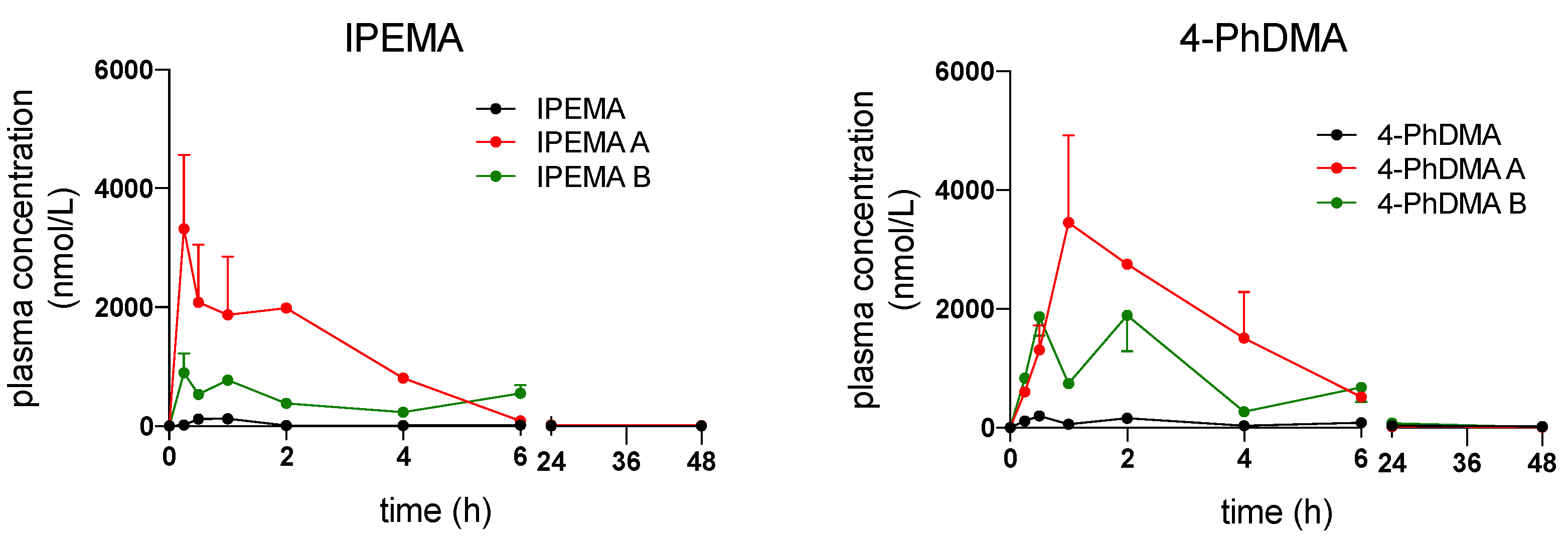
2.3.2. Bioavailability of IPEMA and 4-PhDMA Formulated in SEDDS
3. Materials and Methods
3.1. Chemistry
3.2. Biology
3.2.1. CFTR Functional Rescue
3.2.2. Phototoxicity
3.2.3. Mutagenicity Assay
3.2.4. Cytotoxicity Assay
3.3. Formulation of Lipophilic TMA Derivatives
3.4. Pharmacokinetic Profile
3.4.1. In Vivo Pharmacokinetics
3.4.2. Quantification of TMA Analogs in Plasma Samples by UHPLC-HRMS
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Elborn, J.S. Cystic fibrosis. Lancet 2016, 388, 2519–2531. [Google Scholar] [CrossRef]
- Cutting, G.R. Cystic fibrosis genetics: From molecular understanding to clinical application. Nat. Rev. Genet. 2015, 16, 45–56. [Google Scholar] [CrossRef] [PubMed]
- King, J.A.; Nichols, A.L.; Bentley, S.; Carr, S.B.; Davies, J.C. An Update on CFTR Modulators as New Therapies for Cystic Fibrosis. Paediatr. Drugs 2022, 24, 321–333. [Google Scholar] [CrossRef]
- Deeks, E.D. Lumacaftor/Ivacaftor: A Review in Cystic Fibrosis. Drugs 2016, 76, 1191–1201. [Google Scholar] [CrossRef]
- Guerra, L.; Favia, M.; Di Gioia, S.; Laselva, O.; Bisogno, A.; Casavola, V.; Colombo, C.; Conese, M. The preclinical discovery and development of the combination of ivacaftor + tezacaftor used to treat cystic fibrosis. Expert Opin. Drug Discov. 2020, 15, 873–891. [Google Scholar] [CrossRef]
- Hoy, S.M. Elexacaftor/Ivacaftor/Tezacaftor: First Approval. Drugs 2019, 79, 2001–2007. [Google Scholar] [CrossRef]
- Laselva, O.; Molinski, S.; Casavola, V.; Bear, C.E. The investigational Cystic Fibrosis drug Trimethylangelicin directly modulates CFTR by stabilizing the first membrane-spanning domain. Biochem. Pharmacol. 2016, 119, 85–92. [Google Scholar] [CrossRef]
- Laselva, O.; Molinski, S.; Casavola, V.; Bear, C.E. Correctors of the Major Cystic Fibrosis Mutant Interact through Membrane-Spanning Domains. Mol. Pharmacol. 2018, 93, 612–618. [Google Scholar] [CrossRef]
- Tamanini, A.; Borgatti, M.; Finotti, A.; Piccagli, L.; Bezzerri, V.; Favia, M.; Guerra, L.; Lampronti, I.; Bianchi, N.; Dall’Acqua, F.; et al. Trimethylangelicin reduces IL-8 transcription and potentiates CFTR function. Am. J. Physiol. Lung Cell Mol. Physiol. 2011, 300, L380–L390. [Google Scholar] [CrossRef]
- Europe Medicinal Agency. EU/3/13/1137: Orphan Designation for the Treatment of Cystic Fibrosis. 2020. Available online: https://www.ema.europa.eu/en/medicines/human/orphan-designations/eu3131137 (accessed on 22 August 2022).
- Marzaro, G.; Lampronti, I.; D’Aversa, E.; Sacchetti, G.; Miolo, G.; Vaccarin, C.; Cabrini, G.; Dechecchi, M.C.; Gambari, R.; Chilin, A. Design, synthesis and biological evaluation of novel trimethylangelicin analogues targeting nuclear factor kB (NF-kB). Eur. J. Med. Chem. 2018, 151, 285–293. [Google Scholar] [CrossRef]
- Laselva, O.; Marzaro, G.; Vaccarin, C.; Lampronti, I.; Tamanini, A.; Lippi, G.; Gambari, R.; Cabrini, G.; Bear, C.E.; Chilin, A.; et al. Molecular Mechanism of Action of Trimethylangelicin Derivatives as CFTR Modulators. Front. Pharmacol. 2018, 9, 719. [Google Scholar] [CrossRef]
- Miolo, G.; Dall’Acqua, F.; Moustacchi, E.; Sage, E. Monofunctional angular furocoumarins: Sequence specificity in DNA photobinding of 6,4,4′-trimethylangelicin and other angelicins. Photochem. Photobiol. 1989, 50, 75–84. [Google Scholar] [CrossRef]
- Chimichi, S.; Boccalini, M.; Cosimelli, B.; Viola, G.; Vedaldi, D.; Dall’Acqua, F. A convenient synthesis of psoralens. Tetrahedron 2002, 58, 4859–4863. [Google Scholar] [CrossRef]
- Maron, D.M.; Ames, B.N. Revised methods for the Salmonella mutagenicity test. Mutat. Res. 1983, 113, 173–215. [Google Scholar] [CrossRef]
- Mortelmans, K.; Zeiger, E. The Ames Salmonella/microsome mutagenicity assay. Mutat. Res. 2000, 455, 29–60. [Google Scholar] [CrossRef]
- Holm, R. Bridging the gaps between academic research and industrial product developments of lipid-based formulations. Adv. Drug Deliv. Rev. 2019, 142, 118–127. [Google Scholar] [CrossRef]
- Franceschinis, E.; Roverso, M.; Gabbia, D.; De Martin, S.; Brusegan, M.; Vaccarin, C.; Bogialli, S.; Chilin, A. Self-Emulsifying Formulations to Increase the Oral Bioavailability of 4,6,4′Trimethylangelicin as a Possible Treatment for Cystic Fibrosis. Pharmaceutics 2022, 14, 1806. [Google Scholar] [CrossRef]
- Yin, H.F.; Yin, C.-M.; Ouyang, T.; Sun, S.-D.; Chen, W.-G.; Yang, X.-L.; He, X.; Zhang, C.-F. Self-Nanoemulsifying Drug Delivery System of Genkwanin: A Novel Approach for Anti-Colitis-Associated Colorectal Cancer. Drug Des. Devel. Ther. 2021, 15, 557–576. [Google Scholar] [CrossRef]
- Favia, M.; Mancini, M.T.; Bezzerri, V.; Guerra, L.; Laselva, O.; Abbattiscianni, A.C.; Debellis, L.; Reshkin, S.J.; Gambari, R.; Cabrini, G.; et al. Trimethylangelicin promotes the functional rescue of mutant F508del CFTR protein in cystic fibrosis airway cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2014, 307, L48–L61. [Google Scholar] [CrossRef]
- Pedemonte, N.; Zegarra-Moran, O.; Galietta, L.J. High-throughput screening of libraries of compounds to identify CFTR modulators. Methods Mol. Biol. 2011, 741, 13–21. [Google Scholar] [CrossRef]
- Frión-Herrera, Y.; Gabbia, D.; Cuesta-Rubio, O.; De Martin, S.; Carrara, M. Nemorosone inhibits the proliferation and migration of hepatocellular carcinoma cells. Life Sci. 2019, 235, 116817. [Google Scholar] [CrossRef] [PubMed]
- Guiotto, A.; Rodighiero, P.; Manzini, P.; Pastorini, G.; Bordin, F.; Baccichetti, F.; Carlassare, F.; Vedaldi, D.; Dall’Acqua, F.; Tamaro, M.; et al. 6-Methylangelicins: A new series of potential photochemotherapeutic agents for the treatment of psoriasis. J. Med. Chem. 1984, 27, 959–967. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | R4 | R6 | ID Final Product 1 |
|---|---|---|---|
| 1 | - | Me | - |
| 2 | - | Et | - |
| 3, 8, 11 | cPr | Me | CPDMA |
| 4, 9, 12 | Ph | Me | 4-PhDMA |
| 5, 10, 13 | Ph | Et | 4-PhEMA |
| 3, 14, 17 | cPr | Me | CPMA |
| 6, 15, 18 | iPr | Me | IPMA |
| 7, 16, 19 | iPr | Et | IPEA |
| Compounds | R4 | R6 | ID Final Product 1 |
|---|---|---|---|
| 24 | Me | Ph | 6-PhDMA |
| 25 | Me | p-NH2Ph | pANDMA |
| 26 | Me | m-NH2Ph | mANDMA |
| 27 | Me | Pyridyl | PyDMA |
| 28 | Me | Thienyl | ThiDMA |
| Compounds | R4 | R6 | ID Final Product 1 |
|---|---|---|---|
| 29 | - | Ph | - |
| 30, 32, 34 | iPr | Ph | IPPhMA |
| 31, 33, 35 | cPr | Ph | CPPhMA |
| Compound | t1/2 (h) | Cmax (nmol/L) | Tmax (h) | AUC 0-inf (nmol/L*h) | CL/F (mL/h/g) |
|---|---|---|---|---|---|
| IPEMA | 9.01 ± 1.34 | 120.4 ± 55.3 | 1 ± 0 | 394.50 ± 98.72 | 0.0633 ± 0.005 |
| 4-PhDMA | 21.13 ± 0.98 | 221.77 ± 184.09 | 1.25 ± 1.06 | 2747.36 ± 513.61 | 0.0014 ± 0.0014 |
| pANDMA | 1.91 ± 0.71 | 1523.5 ± 260.9 | 0.5 ± 0 | 2405.4 ± 208.2 | 0.0083 ± 0.0007 |
| pANDMA mesylate | 1.87 ± 0.43 | 3868.3 ± 1065.2 | 0.375 ± 0.177 | 9402.6 ± 742.2 | 0.0091 ± 0.0002 |
| Compound | Formulation | Droplet Size (nm) | PDI (-) |
|---|---|---|---|
| IPEMA | A | 14.62 ± 2.80 | 0.24 |
| IPEMA | B | 15.10 ± 1.50 | 0.18 |
| 4-PhDMA | A | 13.23 ± 2.10 | 0.31 |
| 4-PhDMA | B | 14.56 ± 1.57 | 0.35 |
| Compound/. Formulation | t1/2 (h) | Cmax (nmol/L) | Tmax (h) | AUC 0-inf (nmol/L*h) | CL/F (mL/h/g) |
|---|---|---|---|---|---|
| IPEMA A | 5.99 ± 0.87 | 3184,582 ± 897,45 | 0.25 ± 0 | 12,456.1 ± 1456.3 | 0.0021 ± 0.007 |
| IPEMA B | 6.15 ± 1.09 | 845.73 ± 76.54 | 0.25 ± 0 | 6671.16 ± 432.13 | 0.0037 ± 0.002 |
| 4-PhDMA A | 6.08 ± 4.78 | 4374.8 ± 2296.6 | 1.5 ± 0.71 | 17,699.4 ± 6170.2 | 0.0012 ± 0.0005 |
| 4-PhDMA B | 7.31 ± 4.29 | 2683.0 ± 370.4 | 1.25 ± 1.06 | 13,337.8 ± 6463.4 | 0.0017 ± 0.0008 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vaccarin, C.; Gabbia, D.; Franceschinis, E.; De Martin, S.; Roverso, M.; Bogialli, S.; Sacchetti, G.; Tupini, C.; Lampronti, I.; Gambari, R.; et al. Improved Trimethylangelicin Analogs for Cystic Fibrosis: Design, Synthesis and Preliminary Screening. Int. J. Mol. Sci. 2022, 23, 11528. https://doi.org/10.3390/ijms231911528
Vaccarin C, Gabbia D, Franceschinis E, De Martin S, Roverso M, Bogialli S, Sacchetti G, Tupini C, Lampronti I, Gambari R, et al. Improved Trimethylangelicin Analogs for Cystic Fibrosis: Design, Synthesis and Preliminary Screening. International Journal of Molecular Sciences. 2022; 23(19):11528. https://doi.org/10.3390/ijms231911528
Chicago/Turabian StyleVaccarin, Christian, Daniela Gabbia, Erica Franceschinis, Sara De Martin, Marco Roverso, Sara Bogialli, Gianni Sacchetti, Chiara Tupini, Ilaria Lampronti, Roberto Gambari, and et al. 2022. "Improved Trimethylangelicin Analogs for Cystic Fibrosis: Design, Synthesis and Preliminary Screening" International Journal of Molecular Sciences 23, no. 19: 11528. https://doi.org/10.3390/ijms231911528