

In Vitro/In Vivo Translation of Synergistic Combination of MDM2 and MEK Inhibitors in Melanoma Using PBPK/PD Modelling: Part II

Abstract
:1. Introduction
2. Results
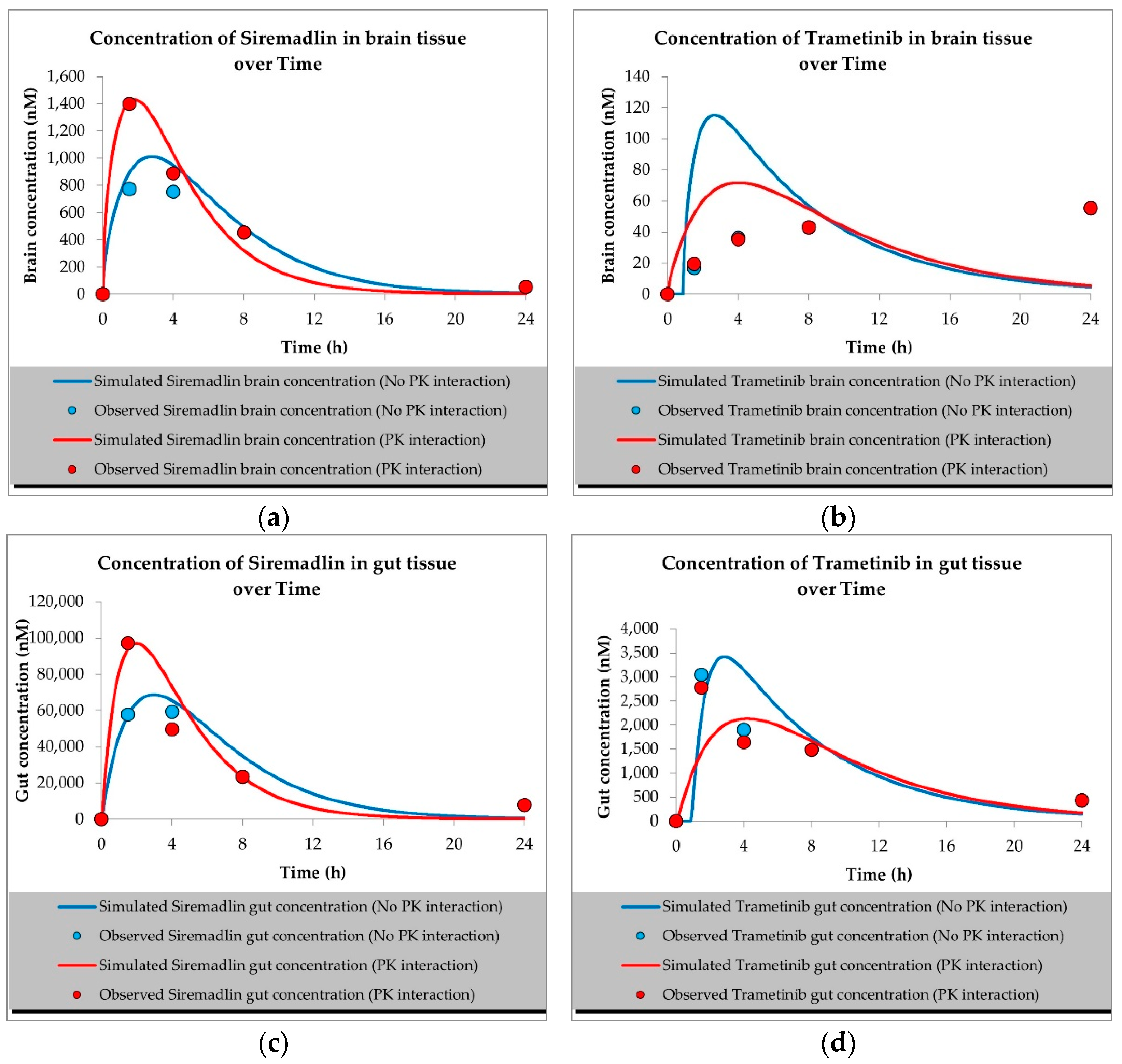
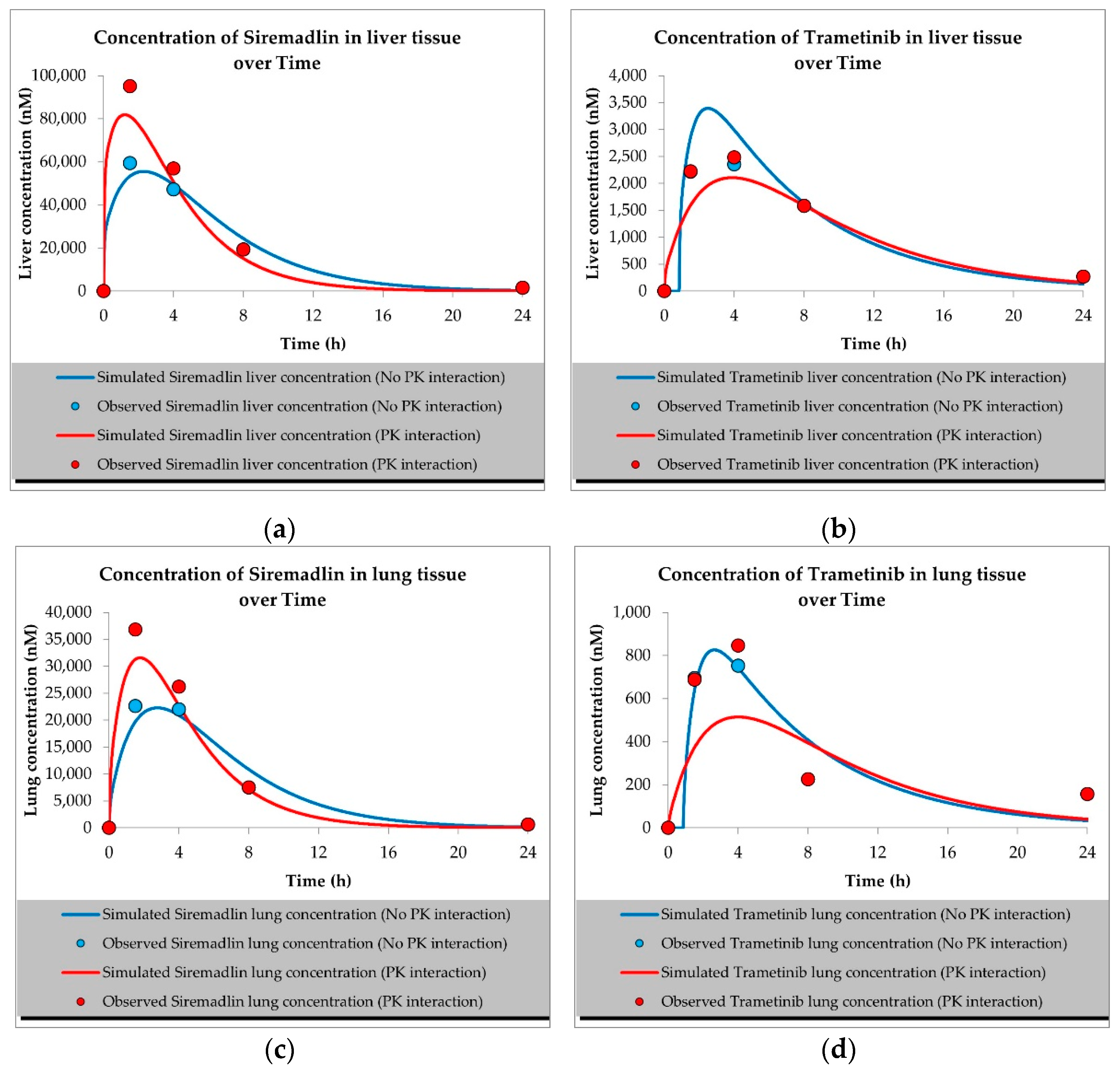
2.1. PBPK Models (with and without PK Interaction)
2.2. PD (TGI) Models
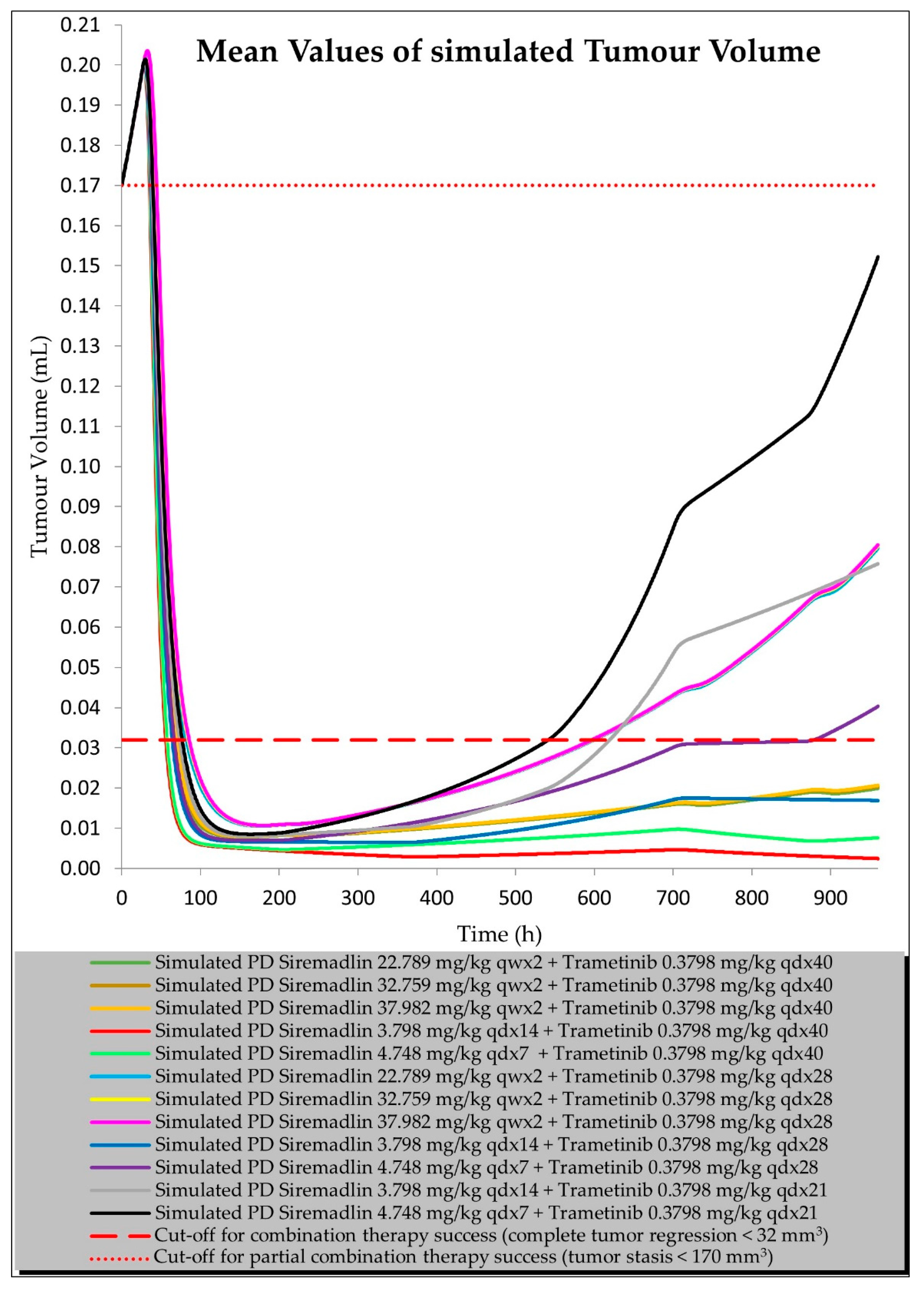
2.3. PBPK/PD Estimation with Universal Model for Drug Combination at Human Equivalent Doses (HEDs)
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Software
4.3. Studies Involving Animals
4.4. Physiologically Based Pharmacokinetic Models
4.4.1. General PBPK Modelling Strategy
4.4.2. Mouse Population
4.4.3. PBPK Model Verification
4.5. Pharmacodynamic Modelling
4.5.1. General PD Modelling Strategy
4.5.2. PD (TGI) Model Development and Verification
4.5.3. TGI Model Parameter Dependence Estimations (Universal Model Development)
4.5.4. Tumour Volume Simulation for Drug Combination at Human Equivalent Doses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Steininger, J.; Gellrich, F.F.; Schulz, A.; Westphal, D.; Beissert, S.; Meier, F. Systemic Therapy of Metastatic Melanoma: On the Road to Cure. Cancers 2021, 13, 1430. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, S.; Tykodi, S.S.; Thompson, J.A. Treatment of Metastatic Melanoma: An Overview. Oncology 2009, 23, 488–496. [Google Scholar] [PubMed]
- Morrison, C.; Pabla, S.; Conroy, J.M.; Nesline, M.K.; Glenn, S.T.; Dressman, D.; Papanicolau-Sengos, A.; Burgher, B.; Andreas, J.; Giamo, V.; et al. Predicting Response to Checkpoint Inhibitors in Melanoma beyond PD-L1 and Mutational Burden. J. Immunother. Cancer 2018, 6, 32. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, V.; Batty, K.; Long, G.V.; Carlino, M.S.; Peters, G.D.; Bhave, P.; Moore, M.A.; Xu, W.; Brown, L.J.; Arneil, M.; et al. Activity and Safety of Third-Line BRAF-Targeted Therapy (TT) Following First-Line TT and Second-Line Immunotherapy (IT) in Advanced Melanoma. J Clin Oncol. 2020, 38, 10049. [Google Scholar] [CrossRef]
- Chatterjee, N.; Bivona, T.G. Polytherapy and Targeted Cancer Drug Resistance. Trends Cancer 2019, 5, 170–182. [Google Scholar] [CrossRef] [PubMed]
- Witkowski, J.; Polak, S.; Rogulski, Z.; Pawelec, D. In Vitro/In Vivo Translation of Synergistic Combination of MDM2 and MEK Inhibitors in Melanoma Using PBPK/PD Modelling: Part I. Int. J. Mol. Sci. 2022, 23, 11939. [Google Scholar] [CrossRef]
- Wu, C.-E.; Koay, T.S.; Esfandiari, A.; Ho, Y.-H.; Lovat, P.; Lunec, J. ATM Dependent DUSP6 Modulation of P53 Involved in Synergistic Targeting of MAPK and P53 Pathways with Trametinib and MDM2 Inhibitors in Cutaneous Melanoma. Cancers 2018, 11, 3. [Google Scholar] [CrossRef] [Green Version]
- Goldblatt, E.M.; Lee, W.-H. From Bench to Bedside: The Growing Use of Translational Research in Cancer Medicine. Am. J. Transl Res. 2010, 2, 1–18. [Google Scholar]
- Niu, J.; Straubinger, R.M.; Mager, D.E. Pharmacodynamic Drug–Drug Interactions. Clin. Pharmacol. Ther. 2019, 105, 1395–1406. [Google Scholar] [CrossRef]
- Berenbaum, M.C. What Is Synergy? Pharmacol. Rev. 1989, 41, 93–141. [Google Scholar]
- Tang, J.; Wennerberg, K.; Aittokallio, T. What Is Synergy? The Saariselkä Agreement Revisited. Front. Pharmacol. 2015, 6, 181. [Google Scholar] [CrossRef] [PubMed]
- Cokol, M.; Chua, H.N.; Tasan, M.; Mutlu, B.; Weinstein, Z.B.; Suzuki, Y.; Nergiz, M.E.; Costanzo, M.; Baryshnikova, A.; Giaever, G.; et al. Systematic Exploration of Synergistic Drug Pairs. Mol. Syst. Biol. 2011, 7, 544. [Google Scholar] [CrossRef]
- Jeay, S.; Ferretti, S.; Holzer, P.; Fuchs, J.; Chapeau, E.A.; Wartmann, M.; Sterker, D.; Romanet, V.; Murakami, M.; Kerr, G.; et al. Dose and Schedule Determine Distinct Molecular Mechanisms Underlying the Efficacy of the P53–MDM2 Inhibitor HDM201. Cancer Res. 2018, 78, 6257–6267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoo, J.Y.; Swanner, J.; Otani, Y.; Nair, M.; Park, F.; Banasavadi-Siddegowda, Y.; Liu, J.; Jaime-Ramirez, A.C.; Hong, B.; Geng, F.; et al. Oncolytic HSV Therapy Increases Trametinib Access to Brain Tumors and Sensitizes Them in Vivo. Neuro Oncol. 2019, 21, 1131–1140. [Google Scholar] [CrossRef] [PubMed]
- Wek, R.C.; Staschke, K.A. How Do Tumours Adapt to Nutrient Stress? EMBO J. 2010, 29, 1946–1947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theile, D.; Wizgall, P. Acquired ABC-Transporter Overexpression in Cancer Cells: Transcriptional Induction or Darwinian Selection? Naunyn-Schmiedeberg’s Arch. Pharmacol. 2021, 394, 1621–1632. [Google Scholar] [CrossRef]
- Huang, Q.; Cai, T.; Bai, L.; Huang, Y.; Li, Q.; Wang, Q.; Chiba, P.; Cai, Y. State of the Art of Overcoming Efflux Transporter Mediated Multidrug Resistance of Breast Cancer. Transl. Cancer Res. 2019, 8, 319–329. [Google Scholar] [CrossRef]
- Chen, J.; Wang, Z.; Gao, S.; Wu, K.; Bai, F.; Zhang, Q.; Wang, H.; Ye, Q.; Xu, F.; Sun, H.; et al. Human Drug Efflux Transporter ABCC5 Confers Acquired Resistance to Pemetrexed in Breast Cancer. Cancer Cell Int. 2021, 21, 136. [Google Scholar] [CrossRef]
- Samimi, G.; Safaei, R.; Katano, K.; Holzer, A.K.; Rochdi, M.; Tomioka, M.; Goodman, M.; Howell, S.B. Increased Expression of the Copper Efflux Transporter ATP7A Mediates Resistance to Cisplatin, Carboplatin, and Oxaliplatin in Ovarian Cancer Cells. Clin. Cancer Res. 2004, 10, 4661–4669. [Google Scholar] [CrossRef] [Green Version]
- Lotz, C.; Kelleher, D.K.; Gassner, B.; Gekle, M.; Vaupel, P.; Thews, O. Role of the Tumor Microenvironment in the Activity and Expression of the P-Glycoprotein in Human Colon Carcinoma Cells. Oncol. Rep. 2007, 17, 239–244. [Google Scholar] [CrossRef]
- Legrand, O.; Simonin, G.; Beauchamp-Nicoud, A.; Zittoun, R.; Marie, J.P. Simultaneous Activity of MRP1 and Pgp Is Correlated with in Vitro Resistance to Daunorubicin and with in Vivo Resistance in Adult Acute Myeloid Leukemia. Blood 1999, 94, 1046–1056. [Google Scholar] [CrossRef] [PubMed]
- Kallinowski, F.; Schlenger, K.H.; Runkel, S.; Kloes, M.; Stohrer, M.; Okunieff, P.; Vaupel, P. Blood Flow, Metabolism, Cellular Microenvironment, and Growth Rate of Human Tumor Xenografts. Cancer Res. 1989, 49, 3759–3764. [Google Scholar] [PubMed]
- Benjaminsen, I.C.; Graff, B.A.; Brurberg, K.G.; Rofstad, E.K. Assessment of Tumor Blood Perfusion by High-Resolution Dynamic Contrast-Enhanced MRI: A Preclinical Study of Human Melanoma Xenografts. Magn. Reason. Med. 2004, 52, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Graff, B.A.; Benjaminsen, I.C.; Melås, E.A.; Brurberg, K.G.; Rofstad, E.K. Changes in Intratumor Heterogeneity in Blood Perfusion in Intradermal Human Melanoma Xenografts during Tumor Growth Assessed by DCE-MRI. Magn. Reason. Imaging 2005, 23, 961–966. [Google Scholar] [CrossRef]
- Howard, G.R.; Johnson, K.E.; Rodriguez Ayala, A.; Yankeelov, T.E.; Brock, A. A Multi-State Model of Chemoresistance to Characterize Phenotypic Dynamics in Breast Cancer. Sci. Rep. 2018, 8, 12058. [Google Scholar] [CrossRef]
- Yoon, N.; Vander Velde, R.; Marusyk, A.; Scott, J.G. Optimal Therapy Scheduling Based on a Pair of Collaterally Sensitive Drugs. Bull. Math. Biol. 2018, 80, 1776–1809. [Google Scholar] [CrossRef]
- Higgins, B.; Glenn, K.; Walz, A.; Tovar, C.; Filipovic, Z.; Hussain, S.; Lee, E.; Kolinsky, K.; Tannu, S.; Adames, V.; et al. Preclinical Optimization of MDM2 Antagonist Scheduling for Cancer Treatment by Using a Model-Based Approach. Clin. Cancer Res. 2014, 20, 3742–3752. [Google Scholar] [CrossRef] [Green Version]
- Guerreiro, N.; Jullion, A.; Ferretti, S.; Fabre, C.; Meille, C. Translational Modeling of Anticancer Efficacy to Predict Clinical Outcomes in a First-in-Human Phase 1 Study of MDM2 Inhibitor HDM201. AAPS J. 2021, 23, 28. [Google Scholar] [CrossRef]
- Flaherty, K.T.; Robert, C.; Hersey, P.; Nathan, P.; Garbe, C.; Milhem, M.; Demidov, L.V.; Hassel, J.C.; Rutkowski, P.; Mohr, P.; et al. Improved Survival with MEK Inhibition in BRAF-Mutated Melanoma. N. Engl. J. Med. 2012, 367, 107–114. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.Q.; Mulford, I.J.; Sharp, F.; Liang, J.; Kurtulus, S.; Trabucco, G.; Quinn, D.S.; Longmire, T.A.; Patel, N.; Patil, R.; et al. Inhibition of MDM2 Promotes Antitumor Responses in P53 Wild-Type Cancer Cells through Their Interaction with the Immune and Stromal Microenvironment. Cancer Res. 2021, 81, 3079–3091. [Google Scholar] [CrossRef]
- Fang, D.D.; Tang, Q.; Kong, Y.; Wang, Q.; Gu, J.; Fang, X.; Zou, P.; Rong, T.; Wang, J.; Yang, D.; et al. MDM2 Inhibitor APG-115 Synergizes with PD-1 Blockade through Enhancing Antitumor Immunity in the Tumor Microenvironment. J. Immuno Ther. Cancer 2019, 7, 327. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Kryczek, I.; Li, S.; Li, X.; Aguilar, A.; Wei, S.; Grove, S.; Vatan, L.; Yu, J.; Yan, Y.; et al. The Ubiquitin Ligase MDM2 Sustains STAT5 Stability to Control T Cell-Mediated Antitumor Immunity. Nat. Immunol. 2021, 22, 460–470. [Google Scholar] [CrossRef] [PubMed]
- Stein, E.M.; DeAngelo, D.J.; Chromik, J.; Chatterjee, M.; Bauer, S.; Lin, C.-C.; Suarez, C.; de Vos, F.; Steeghs, N.; Cassier, P.A.; et al. Results from a First-in-Human Phase I Study of Siremadlin (HDM201) in Patients with Advanced Wild-Type TP53 Solid Tumors and Acute Leukemia. Clin. Cancer Res. 2022, 28, 870–881. [Google Scholar] [CrossRef] [PubMed]
- Mistry, H.B.; Orrell, D.; Eftimie, R. Model Based Analysis of the Heterogeneity in the Tumour Size Dynamics Differentiates Vemurafenib, Dabrafenib and Trametinib in Metastatic Melanoma. Cancer Chemother. Pharm. 2018, 81, 325–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodgers, T.; Leahy, D.; Rowland, M. Physiologically Based Pharmacokinetic Modeling 1: Predicting the Tissue Distribution of Moderate-to-Strong Bases. J. Pharm. Sci. 2005, 94, 1259–1276. [Google Scholar] [CrossRef]
- Jeong, Y.-S.; Jusko, W.J. Consideration of Fractional Distribution Parameter Fd in the Chen and Gross Method for Tissue-to-Plasma Partition Coefficients: Comparison of Several Methods. Pharm Res. 2022, 39, 463–479. [Google Scholar] [CrossRef]
- Lei, Y.; Zhenglin, Y.; Heng, L. MDM2 Inhibitors. U.S. Patent 11,339,171, 24 May 2022. [Google Scholar]
- Hofmann, F. Small Molecule HDM201 Inhibitor HDM201. In Proceedings of the AACR Annual Meeting 2016, New Orleans, LA, USA, 16–20 April 2016. [Google Scholar]
- Pharmaceuticals and Medical Devices Agency (PMDA), Mekinist Initial Approval. Available online: https://www.pmda.go.jp/files/000233741.pdf (accessed on 19 August 2022).
- Food and Drug Administration (FDA), Trametinib Pharmacology Review. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2013/204114Orig1s000PharmR.pdf (accessed on 19 August 2022).
- Tsakalozou, E.; Alam, K.; Babiskin, A.; Zhao, L. Physiologically-Based Pharmacokinetic Modeling to Support Determination of Bioequivalence for Dermatological Drug Products: Scientific and Regulatory Considerations. Clin. Pharmacol. Ther. 2022, 111, 1036–1049. [Google Scholar] [CrossRef]
- Shebley, M.; Sandhu, P.; Emami Riedmaier, A.; Jamei, M.; Narayanan, R.; Patel, A.; Peters, S.A.; Reddy, V.P.; Zheng, M.; de Zwart, L.; et al. Physiologically Based Pharmacokinetic Model Qualification and Reporting Procedures for Regulatory Submissions: A Consortium Perspective. Clin. Pharmacol. Ther. 2018, 104, 88–110. [Google Scholar] [CrossRef] [Green Version]
- Stypmann, J.; Engelen, M.A.; Epping, C.; van Rijen, H.V.M.; Milberg, P.; Bruch, C.; Breithardt, G.; Tiemann, K.; Eckardt, L. Age and Gender Related Reference Values for Transthoracic Doppler-Echocardiography in the Anesthetized CD1 Mouse. Int. J. Cardiovasc. Imaging 2006, 22, 353–362. [Google Scholar] [CrossRef]
- Jeanne, A.; Boulagnon-Rombi, C.; Devy, J.; Théret, L.; Fichel, C.; Bouland, N.; Diebold, M.-D.; Martiny, L.; Schneider, C.; Dedieu, S. Matricellular TSP-1 as a Target of Interest for Impeding Melanoma Spreading: Towards a Therapeutic Use for TAX2 Peptide. Clin. Exp. Metastasis 2016, 33, 637–649. [Google Scholar] [CrossRef]
- Traynard, P. A Library of Tumor Growth and Tumor Growth Inhibition Models for the MonolixSuite. In Proceedings of the Tumor Growth Inhibition Modeling with Monolixsuite, ACoP 11 Virtual Conference, 13 November 2020. [Google Scholar]
- Reig-López, J.; Maldonado, M. del M.; Merino-Sanjuan, M.; Cruz-Collazo, A.M.; Ruiz-Calderón, J.F.; Mangas-Sanjuán, V.; Dharmawardhane, S.; Duconge, J. Physiologically-Based Pharmacokinetic/Pharmacodynamic Model of MBQ-167 to Predict Tumor Growth Inhibition in Mice. Pharmaceutics 2020, 12, 975. [Google Scholar] [CrossRef] [PubMed]
- Simeoni, M.; Magni, P.; Cammia, C.; De Nicolao, G.; Croci, V.; Pesenti, E.; Germani, M.; Poggesi, I.; Rocchetti, M. Predictive Pharmacokinetic-Pharmacodynamic Modeling of Tumor Growth Kinetics in Xenograft Models after Administration of Anticancer Agents. Cancer Res. 2004, 64, 1094–1101. [Google Scholar] [CrossRef] [PubMed]
- Lobo, E.D.; Balthasar, J.P. Pharmacodynamic Modeling of Chemotherapeutic Effects: Application of a Transit Compartment Model to Characterize Methotrexate Effects in Vitro. AAPS J. 2002, 4, 212–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Claret, L.; Girard, P.; Hoff, P.M.; Van Cutsem, E.; Zuideveld, K.P.; Jorga, K.; Fagerberg, J.; Bruno, R. Model-Based Prediction of Phase III Overall Survival in Colorectal Cancer on the Basis of Phase II Tumor Dynamics. J Clin Oncol. 2009, 27, 4103–4108. [Google Scholar] [CrossRef] [PubMed]
- Hahnfeldt, P.; Folkman, J.; Hlatky, L. Minimizing Long-Term Tumor Burden: The Logic for Metronomic Chemotherapeutic Dosing and Its Antiangiogenic Basis. J. Theor. Biol. 2003, 220, 545–554. [Google Scholar] [CrossRef]
- Desmée, S.; Mentré, F.; Veyrat-Follet, C.; Guedj, J. Nonlinear Mixed-Effect Models for Prostate-Specific Antigen Kinetics and Link with Survival in the Context of Metastatic Prostate Cancer: A Comparison by Simulation of Two-Stage and Joint Approaches. AAPS J. 2015, 17, 691–699. [Google Scholar] [CrossRef] [PubMed]
- Jusko, W.J. A Pharmacodynamic Model for Cell-Cycle-Specific Chemotherapeutic Agents. J. Pharmacokinet. Biopharm. 1973, 1, 175–200. [Google Scholar] [CrossRef]
- Mould, D.; Walz, A.-C.; Lave, T.; Gibbs, J.; Frame, B. Developing Exposure/Response Models for Anticancer Drug Treatment: Special Considerations. CPT Pharmacomet. Syst. Pharmacol. 2015, 4, e00016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, A.J.; Arnone, M.R.; Bleam, M.R.; Moss, K.G.; Yang, J.; Fedorowicz, K.E.; Smitheman, K.N.; Erhardt, J.A.; Hughes-Earle, A.; Kane-Carson, L.S.; et al. Dabrafenib; Preclinical Characterization, Increased Efficacy When Combined with Trametinib, While BRAF/MEK Tool Combination Reduced Skin Lesions. PLoS ONE 2013, 8, e67583. [Google Scholar] [CrossRef] [Green Version]
- Gilmartin, A.G.; Bleam, M.R.; Groy, A.; Moss, K.G.; Minthorn, E.A.; Kulkarni, S.G.; Rominger, C.M.; Erskine, S.; Fisher, K.E.; Yang, J.; et al. GSK1120212 (JTP-74057) Is an Inhibitor of MEK Activity and Activation with Favorable Pharmacokinetic Properties for Sustained in Vivo Pathway Inhibition. Clin. Cancer Res. 2011, 17, 989–1000. [Google Scholar] [CrossRef] [Green Version]
- Cubas, R.; Moskalenko, M.; Cheung, J.; Yang, M.; McNamara, E.; Xiong, H.; Hoves, S.; Ries, C.H.; Kim, J.; Gould, S. Chemotherapy Combines Effectively with Anti–PD-L1 Treatment and Can Augment Antitumor Responses. J. Immunol. 2018, 201, 2273–2286. [Google Scholar] [CrossRef] [Green Version]
- Jahchan, N.; Ramoth, H.; Juric, V.; Mayes, E.; Mankikar, S.; Mehta, R.; Binnewies, M.; Dash, S.; Palmer, R.; Pollack, J.; et al. 859 Tuning the Tumor Microenvironment by Reprogramming TREM1+ Myeloid Cells to Unleash Anti-Tumor Immunity in Solid Tumors. J. Immunother. Cancer 2021, 9, A900. [Google Scholar] [CrossRef]
- Houghton, P.J.; Morton, C.L.; Tucker, C.; Payne, D.; Favours, E.; Cole, C.; Gorlick, R.; Kolb, E.A.; Zhang, W.; Lock, R.; et al. The Pediatric Preclinical Testing Program: Description of Models and Early Testing Results. Pediatric Blood Cancer 2007, 49, 928–940. [Google Scholar] [CrossRef]
- Nair, A.B.; Jacob, S. A Simple Practice Guide for Dose Conversion between Animals and Human. J. Basic Clin. Pharm 2016, 7, 27–31. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Initial Tumour Volume (mm3) | Doses (mg/kg) | Dose Schedule | N | Comments |
|---|---|---|---|---|---|
| Vehicle (Adamed) | ~135 | - | q1dx5/q7dx2 | 10 | Adamed reference |
| Siremadlin | ~137 | 25/50 | q1dx5 | 10 | Adamed reference |
| Siremadlin | ~137 | 50/100 | q7dx2 | 10 | Adamed reference |
| Vehicle (current study) | ~162 | - | qdx6 | 11 | Efficacy in current study |
| Siremadlin | ~163–172 | 40/100 | qdx3 | 6 | Efficacy in current study |
| Trametinib | ~167–180 | 0.3/1 | qdx6 | 6 | Efficacy in current study |
| Siremadlin + Trametinib | ~165–169 | 40 + 0.3/40 + 1/100 + 0.3/100 + 1 | qdx3/qdx6 | 6 | Efficacy in current study |
| Siremadlin | ~300 | 100 | qdx1 | 12 | PK in current study |
| Trametinib | ~300 | 1 | qdx1 | 12 | PK in current study |
| Siremadlin + Trametinib | ~300 | 100 + 1 | qdx1 | 12 | PK in current study |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Witkowski, J.; Polak, S.; Rogulski, Z.; Pawelec, D. In Vitro/In Vivo Translation of Synergistic Combination of MDM2 and MEK Inhibitors in Melanoma Using PBPK/PD Modelling: Part II. Int. J. Mol. Sci. 2022, 23, 11939. https://doi.org/10.3390/ijms231911939
Witkowski J, Polak S, Rogulski Z, Pawelec D. In Vitro/In Vivo Translation of Synergistic Combination of MDM2 and MEK Inhibitors in Melanoma Using PBPK/PD Modelling: Part II. International Journal of Molecular Sciences. 2022; 23(19):11939. https://doi.org/10.3390/ijms231911939
Chicago/Turabian StyleWitkowski, Jakub, Sebastian Polak, Zbigniew Rogulski, and Dariusz Pawelec. 2022. "In Vitro/In Vivo Translation of Synergistic Combination of MDM2 and MEK Inhibitors in Melanoma Using PBPK/PD Modelling: Part II" International Journal of Molecular Sciences 23, no. 19: 11939. https://doi.org/10.3390/ijms231911939