
Molecular Pathogenesis of Colorectal Cancer: Impact of Oncogenic Targets Regulated by Tumor Suppressive miR-139-3p
 , , , ,
, , , ,
Abstract
:1. Introduction
2. Results
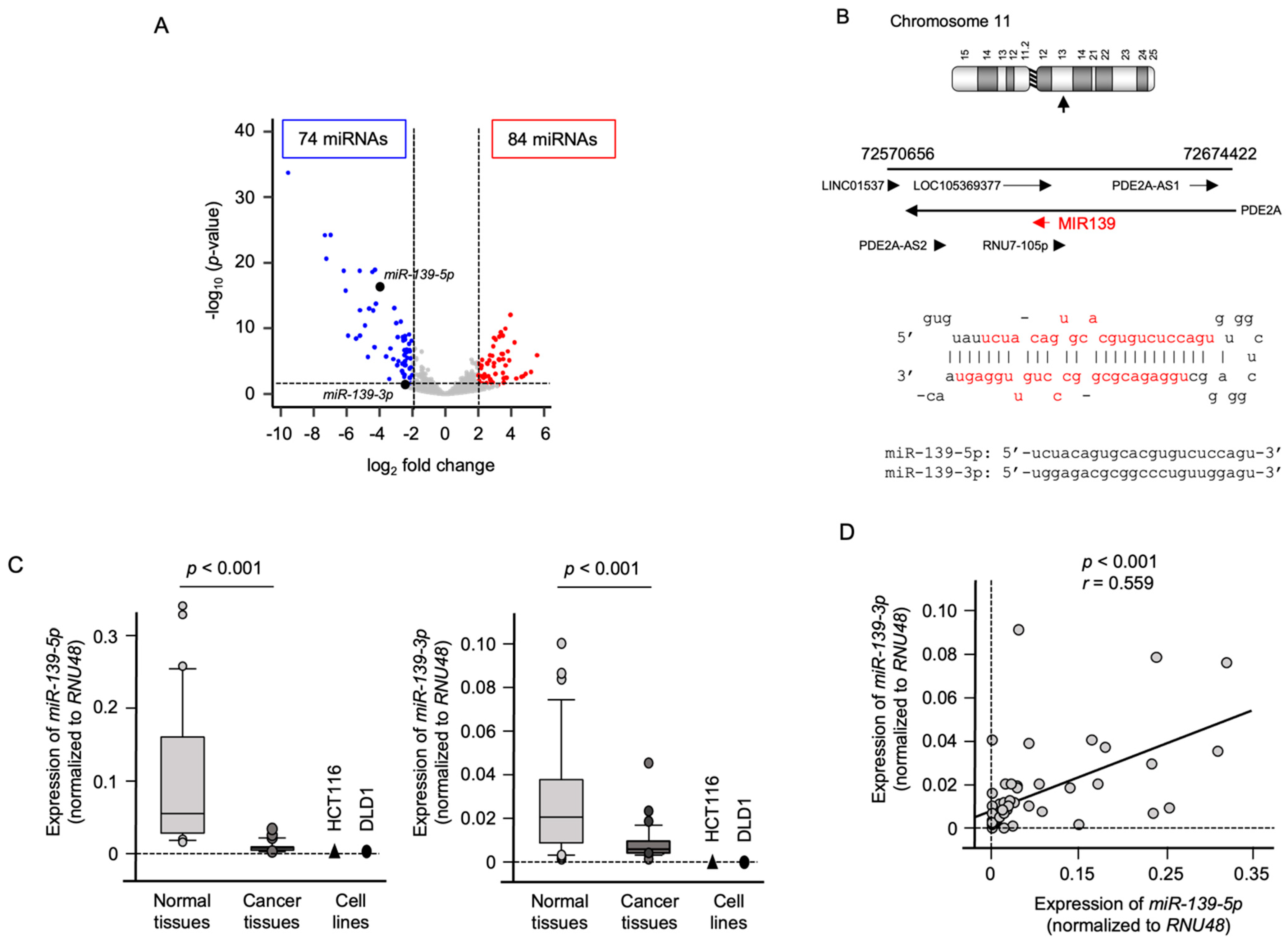
2.1. Expression of miR-139-5p and miR-139-3p in CRC Specimens
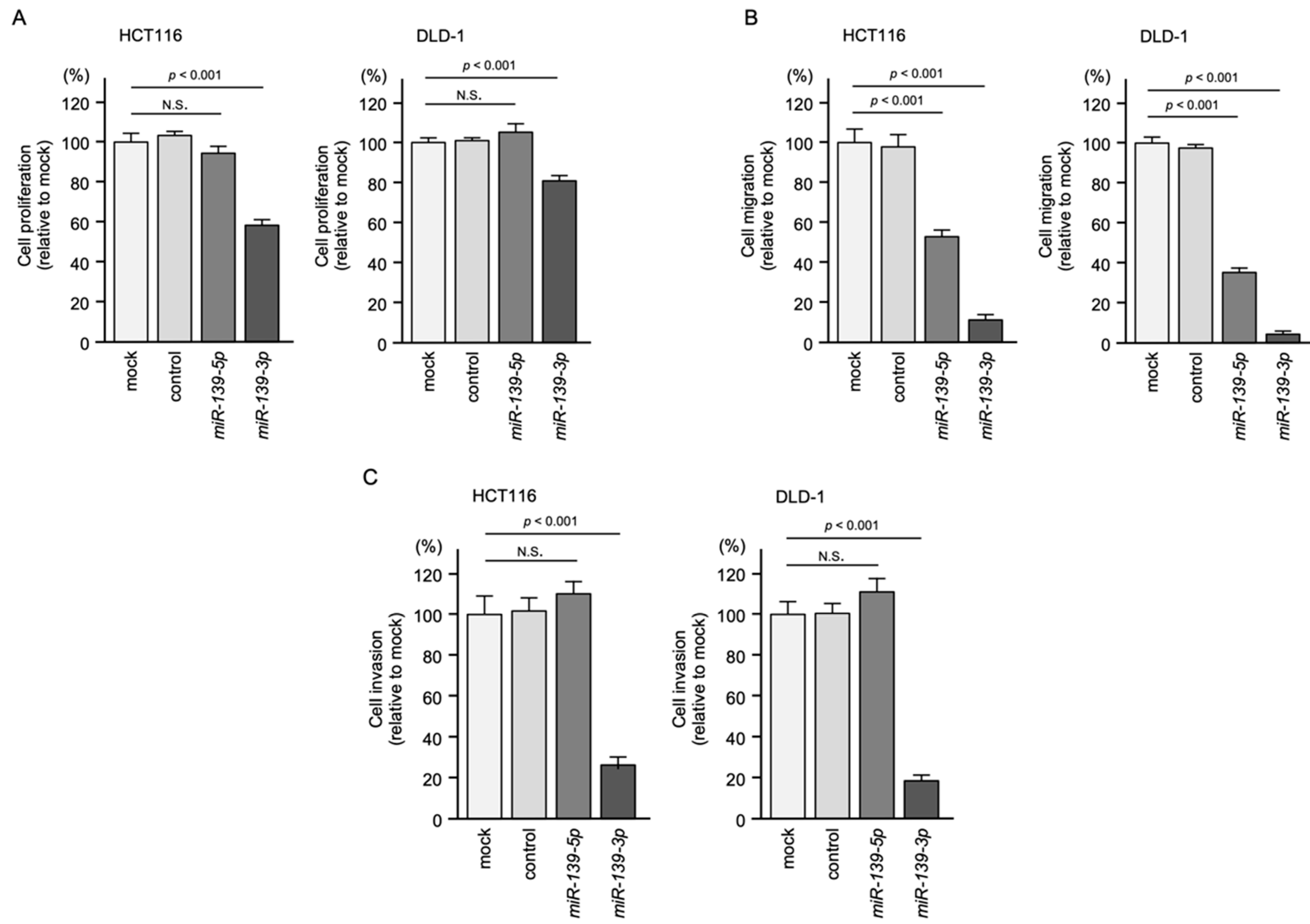
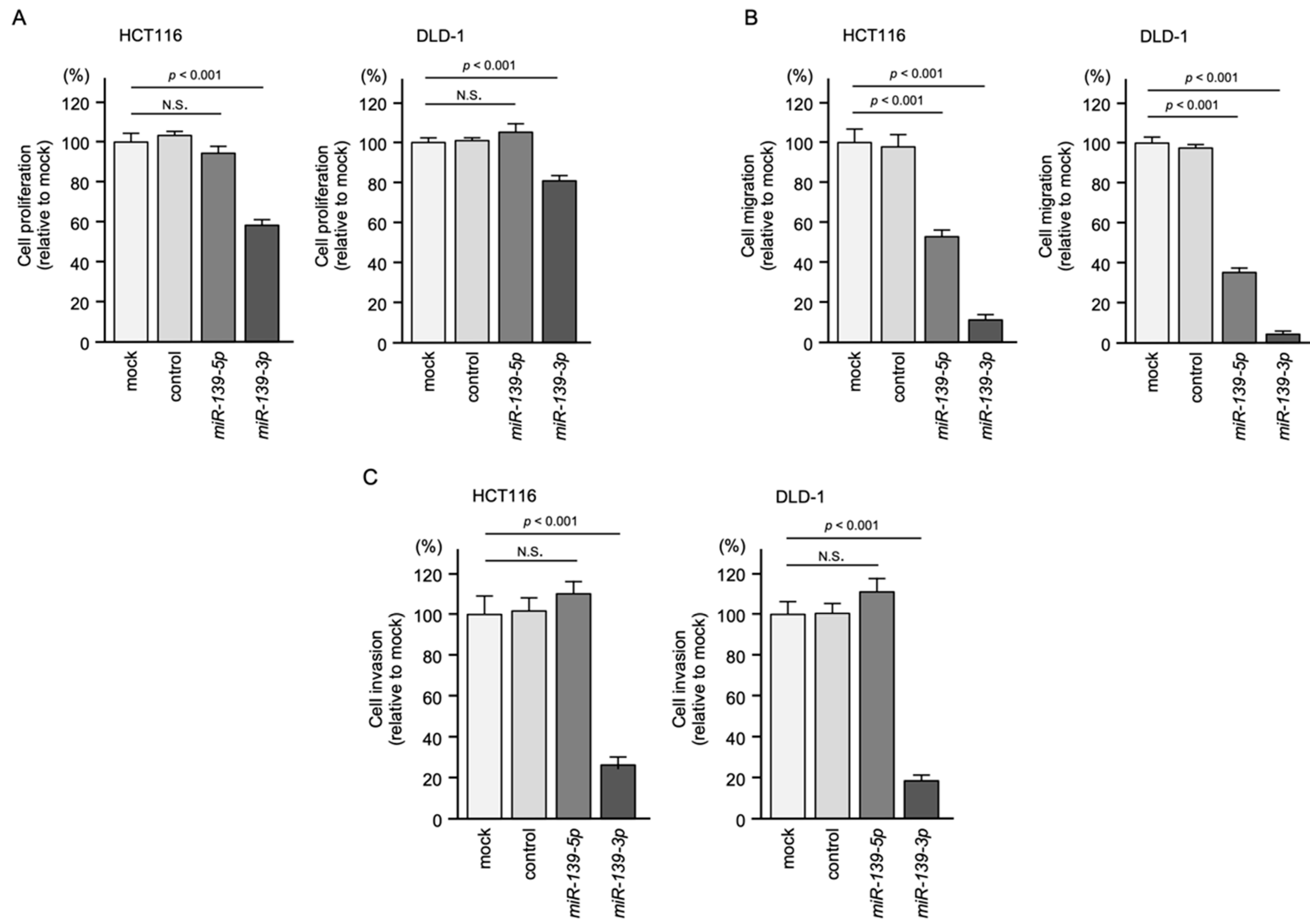
2.2. Ectopic Expression Assays of miR-139-5p and miR-139-3p in CRC Cell Lines
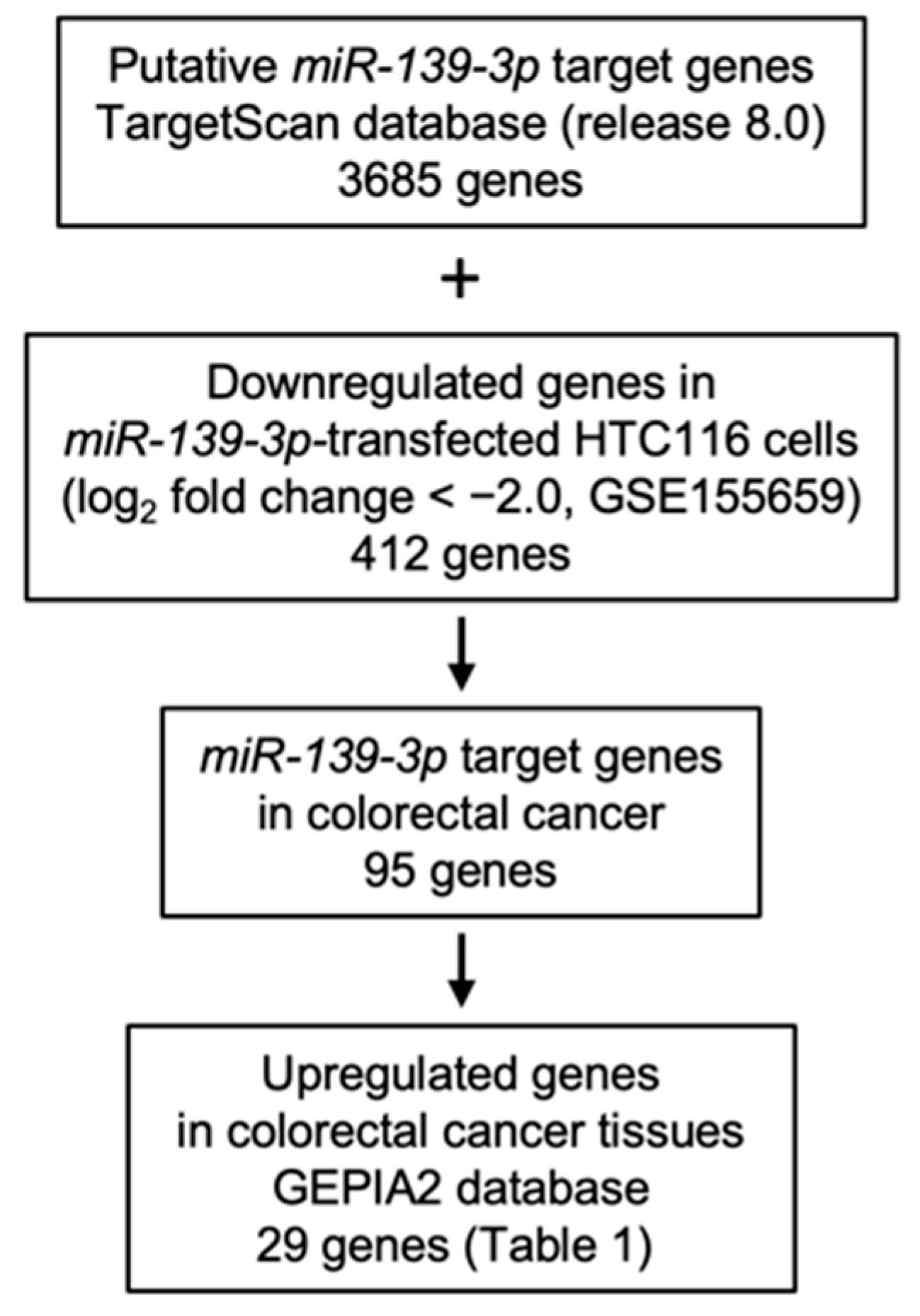
2.3. Identification of Oncogenes Regulated by miR-139-3p in CRC Cells
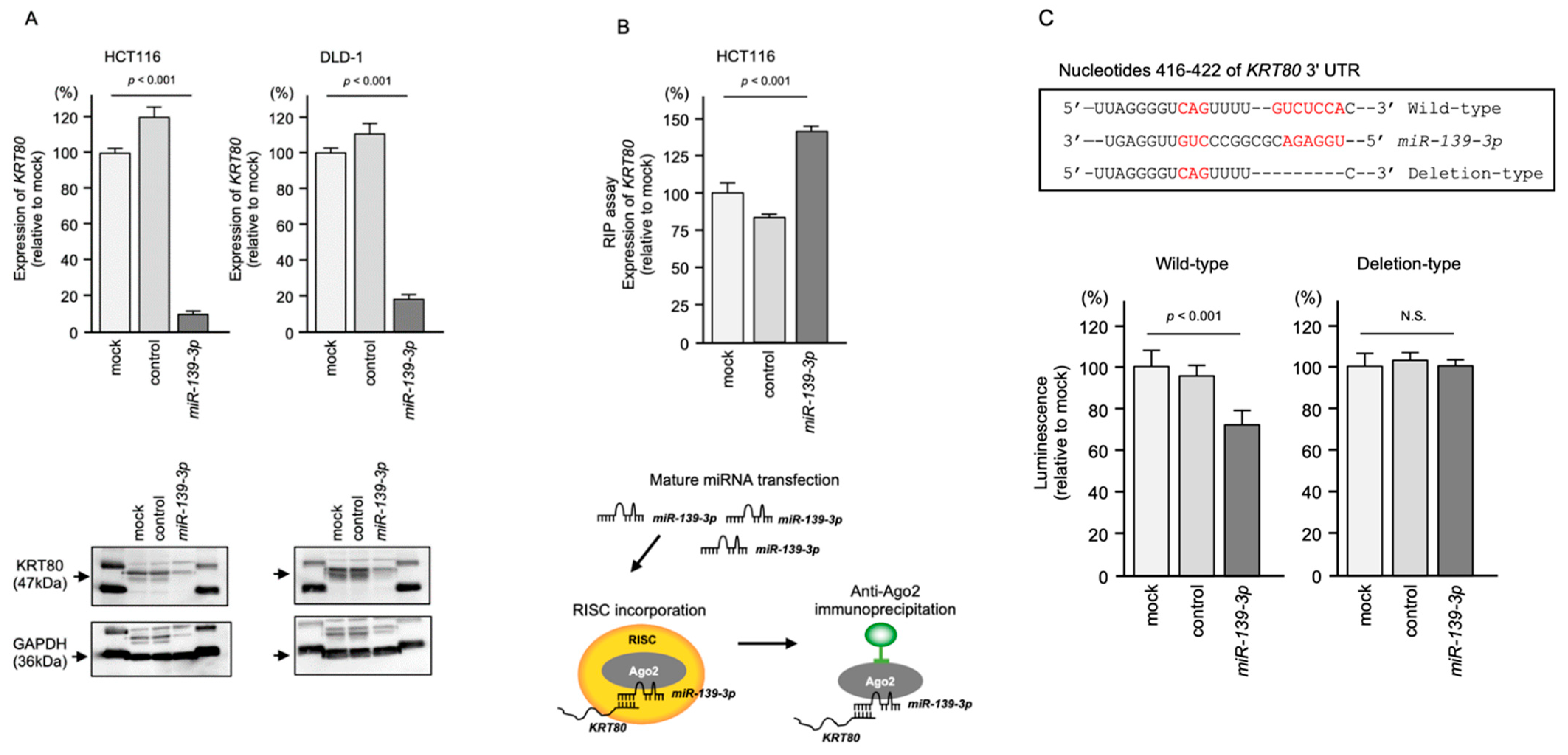
2.4. Direct Regulation of KRT80 by miR-139-3p in CRC Cells
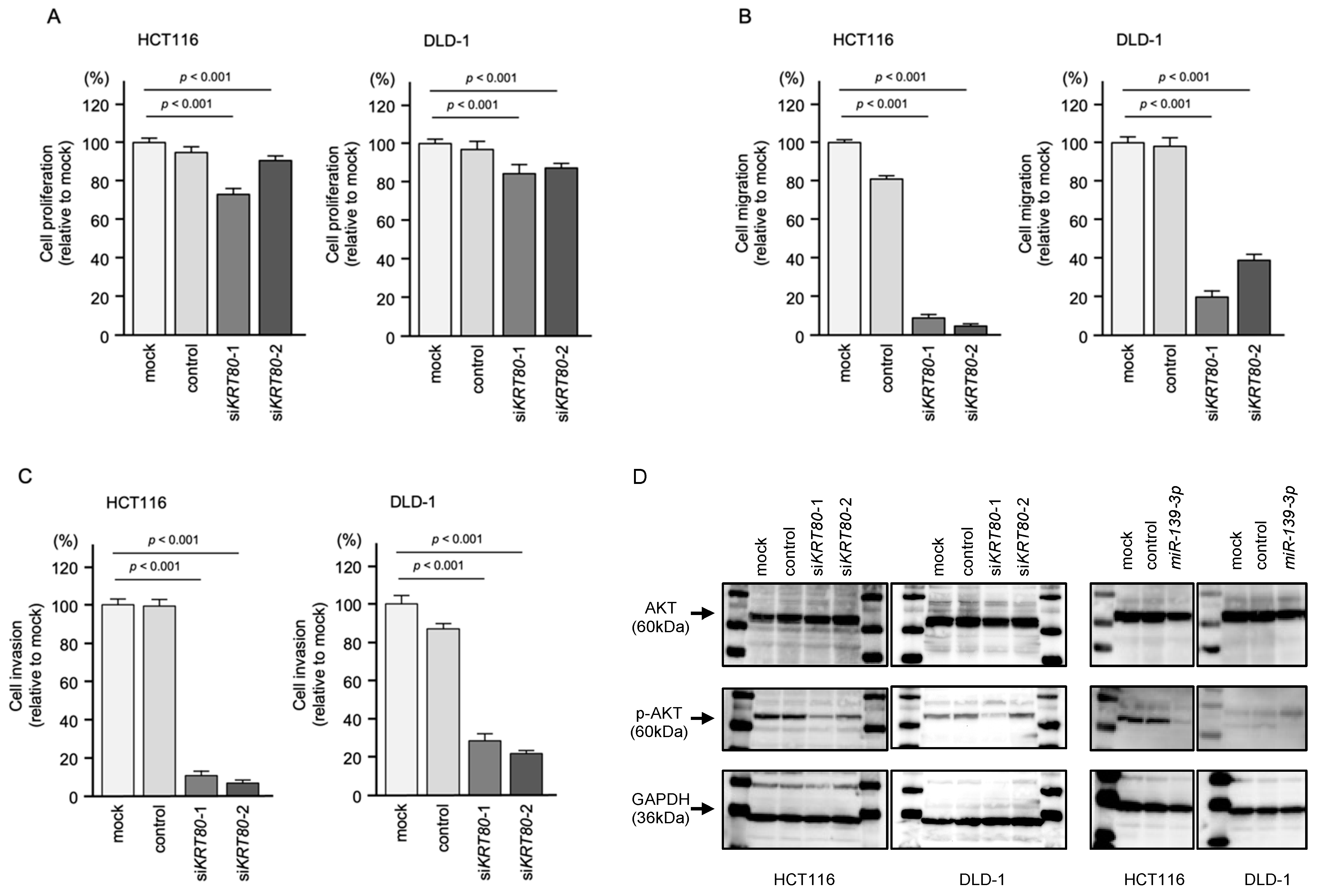
2.5. Knockdown Assays by Small Interfering RNAs (siRNAs) Targeting KRT80 in CRC Cell Lines
2.6. Aberrant Expression of KRT80 Protein in CRC Clinical Specimens
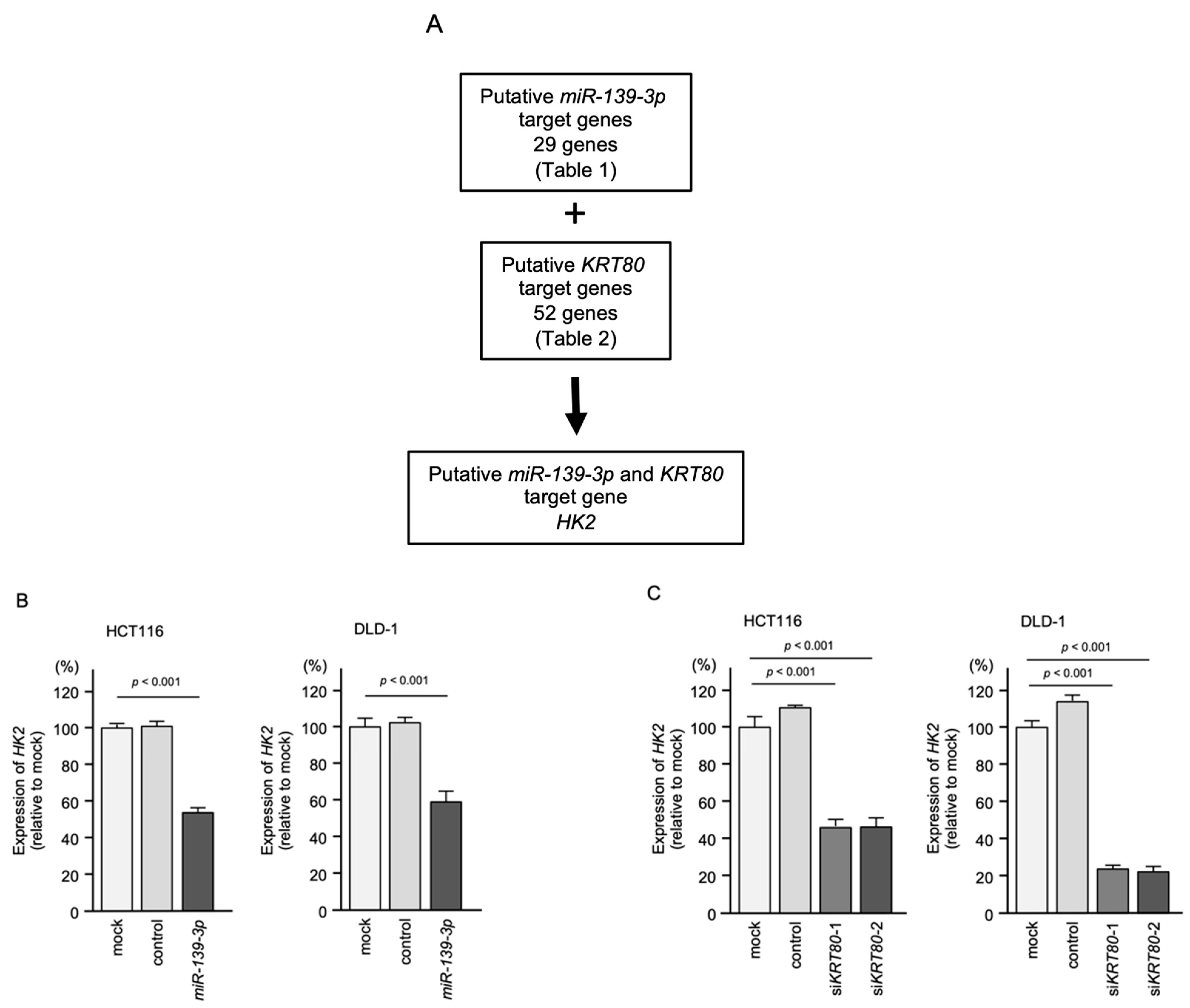
2.7. KRT80-Mediated RNA Networks in CRC Cells
2.8. Expression of Target Genes in Clinical Specimens and Correlation
3. Discussion
4. Materials and Methods
4.1. Clinical Specimens Used to Evaluate miR-139-5p and miR-139-3p Expression
4.2. CRC Cell Lines and Cell Culture
4.3. RNA Extraction and Quantitative Real-Time Reverse-Transcription PCR (qRT-PCR)
4.4. Regulation of miR-139-3p Expression by DNA Demethylation
4.5. Regulation of miR-139-3p Expression by Histone Deacetylation
4.6. Transfection of miRNAs and siRNAs into CRC Cells
4.7. Functional Analyses (Tumor Suppression and Promotion Assays) in CRC Cell Lines
4.8. Identification of Putative Targets Regulated by miR-139-3p in CRC Cells
4.9. RIP Assay
4.10. Dual-Luciferase Reporter Assay
4.11. Western Blot and Immunohistochemical Analyses
4.12. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Goding Sauer, A.; Fedewa, S.A.; Butterly, L.F.; Anderson, J.C.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 145–164. [Google Scholar] [CrossRef] [Green Version]
- Valderrama-Treviño, A.I.; Barrera-Mera, B.; Ceballos-Villalva, J.C.; Montalvo-Javé, E.E. Hepatic Metastasis from Colorectal Cancer. Euroasian J. Hepato-Gastroenterol. 2017, 7, 166–175. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grady, W.M.; Markowitz, S.D. The molecular pathogenesis of colorectal cancer and its potential application to colorectal cancer screening. Dig. Dis. Sci. 2015, 60, 762–772. [Google Scholar] [CrossRef] [Green Version]
- Leslie, A.; Carey, F.A.; Pratt, N.R.; Steele, R.J. The colorectal adenoma-carcinoma sequence. Br. J. Surg. 2002, 89, 845–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guinney, J.; Dienstmann, R.; Wang, X.; De Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef] [PubMed]
- Anfossi, S.; Babayan, A.; Pantel, K.; Calin, G.A. Clinical utility of circulating non-coding RNAs—An update. Nat. Rev. Clin. Oncol. 2018, 15, 541–563. [Google Scholar] [CrossRef]
- Esteller, M. Non-coding RNAs in human disease. Nat. Rev. Genet. 2011, 12, 861–874. [Google Scholar] [CrossRef]
- Panni, S.; Lovering, R.C.; Porras, P.; Orchard, S. Non-coding RNA regulatory networks. Biochim. Biophys. Acta Gene Regul. Mech. 2020, 1863, 194417. [Google Scholar] [CrossRef]
- Anastasiadou, E.; Jacob, L.S.; Slack, F.J. Non-coding RNA networks in cancer. Nat. Rev. Cancer 2018, 18, 5–18. [Google Scholar] [CrossRef]
- Ha, M.; Kim, V.N. Regulation of microRNA biogenesis. Nat. Rev. Mol. Cell Biol. 2014, 15, 509–524. [Google Scholar] [CrossRef]
- He, L.; Hannon, G.J. MicroRNAs: Small RNAs with a big role in gene regulation. Nat. Rev. Genet. 2004, 5, 522–531. [Google Scholar] [CrossRef]
- Ding, L.; Lan, Z.; Xiong, X.; Ao, H.; Feng, Y.; Gu, H.; Yu, M.; Cui, Q. The Dual Role of MicroRNAs in Colorectal Cancer Progression. Int. J. Mol. Sci. 2018, 19, 2791. [Google Scholar] [CrossRef] [Green Version]
- Schetter, A.J.; Okayama, H.; Harris, C.C. The role of microRNAs in colorectal cancer. Cancer J. 2012, 18, 244–252. [Google Scholar] [CrossRef] [Green Version]
- Xie, N.; Meng, Q.; Zhang, Y.; Luo, Z.; Xue, F.; Liu, S.; Li, Y.; Huang, Y. MicroRNA-142-3p suppresses cell proliferation, invasion and epithelial-to-mesenchymal transition via RAC1-ERK1/2 signaling in colorectal cancer. Mol. Med. Rep. 2021, 24, 568. [Google Scholar] [CrossRef]
- Hozaka, Y.; Kita, Y.; Yasudome, R.; Tanaka, T.; Wada, M.; Idichi, T.; Tanabe, K.; Asai, S.; Moriya, S.; Toda, H.; et al. RNA-Sequencing Based microRNA Expression Signature of Colorectal Cancer: The Impact of Oncogenic Targets Regulated by miR-490-3p. Int. J. Mol. Sci. 2021, 22, 9876. [Google Scholar] [CrossRef]
- Shimomura, H.; Okada, R.; Tanaka, T.; Hozaka, Y.; Wada, M.; Moriya, S.; Idichi, T.; Kita, Y.; Kurahara, H.; Ohtsuka, T.; et al. Role of miR-30a-3p Regulation of Oncogenic Targets in Pancreatic Ductal Adenocarcinoma Pathogenesis. Int. J. Mol. Sci. 2020, 21, 6459. [Google Scholar] [CrossRef]
- Shimonosono, M.; Idichi, T.; Seki, N.; Yamada, Y.; Arai, T.; Arigami, T.; Sasaki, K.; Omoto, I.; Uchikado, Y.; Kita, Y.; et al. Molecular pathogenesis of esophageal squamous cell carcinoma: Identification of the antitumor effects of miR1453p on gene regulation. Int. J. Oncol. 2019, 54, 673–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shinden, Y.; Hirashima, T.; Nohata, N.; Toda, H.; Okada, R.; Asai, S.; Tanaka, T.; Hozaka, Y.; Ohtsuka, T.; Kijima, Y.; et al. Molecular pathogenesis of breast cancer: Impact of miR-99a-5p and miR-99a-3p regulation on oncogenic genes. J. Hum. Genet. 2021, 66, 519–534. [Google Scholar] [CrossRef]
- Wada, M.; Goto, Y.; Tanaka, T.; Okada, R.; Moriya, S.; Idichi, T.; Noda, M.; Sasaki, K.; Kita, Y.; Kurahara, H.; et al. RNA sequencing-based microRNA expression signature in esophageal squamous cell carcinoma: Oncogenic targets by antitumor miR-143-5p and miR-143-3p regulation. J. Hum. Genet. 2020, 65, 1019–1034. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Liu, X.; Liu, Y.; Liu, X.; Wang, R.; Liao, J.; Wu, S.; Fan, J.; Peng, Z.; Li, B.; et al. Keratin 80 promotes migration and invasion of colorectal carcinoma by interacting with PRKDC via activating the AKT pathway. Cell Death Dis. 2018, 9, 1009. [Google Scholar] [CrossRef] [Green Version]
- Mossman, D.; Kim, K.T.; Scott, R.J. Demethylation by 5-aza-2′-deoxycytidine in colorectal cancer cells targets genomic DNA whilst promoter CpG island methylation persists. BMC Cancer 2010, 10, 366. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Gao, H.; Ren, L.; Gu, J.; Zhang, Y.; Zhang, Y. Demethylation of the miR-146a promoter by 5-Aza-2′-deoxycytidine correlates with delayed progression of castration-resistant prostate cancer. BMC Cancer 2014, 14, 308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, K.; Amano, Y.; Ishikawa, R.; Sunohara, M.; Kage, H.; Ichinose, J.; Sano, A.; Nakajima, J.; Fukayama, M.; Yatomi, Y.; et al. Histone methylation-mediated silencing of miR-139 enhances invasion of non-small-cell lung cancer. Cancer Med. 2015, 4, 1573–1582. [Google Scholar] [CrossRef] [Green Version]
- Desjobert, C.; Carrier, A.; Delmas, A.; Marzese, D.M.; Daunay, A.; Busato, F.; Pillon, A.; Tost, J.; Riond, J.; Favre, G.; et al. Demethylation by low-dose 5-aza-2′-deoxycytidine impairs 3D melanoma invasion partially through miR-199a-3p expression revealing the role of this miR in melanoma. Clin. Epigenet. 2019, 11, 9. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Okada, R.; Hozaka, Y.; Wada, M.; Moriya, S.; Satake, S.; Idichi, T.; Kurahara, H.; Ohtsuka, T.; Seki, N. Molecular Pathogenesis of Pancreatic Ductal Adenocarcinoma: Impact of miR-30c-5p and miR-30c-2-3p Regulation on Oncogenic Genes. Cancers 2020, 12, 2731. [Google Scholar] [CrossRef]
- Toda, H.; Seki, N.; Kurozumi, S.; Shinden, Y.; Yamada, Y.; Nohata, N.; Moriya, S.; Idichi, T.; Maemura, K.; Fujii, T.; et al. RNA-sequence-based microRNA expression signature in breast cancer: Tumor-Suppressive miR-101-5p regulates molecular pathogenesis. Mol. Oncol. 2020, 14, 426–446. [Google Scholar] [CrossRef] [Green Version]
- Koma, A.; Asai, S.; Minemura, C.; Oshima, S.; Kinoshita, T.; Kikkawa, N.; Koshizuka, K.; Moriya, S.; Kasamatsu, A.; Hanazawa, T.; et al. Impact of Oncogenic Targets by Tumor-Suppressive miR-139-5p and miR-139-3p Regulation in Head and Neck Squamous Cell Carcinoma. Int. J. Mol. Sci. 2021, 22, 9947. [Google Scholar] [CrossRef]
- Okada, R.; Goto, Y.; Yamada, Y.; Kato, M.; Asai, S.; Moriya, S.; Ichikawa, T.; Seki, N. Regulation of Oncogenic Targets by the Tumor-Suppressive miR-139 Duplex (miR-139-5p and miR-139-3p) in Renal Cell Carcinoma. Biomedicines 2020, 8, 599. [Google Scholar] [CrossRef]
- Yonemori, M.; Seki, N.; Yoshino, H.; Matsushita, R.; Miyamoto, K.; Nakagawa, M.; Enokida, H. Dual tumor-suppressors miR-139-5p and miR-139-3p targeting matrix metalloprotease 11 in bladder cancer. Cancer Sci. 2016, 107, 1233–1242. [Google Scholar] [CrossRef] [Green Version]
- Kim, A.; Lee, J.E.; Lee, S.S.; Kim, C.; Lee, S.J.; Jang, W.S.; Park, S. Coexistent mutations of KRAS and PIK3CA affect the efficacy of NVP-BEZ235, a dual PI3K/MTOR inhibitor, in regulating the PI3K/MTOR pathway in colorectal cancer. Int. J. Cancer 2013, 133, 984–996. [Google Scholar] [CrossRef]
- Lee, J.; Hong, H.K.; Peng, S.B.; Kim, T.W.; Lee, W.Y.; Yun, S.H.; Kim, H.C.; Liu, J.; Ebert, P.J.; Aggarwal, A.; et al. Identifying metastasis-initiating miRNA-target regulations of colorectal cancer from expressional changes in primary tumors. Sci. Rep. 2020, 10, 14919. [Google Scholar] [CrossRef]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018, 173, 321–337.e310. [Google Scholar] [CrossRef] [Green Version]
- Stefani, C.; Miricescu, D.; Stanescu, S., II; Nica, R.I.; Greabu, M.; Totan, A.R.; Jinga, M. Growth Factors, PI3K/AKT/mTOR and MAPK Signaling Pathways in Colorectal Cancer Pathogenesis: Where Are We Now? Int. J. Mol. Sci. 2021, 22, 10260. [Google Scholar] [CrossRef]
- Canavese, M.; Ngo, D.T.; Maddern, G.J.; Hardingham, J.E.; Price, T.J.; Hauben, E. Biology and therapeutic implications of VEGF-A splice isoforms and single-nucleotide polymorphisms in colorectal cancer. Int. J. Cancer 2017, 140, 2183–2191. [Google Scholar] [CrossRef] [Green Version]
- Napolitano, S.; Matrone, N.; Muddassir, A.L.; Martini, G.; Sorokin, A.; De Falco, V.; Giunta, E.F.; Ciardiello, D.; Martinelli, E.; Belli, V.; et al. Triple blockade of EGFR, MEK and PD-L1 has antitumor activity in colorectal cancer models with constitutive activation of MAPK signaling and PD-L1 overexpression. J. Exp. Clin. Cancer Res. 2019, 38, 492. [Google Scholar] [CrossRef] [Green Version]
- Therkildsen, C.; Bergmann, T.K.; Henrichsen-Schnack, T.; Ladelund, S.; Nilbert, M. The predictive value of KRAS, NRAS, BRAF, PIK3CA and PTEN for anti-EGFR treatment in metastatic colorectal cancer: A systematic review and meta-analysis. Acta Oncol. 2014, 53, 852–864. [Google Scholar] [CrossRef] [Green Version]
- Moafian, Z.; Maghrouni, A.; Soltani, A.; Hashemy, S.I. Cross-talk between non-coding RNAs and PI3K/AKT/mTOR pathway in colorectal cancer. Mol. Biol. Rep. 2021, 48, 4797–4811. [Google Scholar] [CrossRef]
- Song, M.; Bode, A.M.; Dong, Z.; Lee, M.H. AKT as a Therapeutic Target for Cancer. Cancer Res. 2019, 79, 1019–1031. [Google Scholar] [CrossRef] [Green Version]
- Fagerberg, L.; Hallstrom, B.M.; Oksvold, P.; Kampf, C.; Djureinovic, D.; Odeberg, J.; Habuka, M.; Tahmasebpoor, S.; Danielsson, A.; Edlund, K.; et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell Proteom. 2014, 13, 397–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, O.; Wang, C.; Wang, S.; Hu, Y.; Gou, R.; Dong, H.; Li, S.; Li, X.; Lin, B. Keratin 80 regulated by miR-206/ETS1 promotes tumor progression via the MEK/ERK pathway in ovarian cancer. J. Cancer 2021, 12, 6835–6850. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Xu, Y.; Xu, T.; Fan, R.; Jiang, T.; Cao, M.; Shi, L.; Song, J. CircPIP5K1A activates KRT80 and PI3K/AKT pathway to promote gastric cancer development through sponging miR-671-5p. Biomed. Pharmacother. 2020, 126, 109941. [Google Scholar] [CrossRef] [PubMed]
- John, S.; Weiss, J.N.; Ribalet, B. Subcellular localization of hexokinases I and II directs the metabolic fate of glucose. PLoS ONE 2011, 6, e17674. [Google Scholar] [CrossRef] [Green Version]
- Mathupala, S.P.; Ko, Y.H.; Pedersen, P.L. Hexokinase II: Cancer’s double-edged sword acting as both facilitator and gatekeeper of malignancy when bound to mitochondria. Oncogene 2006, 25, 4777–4786. [Google Scholar] [CrossRef] [Green Version]
- Roberts, D.J.; Tan-Sah, V.P.; Smith, J.M.; Miyamoto, S. Akt phosphorylates HK-II at Thr-473 and increases mitochondrial HK-II association to protect cardiomyocytes. J. Biol. Chem. 2013, 288, 23798–23806. [Google Scholar] [CrossRef] [Green Version]
- Bhaskar, P.T.; Nogueira, V.; Patra, K.C.; Jeon, S.M.; Park, Y.; Robey, R.B.; Hay, N. mTORC1 hyperactivity inhibits serum deprivation-induced apoptosis via increased hexokinase II and GLUT1 expression, sustained Mcl-1 expression, and glycogen synthase kinase 3beta inhibition. Mol. Cell Biol. 2009, 29, 5136–5147. [Google Scholar] [CrossRef] [Green Version]
- Franke, T.F.; Yang, S.I.; Chan, T.O.; Datta, K.; Kazlauskas, A.; Morrison, D.K.; Kaplan, D.R.; Tsichlis, P.N. The protein kinase encoded by the Akt proto-oncogene is a target of the PDGF-activated phosphatidylinositol 3-kinase. Cell 1995, 81, 727–736. [Google Scholar] [CrossRef] [Green Version]
- Robey, R.B.; Hay, N. Mitochondrial hexokinases, novel mediators of the antiapoptotic effects of growth factors and Akt. Oncogene 2006, 25, 4683–4696. [Google Scholar] [CrossRef] [Green Version]
- Pusapati, R.V.; Daemen, A.; Wilson, C.; Sandoval, W.; Gao, M.; Haley, B.; Baudy, A.R.; Hatzivassiliou, G.; Evangelista, M.; Settleman, J. mTORC1-Dependent Metabolic Reprogramming Underlies Escape from Glycolysis Addiction in Cancer Cells. Cancer Cell 2016, 29, 548–562. [Google Scholar] [CrossRef] [Green Version]
- Idichi, T.; Seki, N.; Kurahara, H.; Fukuhisa, H.; Toda, H.; Shimonosono, M.; Yamada, Y.; Arai, T.; Kita, Y.; Kijima, Y.; et al. Involvement of anti-tumor miR-124-3p and its targets in the pathogenesis of pancreatic ductal adenocarcinoma: Direct regulation of ITGA3 and ITGB1 by miR-124-3p. Oncotarget 2018, 9, 28849–28865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yonemori, K.; Seki, N.; Idichi, T.; Kurahara, H.; Osako, Y.; Koshizuka, K.; Arai, T.; Okato, A.; Kita, Y.; Arigami, T.; et al. The microRNA expression signature of pancreatic ductal adenocarcinoma by RNA sequencing: Anti-Tumour functions of the microRNA-216 cluster. Oncotarget 2017, 8, 70097–70115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwal, V.; Bell, G.W.; Nam, J.W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. eLife 2015, 4, e05005. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Kang, B.; Li, C.; Chen, T.; Zhang, Z. GEPIA2: An enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 2019, 47, W556–W560. [Google Scholar] [CrossRef] [Green Version]
- Minemura, C.; Asai, S.; Koma, A.; Kikkawa, N.; Kato, M.; Kasamatsu, A.; Uzawa, K.; Hanazawa, T.; Seki, N. Identification of Antitumor miR-30e-5p Controlled Genes; Diagnostic and Prognostic Biomarkers for Head and Neck Squamous Cell Carcinoma. Genes 2022, 13, 1225. [Google Scholar] [CrossRef]
- Sayers, E.W.; Beck, J.; Bolton, E.E.; Bourexis, D.; Brister, J.R.; Canese, K.; Comeau, D.C.; Funk, K.; Kim, S.; Klimke, W.; et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2021, 49, D10–D17. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entrez Gene ID | Gene Symbol | Gene Name | No. of Binding Sites | miR-139-3p-Transfected HCT116 Cells log2 FC < −2 |
|---|---|---|---|---|
| 9768 | KIAA0101 | KIAA0101 | 2 | −3.773 |
| 6541 | SLC7A1 | Solute carrier family 7 (cationic amino acid transporter, y+ system), member 1 | 4 | −3.072 |
| 23094 | SIPA1L3 | Signal-induced proliferation-associated 1 like 3 | 2 | −2.961 |
| 79628 | SH3TC2 | SH3 domain and tetratricopeptide repeats 2 | 1 | −2.921 |
| 201232 | SLC16A13 | Solute carrier family 16, member 13 | 1 | −2.905 |
| 27286 | SRPX2 | Sushi-repeat containing protein, X-linked 2 | 1 | −2.901 |
| 118932 | ANKRD22 | Ankyrin repeat domain 22 | 1 | −2.901 |
| 3099 | HK2 | Hexokinase 2 | 1 | −2.729 |
| 57116 | ZNF695 | Zinc finger protein 695 | 2 | −2.525 |
| 3352 | HTR1D | 5-hydroxytryptamine (serotonin) receptor 1D, G protein-coupled | 1 | −2.510 |
| 140893 | RBBP8NL | RBBP8 N-terminal like | 1 | −2.509 |
| 4171 | MCM2 | Minichromosome maintenance complex component 2 | 1 | −2.475 |
| 201266 | SLC39A11 | Solute carrier family 39, member 11 | 1 | −2.474 |
| 90861 | HN1L | Hematological and neurological expressed 1-like | 1 | −2.441 |
| 57402 | S100A14 | S100 calcium binding protein A14 | 1 | −2.394 |
| 8884 | SLC5A6 | Solute carrier family 5 (sodium/multivitamin and iodide cotransporter), member 6 | 1 | −2.371 |
| 55612 | FERMT1 | Fermitin family member 1 | 1 | −2.359 |
| 9721 | GPRIN2 | G protein regulated inducer of neurite outgrowth 2 | 1 | −2.358 |
| 54552 | GNL3L | Guanine nucleotide binding protein-like 3 (nucleolar)-like | 1 | −2.252 |
| 5653 | KLK6 | Kallikrein-related peptidase 6 | 1 | −2.251 |
| 157285 | SGK223 | Tyrosine-protein kinase sgk223 | 1 | −2.224 |
| 144501 | KRT80 | Keratin 80 | 1 | −2.193 |
| 154796 | AMOT | Angiomotin | 1 | −2.126 |
| 9052 | GPRC5A | G protein-coupled receptor, family C, group 5, member A | 1 | −2.114 |
| 54815 | GATAD2A | GATA zinc finger domain containing 2A | 1 | −2.113 |
| 3898 | LAD1 | Ladinin 1 | 1 | −2.046 |
| 414 | ARSD | Arylsulfatase D | 1 | −2.023 |
| 90381 | TICRR | TOPBP1-interacting checkpoint and replication regulator | 1 | −2.022 |
| 10189 | ALYREF | Aly/REF export factor | 1 | −2.006 |
| Entrez Gene ID | Gene Symbol | Gene Name | siKRT80-1-Transfected HCT116 Cells log2 FC < −1 | siKRT80-2-Transfected HCT116 Cells log2 FC < −1 |
|---|---|---|---|---|
| 4155 | MBP | Myelin basic protein | −4.259 | −3.823 |
| 5027 | P2RX7 | Purinergic receptor P2X 7 | −4.078 | −1.151 |
| 7274 | TTPA | Alpha tocopherol transfer protein | −2.983 | −1.257 |
| 51339 | DACT1 | Disheveled binding antagonist of beta catenin 1 | −2.745 | −1.248 |
| 5163 | PDK1 | Pyruvate dehydrogenase kinase 1 | −2.686 | −2.183 |
| 114088 | TRIM9 | Tripartite motif containing 9 | −2.641 | −1.383 |
| 54434 | SSH1 | Slingshot protein phosphatase 1 | −2.465 | −1.668 |
| 148418 | SAMD13 | Sterile alpha motif domain containing 13 | −2.444 | −1.292 |
| 284716 | RIMKLA | Ribosomal modification protein rimk like family member A | −2.323 | −1.662 |
| 144501 | KRT80 | Keratin 80 | −2.321 | −1.878 |
| 285735 | LINC00326 | Long intergenic non-protein coding RNA 326 | −2.257 | −1.868 |
| 9194 | SLC16A7 | Solute carrier family 16 member 7 | −2.221 | −1.022 |
| 112399 | EGLN3 | egl-9 family hypoxia inducible factor 3 | −2.209 | −1.350 |
| 256435 | ST6GALNAC3 | ST6 N-acetylgalactosaminide alpha-2,6-sialyltransferase 3 | −2.185 | −1.875 |
| 4907 | NT5E | 5′-nucleotidase ecto | −2.087 | −1.564 |
| 100287314 | LINC00941 | Long intergenic non-protein coding RNA 941 | −2.077 | −1.532 |
| 254128 | NIFK-AS1 | NIFK antisense RNA 1 | −2.049 | −1.204 |
| 3099 | HK2 | Hexokinase 2 | −1.916 | −3.518 |
| 1956 | EGFR | Epidermal growth factor receptor | −1.872 | −1.096 |
| 7378 | UPP1 | Uridine phosphorylase 1 | −1.854 | −1.023 |
| 51384 | WNT16 | Wnt family member 16 | −1.835 | −1.759 |
| 115330 | GPR146 | G protein-coupled receptor 146 | −1.827 | −1.710 |
| 170384 | FUT11 | fucosyltransferase 11 | −1.818 | −1.129 |
| 2113 | ETS1 | ETS proto-oncogene 1, transcription factor | −1.754 | −1.284 |
| 861 | RUNX1 | RUNX family transcription factor 1 | −1.720 | −1.209 |
| 10397 | NDRG1 | N-myc downstream regulated 1 | −1.666 | −1.856 |
| 22989 | MYH15 | Myosin heavy chain 15 | −1.634 | −1.744 |
| N.A. | lnc-OR10H1-1 | lnc-OR10H1-1:1 | −1.576 | −1.277 |
| 644316 | FLJ43315 | asparagine synthetase pseudogene | −1.568 | −1.405 |
| 4781 | NFIB | nuclear factor i b | −1.565 | −1.333 |
| 25886 | POC1A | POC1 centriolar protein A | −1.549 | −1.338 |
| N.A. | lnc-CLEC2D-7 | lnc-CLEC2D-7:1 | −1.447 | −1.571 |
| 51175 | TUBE1 | Tubulin epsilon 1 | −1.383 | −1.488 |
| 10549 | PRDX4 | Peroxiredoxin 4 | −1.373 | −1.158 |
| 843 | CASP10 | Caspase 10 | −1.347 | −1.054 |
| 3613 | IMPA2 | Inositol monophosphatase 2 | −1.320 | −1.712 |
| 100505933 | ADD3-AS1 | ADD3 antisense RNA 1 | −1.296 | −1.095 |
| 4783 | NFIL3 | Nuclear factor, interleukin 3 regulated | −1.267 | −1.184 |
| 57834 | CYP4F11 | Cytochrome P450 family 4 subfamily F member 11 | −1.265 | −1.522 |
| 11199 | ANXA10 | Annexin A10 | −1.262 | −2.649 |
| 64946 | CENPH | Centromere protein H | −1.251 | −1.236 |
| 8614 | STC2 | Stanniocalcin 2 | −1.243 | −1.373 |
| 286144 | TRIQK | Triple qxxk/R motif containing | −1.194 | −1.351 |
| 2035 | EPB41 | Erythrocyte membrane protein band 4.1 | −1.167 | −1.156 |
| 28996 | HIPK2 | Homeodomain interacting protein kinase 2 | −1.138 | −1.166 |
| 4233 | MET | MET proto-oncogene, receptor tyrosine kinase | −1.130 | −1.247 |
| 100506211 | MIR210HG | MIR210 host gene | −1.109 | −1.795 |
| 23015 | GOLGA8A | Golgin A8 family member A | −1.068 | −1.087 |
| 23516 | SLC39A14 | Solute carrier family 39 member 14 | −1.063 | −1.441 |
| 84986 | ARHGAP19 | Rho gtpase activating protein 19 | −1.062 | −1.035 |
| 255082 | CASC2 | Cancer susceptibility 2 | −1.045 | −1.546 |
| 100507065 | LOC100507065 | Uncharacterized LOC100507065 | −1.032 | −1.425 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yasudome, R.; Seki, N.; Asai, S.; Goto, Y.; Kita, Y.; Hozaka, Y.; Wada, M.; Tanabe, K.; Idichi, T.; Mori, S.; et al. Molecular Pathogenesis of Colorectal Cancer: Impact of Oncogenic Targets Regulated by Tumor Suppressive miR-139-3p. Int. J. Mol. Sci. 2022, 23, 11616. https://doi.org/10.3390/ijms231911616
Yasudome R, Seki N, Asai S, Goto Y, Kita Y, Hozaka Y, Wada M, Tanabe K, Idichi T, Mori S, et al. Molecular Pathogenesis of Colorectal Cancer: Impact of Oncogenic Targets Regulated by Tumor Suppressive miR-139-3p. International Journal of Molecular Sciences. 2022; 23(19):11616. https://doi.org/10.3390/ijms231911616
Chicago/Turabian StyleYasudome, Ryutaro, Naohiko Seki, Shunichi Asai, Yusuke Goto, Yoshiaki Kita, Yuto Hozaka, Masumi Wada, Kan Tanabe, Tetsuya Idichi, Shinichiro Mori, and et al. 2022. "Molecular Pathogenesis of Colorectal Cancer: Impact of Oncogenic Targets Regulated by Tumor Suppressive miR-139-3p" International Journal of Molecular Sciences 23, no. 19: 11616. https://doi.org/10.3390/ijms231911616