

Conuping BSA-Seq and RNA-Seq Reveal the Molecular Pathway and Genes Associated with the Plant Height of Foxtail Millet (Setaria italica)
 , ,
, ,
Abstract
:1. Introduction
2. Results
2.1. Statistics and Analysis of Plant Height
2.2. Bulked Segregant Analysis Sequencing
2.2.1. Evaluation of BSA-Seq Quality
2.2.2. Mapping Analysis, Detection, and Annotation of SNP
2.2.3. Location of Candidate Regions and Screening of Genes
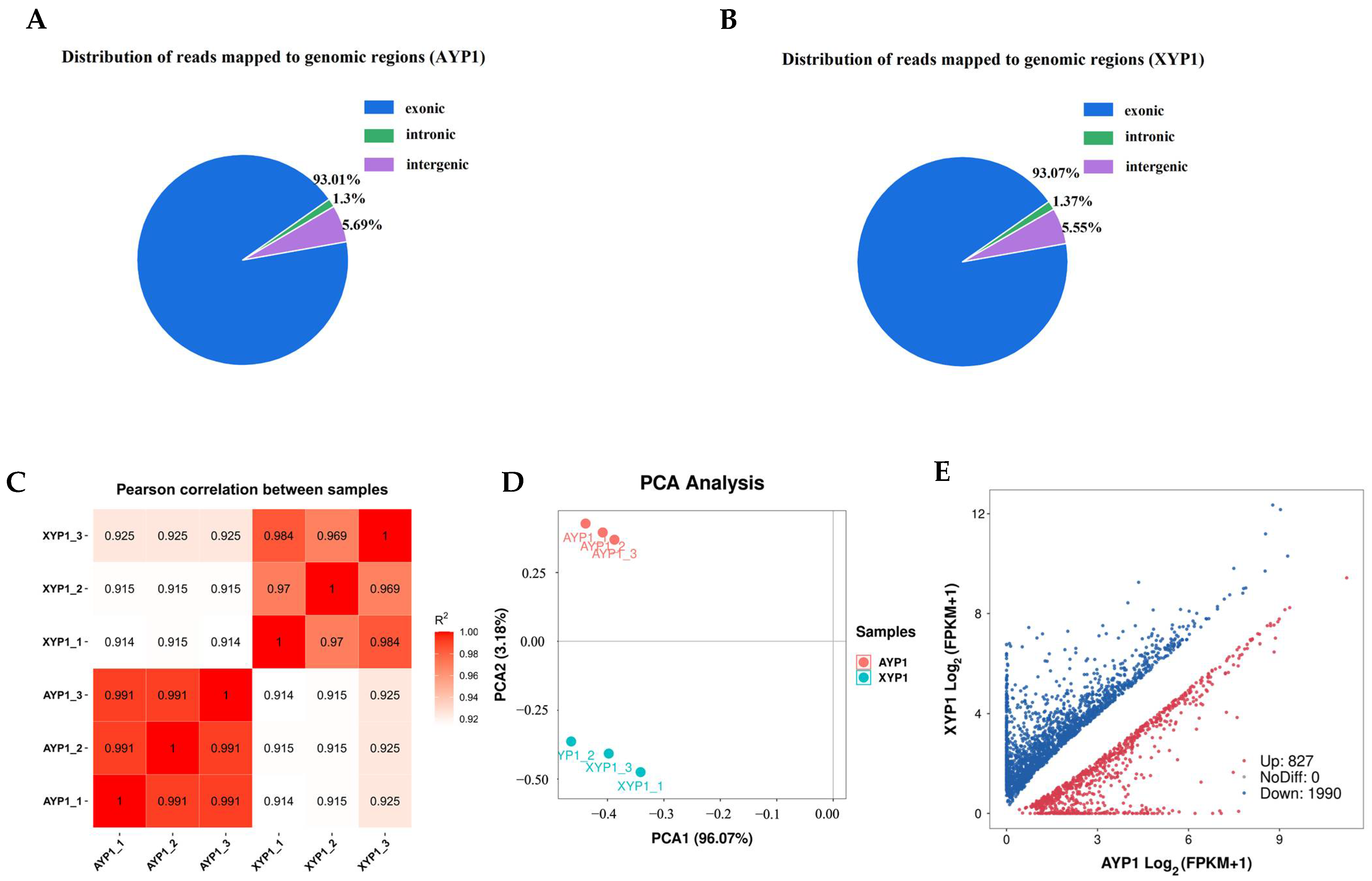
2.3. Results of RNA-Seq
2.3.1. Quality Control of Transcript Sequencing Data
2.3.2. Quality Assessment of RNA-Seq
2.3.3. Identification of Differentially Expressed Genes Related to Plant Height
2.3.4. GO Classification and Enrichment Analysis of DEGs
2.3.5. KEGG Enrichment Analysis of DEGs
2.4. Combined Analysis of BSA-Seq and RNA-Seq
2.4.1. Identify the Key Genes Related to Plant Height
2.4.2. Functional Analysis of Candidate Genes
2.5. Verification of RNA-Seq and Candidate Genes by qRT-PCR
3. Discussion
3.1. Excavation of Key Genes in Plant Height Contributes to the Promotion of Foxtail Millet
3.2. New Genes Controlling Plant Height on Chromosomes I and IX
3.3. Metabolism and the MAPK Signaling Pathway Play Important Roles in the Plant Height Formation of Foxtail Millet
3.4. Nitrate Transporter Gene and Hormone Transport Gene Were Key Genes Controlling Plant Height Formation in Foxtail Millet
4. Materials and Methods
4.1. Technical Route
4.2. Plant Materials
4.3. Investigation and Statistics of Plant Height
4.4. BSA-Seq
4.4.1. DNA Extraction, Library Construction, and Sequencing
4.4.2. Comparison with Reference Genomes, Detection, and Annotation of Single-Nucleotide Polymorphisms
4.4.3. Identification of Candidate Regions and Genes
4.5. RNA-Seq
4.5.1. RNA Extraction, Library Construction, and Sequencing
4.5.2. Sequencing Data Quality Control and Reads Mapping
4.5.3. Gene Expression Level Analysis
4.5.4. Functional Analysis of DEGs
4.6. Combined Analysis of BSA-Seq and RNA-Seq
4.7. Validation of Real-Time PCR
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cappelli, G.; Yamac, S.S.; Stella, T.; Francone, C.; Paleari, L.; Negri, M.; Confalonieri, R. Are advantages from the partial replacement of corn with second-generation energy crops undermined by climate change? A case study for giant reed in northern Italy. Biomass Bioenergy 2015, 80, 85–93. [Google Scholar] [CrossRef]
- Gupta, A.; Rico-Medina, A.; Cao-Delgado, A.I. The physiology of plant responses to drought. Science 2020, 368, 266–269. [Google Scholar] [CrossRef] [PubMed]
- Sambo, B.E. Endangered, Neglected, Indigenous Resilient Crops: A Potential against Climate Change Impact for Sustainable Crop Productivity and Food Security. IOSR J. Agric. Vet. Sci. 2014, 7, 34–41. [Google Scholar] [CrossRef]
- Doust, A.N.; Kellogg, E.A.; Devos, K.M.; Bennetzen, J.L. Foxtail millet: A sequence-driven grass model system. Plant Physiol. 2009, 149, 137–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lata, C.; Gupta, S.; Prasad, M. Foxtail millet: A model crop for genetic and genomic studies in bioenergy grasses. Crit. Rev. Biotechnol. 2013, 33, 328–343. [Google Scholar] [CrossRef]
- Gupta, S.M.; Arora, S.; Mirza, N.; Pande, A.; Lata, C.; Puranik, S.; Kumar, J.; Kumar, A. Finger millet: A “certain” crop for an “uncertain” future and a solution to food insecurity and hidden hunger under stressful environments. Front. Plant Sci. 2017, 8, 643. [Google Scholar] [CrossRef] [Green Version]
- Tian, B.; Wang, J.; Zhang, L.; Li, Y.; Wang, S.; Li, H. Assessment of resistance to lodging of landrace and improved cultivars in foxtail millet. Euphytica 2009, 172, 295–302. [Google Scholar] [CrossRef]
- Gooding, M.J.; Addisu, M.; Uppal, R.K.; Snape, J.W.; Jones, H.E. Effect of wheat dwarfing genes on nitrogen-use efficiency. J. Agric. Sci. 2011, 150, 3–22. [Google Scholar] [CrossRef] [Green Version]
- Lisa, M.; Noriyuki, K.; Rika, Y.; Junko, S.; Haruka, M.; Yumiko, M.; Masao, T.; Mizuho, S.; Shinobu, N.; Yuzo, M. Positional Cloning of Rice Semidwarfing Gene, sd-1: Rice "Green Revolution Gene" Encodes a Mutant Enzyme Involved in Gibberellin Synthesis. Dna Res. 2002, 9, 11–17. [Google Scholar]
- Jia, X.P.; Dong, P.H.; Zhang, H.X.; Quan, J.Z.; Dong, Z.P. Correlation Study of Lodging Resistance and Plant Height, Panicle Traits in Foxtail Millet. J. Plant Genet. Resour. 2015, 16, 6. [Google Scholar]
- Wang, J.; Wang, Z.; Du, X.; Yang, H.; Han, F.; Han, Y.; Yuan, F.; Zhang, L.; Peng, S.; Guo, E. A high-density genetic map and QTL analysis of agronomic traits in foxtail millet [Setaria italica (L.) P. Beauv.] using RAD-seq. PLoS ONE 2017, 12, e0179717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, K.; Fan, G.; Zhang, X.; Zhao, F.; Wei, W.; Du, G.; Feng, X.; Wang, X.; Wang, F.; Song, G.; et al. Identification of QTLs for 14 Agronomically Important Traits in Setaria italica Based on SNPs Generated from High-Throughput Sequencing. G3 2017, 7, 1587–1594. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wang, J.; Peng, J.; Du, X.; Jiang, M.; Li, Y.; Han, F.; Du, G.; Yang, H.; Lian, S.; et al. QTL mapping for 11 agronomic traits based on a genome-wide Bin-map in a large F2 population of foxtail millet (Setaria italica (L.) P. Beauv). Mol. Breed. 2019, 39, 18. [Google Scholar] [CrossRef]
- Zhao, M.; Zhi, H.; Zhang, X.; Jia, G.; Diao, X. Retrotransposon-mediated DELLA transcriptional reprograming underlies semi-dominant dwarfism in foxtail millet. Crop J. 2019, 7, 458–468. [Google Scholar] [CrossRef]
- He, Q.; Zhi, H.; Tang, S.; Xing, L.; Wang, S.; Wang, H.; Zhang, A.; Li, Y.; Gao, M.; Zhang, H.; et al. QTL mapping for foxtail millet plant height in multi-environment using an ultra-high density bin map. Appl. Genet. 2021, 134, 557–572. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Wang, Z.; Han, K.; Lian, S.; Li, Y.; Zhang, L.; Guo, E.; Wang, J. Fine mapping of qPH9, a major quantitative trait locus, responsible for plant height in foxtail millet [Setaria italica (L.) P. Beauv.]. Mol. Breed. 2021, 41, 77. [Google Scholar] [CrossRef]
- Michelmore, R.W.; Paran, I.; Kesseli, R. Identification of markers linked to disease-resistance genes by bulked segregant analysis: A rapid method to detect markers in specific genomic regions by using segregating populations. Proc. Natl. Acad. Sci. USA 1991, 88, 9828–9832. [Google Scholar] [CrossRef] [Green Version]
- Wang, M. A Gain-of-Function Mutant of IAA7 Inhibits Stem Elongation by Transcriptional Repression of EXPA5 Genes in Brassica napus. Int. J. Mol. Sci. 2021, 22, 9018. [Google Scholar]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Li, Z.; Xu, Y. Bulk segregation analysis in the NGS era: A review of its teenage years. Plant J. 2022, 109, 1355–1374. [Google Scholar] [CrossRef]
- Guo, Z.; Cai, L.; Chen, Z.; Wang, R.; Zhang, L.; Guan, S.; Zhang, S.; Ma, W.; Liu, C.; Pan, G. Identification of candidate genes controlling chilling tolerance of rice in the cold region at the booting stage by BSA-Seq and RNA-Seq. R. Soc. Open Sci. 2020, 7, 201081. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Hou, Z.; Gao, L.; Xiao, D.; Hou, X.; Zhang, C.; Yan, J.; Song, L. Conjunctive Analyses of BSA-Seq and BSR-Seq to Reveal the Molecular Pathway of Leafy Head Formation in Chinese Cabbage. Plants 2019, 8, 603. [Google Scholar] [CrossRef] [Green Version]
- Hussin, S.H.; Wang, H.; Tang, S.; Zhi, H.; Tang, C.; Zhang, W.; Jia, G.; Diao, X. SiMADS34, an E-class MADS-box transcription factor, regulates inflorescence architecture and grain yield in Setaria italica. Plant Mol. Biol. 2021, 105, 419–434. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Kumari, K.; Muthamilarasan, M.; Parida, S.K.; Prasad, M. Population structure and association mapping of yield contributing agronomic traits in foxtail millet. Plant Cell Rep. 2014, 33, 881–893. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Gao, S.; Yang, M.; Zhang, M. Researches on Breeding of New Types Good Quality Millet. J. Northeast Agric. Sci. 2008, 33, 2. [Google Scholar]
- Jia, X.; Zhang, B.; Quan, J.; Li, J.; Wang, Y.; Yuan, Y. Genome-wide Association Analysis of Plant Heightin Foxtail Millet under Different Photoperiod Conditions. Acta Agric. Boreali Sin. 2019, 34, 8. [Google Scholar]
- Wang, F.; Yoshida, H.; Matsuoka, M. Making the ‘Green Revolution’ Truly Green: Improving Crop Nitrogen Use Efficiency. Plant Cell Physiol. 2021, 62, 942–947. [Google Scholar] [CrossRef]
- Jiang, H.; Guo, L.B.; Qian, Q. Recent Progress on Rice Genetics in China. J. Integr. Plant Biol. 2007, 49, 15. [Google Scholar] [CrossRef]
- Zhong, M.; Wei, S.; Peng, Z.; Yang, Z. A Review of the Research and Application of Rht Dwarf Genes in Wheat. Mol. Plant Breed. 2018, 16, 8. [Google Scholar]
- Li, J.; Yang, G.; Meng, Z.; Fan, Y.; Wei, L. Innovation of Plant Type Structure Source, High Quality Source and Resistance Source in Millet Breeding. Crops 2008, 2, 108–110. [Google Scholar]
- Dong, X.; Li, Z.; Ma, J.; Li, X.; Sun, G.; Zheng, Y. Research Status and Control Strategies of Maize Stem Rot. Heilongjiang Agric. Sci. 2022, 3, 76–80. [Google Scholar]
- Bennetzen, J.L.; Schmutz, J.; Wang, H.; Percifield, R.; Hawkins, J.; Pontaroli, A.C.; Estep, M.; Feng, L.; Vaughn, J.N.; Grimwood, J. Reference genome sequence of the model plant Setaria. Nat. Biotechnol. 2012, 30, 555–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Diao, X.; Wang, J.; Wang, C.; Wang, G.; Hao, X.; Liang, Z.; Wang, X.; Zhao, F. Construction of Genetic Map and QTL Analysis of Some Main Agronomic Traits in Millet. J. Plant Genet. Resour. 2013, 14, 871–878. [Google Scholar]
- Zhao, M. Map-based Cloning and Duplication Mechanism Analysis of Foxtail Millet Semidominant Dwarf Gene SiDw1; Chinese Academy of Agricultural Sciences: Beijing, China, 2013. [Google Scholar]
- Zhao, B.; Wang, B.; Li, Z.; Guo, T.; Zhao, J.; Guan, Z.; Liu, K. Identification and characterization of a new dwarf locus DS-4 encoding an Aux/IAA7 protein in Brassica napus. Appl. Genet. 2019, 132, 1435–1449. [Google Scholar] [CrossRef]
- Song, Q.; Yu, J.; Liu, J.; Yin, Y.; Zhang, Q.; Wang, F.; Liu, L.; Wan, H. Research Progress of Dwarfing Genes in Plants. Guangdong Agric. Sci. 2021, 048, 19–28. [Google Scholar]
- Dong, N.Q.; Lin, H.X. Contribution of phenylpropanoid metabolism to plant development and plant–environment interactions. J. Integr. Plant Biol. 2021, 63, 180–209. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Pérez, F.; Pomar, F.; Pedreo, M.A.; Novo-Uzal, E. Suppression of Arabidopsis peroxidase 72 alters cell wall and phenylpropanoid metabolism. Plant Sci. 2015, 239, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Li, C.; Zhang, R.; Zhao, D.; Gao, Z.; Wang, F.; Yang, Y.; Wang, Z.; Wang, M. Transcriptome sequencing analysis of dwarfing traits in Brassica napus line DW871. Chin. J. Oil Crop Sci. 2022, 44, 11. [Google Scholar]
- Huang, P.; Yoshida, H.; Yano, K.; Kinoshita, S.; Kawai, K.; Koketsu, E.; Hattori, M.; Takehara, S.; Huang, J.; Hirano, K.; et al. OsIDD2, a zinc finger and INDETERMINATE DOMAIN protein, regulates secondary cell wall formation. J. Integr. Plant Biol. 2018, 60, 130–143. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Xiao, X.; Wang, X.; Zeng, J.; Kang, H.; Fan, X.; Sha, L.; Zhang, H.; Zhou, Y. RNA-Seq and iTRAQ Reveal the Dwarfing Mechanism of Dwarf Polish Wheat (Triticum polonicum L.). Int. J. Biol. Sci. 2016, 12, 653–666. [Google Scholar] [CrossRef] [Green Version]
- Lam, T.; Iiyama, K. Role of ferulic acid bridges between lignin and polysaccharides in the growth of wheat (Triticum aestivum) internodes. J. Jpn. Wood Res. Soc. 1996, 42, 157–162. [Google Scholar]
- Qiang, Z.; Sun, H.; Ge, F.; Li, W.; Li, C.; Wang, S.; Zhang, B.; Zhu, L.; Zhang, S.; Wang, X.; et al. The transcription factor ZmMYB69 represses lignin biosynthesis by activating ZmMYB31/42 expression in maize. Plant Physiol. 2022, 189, 1916–1919. [Google Scholar] [CrossRef] [PubMed]
- Widmann, C.; Gibson, S.; Jarpe, M.B.; Johnson, G.L. Mitogen-activated protein kinase: Conservation of a three-kinase module from yeast to human. Physiol. Rev. 1999, 79, 143–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ichimura, K.; Shinozaki, K.; Tena, G.; Sheen, J.; Henry, Y.; Champion, A.; Kreis, M.; Zhang, S.; Hirt, H.; Wilson, C. Mitogen-activated protein kinase cascades in plants: A new nomenclature. Trends Plant Sci. 2002, 7, 301–308. [Google Scholar]
- Zhang, S. Mitogen-Activated Protein Kinase Cascades in Plant Signaling. In Annual Plant Reviews Online; Wiley: Hoboken, NJ, USA, 2018; pp. 100–136. [Google Scholar]
- Zhang, M.; Su, J.; Zhang, Y.; Xu, J.; Zhang, S. Conveying endogenous and exogenous signals: MAPK cascades in plant growth and defense. Curr. Opin. Plant Biol. 2018, 45, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Komis, G.; Samajova, O.; Ovecka, M.; Samaj, J. Cell and Developmental Biology of Plant Mitogen-Activated Protein Kinases. Annu. Rev. Plant Biol. 2018, 69, 237–265. [Google Scholar] [CrossRef]
- Shpak, E.D. Diverse roles of ERECTA family genes in plant development. J. Integr. Plant Biol. 2013, 55, 1238–1250. [Google Scholar] [CrossRef]
- Zuch, D.T.; Doyle, S.M.; Majda, M.; Smith, R.S.; Robert, S.; Torii, K.U. Cell biology of the leaf epidermis: Fate specification, morphogenesis, and coordination. Plant Cell 2022, 34, 209–227. [Google Scholar] [CrossRef]
- Takagi, M.; Hamano, K.; Takagi, H.; Morimoto, T.; Akimitsu, K.; Terauchi, R.; Shirasu, K.; Ichimura, K. Disruption of the MAMP-Induced MEKK1-MKK1/MKK2-MPK4 Pathway Activates the TNL Immune Receptor SMN1/RPS6. Plant Cell Physiol. 2019, 60, 778–787. [Google Scholar] [CrossRef]
- Jia, W.; Li, B.; Li, S.; Liang, Y.; Wu, X.; Ma, M.; Wang, J.; Gao, J.; Cai, Y.; Zhang, Y.; et al. Mitogen-Activated Protein Kinase Cascade MKK7-MPK6 Plays Important Roles in Plant Development and Regulates Shoot Branching by Phosphorylating PIN1 in Arabidopsis. PLoS Biol. 2016, 14, e1002550. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Wu, H.; Su, J.; Wang, H.; Zhu, Q.; Liu, Y.; Xu, J.; Lukowitz, W.; Zhang, S. Maternal control of embryogenesis by MPK6 and its upstream MKK4/MKK5 in Arabidopsis. Plant J. 2017, 92, 1005–1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smekalova, V.; Luptovciak, I.; Komis, G.; Samajova, O.; Ovecka, M.; Doskocilova, A.; Takac, T.; Vadovic, P.; Novak, O.; Pechan, T.; et al. Involvement of YODA and mitogen activated protein kinase 6 in Arabidopsis post-embryogenic root development through auxin up-regulation and cell division plane orientation. New Phytol. 2014, 203, 1175–1193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, C.; Box, M.S.; Thiruppathi, D.; Hu, H.; Yu, Y.; Martin, C.; Doust, A.N.; McSteen, P.; Kellogg, E.A. Pleiotropic and nonredundant effects of an auxin importer in Setaria and maize. Plant Physiol. 2022, 189, 715–734. [Google Scholar] [CrossRef] [PubMed]
- Wen, B.Q.; Xing, M.Q.; Zhang, H.; Dai, C.; Xue, H.W. Rice homeobox transcription factor HOX1a positively regulates gibberellin responses by directly suppressing EL1. J. Integr. Plant Biol. 2011, 53, 869–878. [Google Scholar] [CrossRef]
- Ikeda, T.; Tanaka, W.; Toriba, T.; Suzuki, C.; Maeno, A.; Tsuda, K.; Shiroishi, T.; Kurata, T.; Sakamoto, T.; Murai, M.; et al. BELL1-like homeobox genes regulate inflorescence architecture and meristem maintenance in rice. Plant J. 2019, 98, 465–478. [Google Scholar] [CrossRef]
- Bencivenga, S.; Simonini, S.; Benkova, E.; Colombo, L. The transcription factors BEL1 and SPL are required for cytokinin and auxin signaling during ovule development in Arabidopsis. Plant Cell 2012, 24, 2886–2897. [Google Scholar] [CrossRef] [Green Version]
- Fang, J.; Chai, C.; Qian, Q.; Li, C.; Tang, J.; Sun, L.; Huang, Z.; Guo, X.; Sun, C.; Liu, M.; et al. Mutations of genes in synthesis of the carotenoid precursors of ABA lead to pre-harvest sprouting and photo-oxidation in rice. Plant J. 2008, 54, 177–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamiru, M.; Abe, A.; Utsushi, H.; Yoshida, K.; Takagi, H.; Fujisaki, K.; Undan, J.R.; Rakshit, S.; Takaichi, S.; Jikumaru, Y.; et al. The tillering phenotype of the rice plastid terminal oxidase (PTOX) loss-of-function mutant is associated with strigolactone deficiency. New Phytol. 2014, 202, 116–131. [Google Scholar] [CrossRef]
- Zhou, H.; Yang, M.; Zhao, L.; Zhu, Z.; Liu, F.; Sun, H.; Sun, C.; Tan, L. HIGH-TILLERING AND DWARF 12 modulates photosynthesis and plant architecture by affecting carotenoid biosynthesis in rice. J. Exp. Bot. 2021, 72, 1212–1224. [Google Scholar] [CrossRef]
- Cazzonelli, C.I.; Pogson, B.J. Source to sink: Regulation of carotenoid biosynthesis in plants. Trends Plant Sci. 2010, 15, 266–274. [Google Scholar] [CrossRef]
- Qin, G.; Gu, H.; Ma, L.; Peng, Y.; Deng, X.W.; Chen, Z.; Qu, L.J. Disruption of phytoene desaturase gene results in albino and dwarf phenotypes in Arabidopsis by impairing chlorophyll, carotenoid, and gibberellin biosynthesis. Cell Res. 2007, 17, 471–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tu, X.; Wang, Q.; Xie, C.; Li, Y.; Li, R.; Liu, A.; University, H.A. Regulatory Mechanism of the Photosynthetic Pathway in Dwarfed Upland Cotton (Gossypium hirsutum L.). Cotton Sci. 2019, 3, 201–209. [Google Scholar]
- Gibson, S.I. Plant Sugar-Response Pathways. Part of a Complex Regulatory Web 1. Plant Physiol. 2000, 124, 1532–1539. [Google Scholar] [CrossRef] [Green Version]
- Bel, A. Strategies of Phloem Loading. Annu. Rev. Plant Biol. 2003, 44, 253–281. [Google Scholar]
- Rennie, E.A.; Turgeon, R. A comprehensive picture of phloem loading strategies. Proc. Natl. Acad. Sci. USA 2009, 106, 14162–14167. [Google Scholar] [CrossRef] [PubMed]
- Riesmeier, J.W.; Willmitzer, L.; Frommer, W.B. Evidence for an essential role of the sucrose transporter in phloem loading and assimilate partitioning. Embo. J. 1994, 13, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Gottwald, J.R.; Krysan, P.J. Genetic evidence for the in planta role of phloem-specific plasma membrane sucrose transporters. Proc. Natl. Acad. Sci. USA 2000, 97, 13979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Zhang, Y.; Yang, C.; Tian, Z.; Li, J. AtSWEET4, a hexose facilitator, mediates sugar transport to axial sinks and affects plant development. Sci. Rep. 2016, 6, 24563. [Google Scholar] [CrossRef] [Green Version]
- Yao, L.; Ding, C.; Hao, X.; Zeng, J.; Yang, Y.; Wang, X.; Wang, L. CsSWEET1a and CsSWEET17 Mediate Growth and Freezing Tolerance by Promoting Sugar Transport across the Plasma Membrane. Plant Cell Physiol. 2020, 61, 1669–1682. [Google Scholar] [CrossRef]
- Chen, G.; Zhang, Y.; Ruan, B.; Guo, L.; Zeng, D.; Gao, Z.; Zhu, L.; Hu, J.; Ren, D.; Yu, L.; et al. OsHAK1 controls the vegetative growth and panicle fertility of rice by its effect on potassium-mediated sugar metabolism. Plant Sci. 2018, 274, 261–270. [Google Scholar] [CrossRef]
- Li, S.; Tian, Y.; Wu, K.; Ye, Y.; Yu, J.; Zhang, J.; Liu, Q.; Hu, M.; Li, H.; Tong, Y.; et al. Modulating plant growth-metabolism coordination for sustainable agriculture. Nature 2018, 560, 595–600. [Google Scholar] [CrossRef] [PubMed]
- Parker, J.L.; Newstead, S. Molecular basis of nitrate uptake by the plant nitrate transporter NRT1.1. Nature 2014, 507, 68–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hachiya, T.; Mizokami, Y.; Miyata, K.; Tholen, D.; Watanabe, C.K.; Noguchi, K. Evidence for a nitrate-independent function of the nitrate sensor NRT1.1 in Arabidopsis thaliana. J. Plant Res. 2011, 124, 425–430. [Google Scholar] [CrossRef]
- Guo, T.; Xuan, H.; Yang, Y.; Wang, L.; Wei, L.; Wang, Y.; Kang, G. Transcription Analysis of Genes Encoding the Wheat Root Transporter NRT1 and NRT2 Families During Nitrogen Starvation. J. Plant Growth Regul. 2014, 33, 837–848. [Google Scholar] [CrossRef]
- Wang, Y.; Shen, Y.; Qian, J.; Liu, W.; Zhao, G.; Sun, S. Effects of Nitrogen Forms on Plant Growth, Expression of Nitrate Transporter Gene MtNRT1.3,and Nitrogen Absorption in Medicago sativa L. Acta Agrestia Sin. 2019, 27, 9. [Google Scholar]
- Corratge-Faillie, C.; Lacombe, B. Substrate (un)specificity of Arabidopsis NRT1/PTR FAMILY (NPF) proteins. J. Exp. Bot. 2017, 68, 3107–3113. [Google Scholar] [CrossRef] [PubMed]
- Bagchi, R.; Salehin, M.; Adeyemo, O.S.; Salazar, C.; Shulaev, V.; Sherrier, D.J.; Dickstein, R. Functional assessment of the Medicago truncatula NIP/LATD protein demonstrates that it is a high-affinity nitrate transporter. Plant Physiol. 2012, 160, 906–916. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Nong, B.; Chen, C.; Wang, J.; Xia, X.; Zhang, Z.; Wei, Y.; Zeng, Y.; Feng, R.; Wu, Y.; et al. OsNPF3.1, a member of the NRT1/PTR family, increases nitrogen use efficiency and biomass production in rice. Crop J. 2022. [Google Scholar] [CrossRef]
- Chen, X.; Wang, R.; Guo-Hua, D. Methods and Progress on Plant DNA Methylation. North. Hortic. 2013, 12, 188–191. [Google Scholar]
- Sung, D.Y.; Kim, T.H.; Komives, E.A.; Mendoza-Cozatl, D.G.; Schroeder, J.I. ARS5 is a component of the 26S proteasome complex, and negatively regulates thiol biosynthesis and arsenic tolerance in Arabidopsis. Plant J. 2009, 59, 802–813. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Du, L.; Ma, Q.; Yuan, Y.; Liu, J.; Song, H.; Feng, B. Conjunctive Analyses of Bulk Segregant Analysis Sequencing and Bulk Segregant RNA Sequencing to Identify Candidate Genes Controlling Spikelet Sterility of Foxtail Millet. Front. Plant Sci. 2022, 13, 842336. [Google Scholar] [CrossRef] [PubMed]
- Doyle, J.; Doyle, J.L. Isolation of Plant DNA from fresh tissue. Focus 1990, 12, 13–15. [Google Scholar]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Mapped Reads | Total Reads | Mapping Rate (%) | Average Depth (×) | Coverage 1 × (%) | Coverage 4 × (%) |
|---|---|---|---|---|---|---|
| XAP1 | 36,115,445 | 36,886,738 | 97.91 | 10.57 | 95.49 | 90.82 |
| AYP1 | 32,975,648 | 33,284,924 | 99.07 | 9.94 | 96.5 | 91.38 |
| F2H | 191,560,590 | 194,204,816 | 98.64 | 48.92 | 98.93 | 98.13 |
| F2L | 192,966,265 | 195,334,940 | 98.79 | 51.7 | 98.92 | 98.14 |
| Sample Name | Total Reads | Total Mapped Rate (%) | Multiple Mapped Rate (%) | Uniquely Mapped | Reads Map to ‘+’ (%) | Reads Map to ‘−’ (%) | Non-Splice Reads | Splice Reads |
|---|---|---|---|---|---|---|---|---|
| AYP1_1 | 51,123,404 | 94.75 | 3.18 | 91.57 | 45.78 | 45.78 | 55.73 | 35.84 |
| AYP1_2 | 47,571,968 | 94.72 | 3.24 | 91.48 | 45.74 | 45.74 | 55.74 | 35.74 |
| AYP1_3 | 45,527,652 | 94.89 | 3.24 | 91.66 | 45.83 | 45.83 | 55.91 | 35.74 |
| XYP1_1 | 44,074,408 | 95.47 | 2.73 | 92.74 | 46.37 | 46.37 | 57.11 | 35.63 |
| XYP1_2 | 55,221,554 | 94.92 | 3.09 | 91.83 | 45.91 | 45.91 | 55.89 | 35.93 |
| XYP1_3 | 50,557,676 | 95.10 | 3.07 | 92.03 | 46.02 | 46.02 | 56.02 | 36.01 |
| Gene Name | Chromosome Location | Description |
|---|---|---|
| SETIT034720mg | IX: 57,492,650–57,497,221 | Domain associated with HOX domains; homeobox protein BEL1 homolog |
| SETIT034843mg | IX: 51,772,544–51,7773,35 | protein NRT1/PTR FAMILY 8.3 |
| SETIT035219mg | IX: 58,779,433–58,782,708 | Belongs to the major facilitator superfamily. |
| Sugar transporter (TC 2. A. 1. 1) family | ||
| SETIT040190mg | IX: 51,799,242–51,801,341 | protein NRT1/PTR FAMILY 8.3 |
| SETIT033879mg | IX: 51,916,122–51,925,937 | C-5 cytosine-specific DNA methylase |
| SETIT034904mg | IX: 57,439,447–57,442,618 | Belongs to the sterol desaturase family; Fatty acid hydroxylase domain-containing protein |
| SETIT019635mg | I: 37,589,455–37,591,340 | E2F/DP family winged-helix DNA-binding domain |
| SETIT017539mg | I: 36,542,422–36,544,052 | Belongs to the peptidase A1 family |
| SETIT020559mg | I: 37,443,258–37,443,787 | hypothetical proteins |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, Y.; Yuan, Y.; Zhang, X.; Song, H.; Yang, Q.; Yang, P.; Gao, X.; Gao, J.; Feng, B. Conuping BSA-Seq and RNA-Seq Reveal the Molecular Pathway and Genes Associated with the Plant Height of Foxtail Millet (Setaria italica). Int. J. Mol. Sci. 2022, 23, 11824. https://doi.org/10.3390/ijms231911824
Gao Y, Yuan Y, Zhang X, Song H, Yang Q, Yang P, Gao X, Gao J, Feng B. Conuping BSA-Seq and RNA-Seq Reveal the Molecular Pathway and Genes Associated with the Plant Height of Foxtail Millet (Setaria italica). International Journal of Molecular Sciences. 2022; 23(19):11824. https://doi.org/10.3390/ijms231911824
Chicago/Turabian StyleGao, Yongbin, Yuhao Yuan, Xiongying Zhang, Hui Song, Qinghua Yang, Pu Yang, Xiaoli Gao, Jinfeng Gao, and Baili Feng. 2022. "Conuping BSA-Seq and RNA-Seq Reveal the Molecular Pathway and Genes Associated with the Plant Height of Foxtail Millet (Setaria italica)" International Journal of Molecular Sciences 23, no. 19: 11824. https://doi.org/10.3390/ijms231911824