
Desmin Knock-Out Cardiomyopathy: A Heart on the Verge of Metabolic Crisis


 , , , , , , and
, , , , , , and
Abstract
:1. Introduction
2. Results
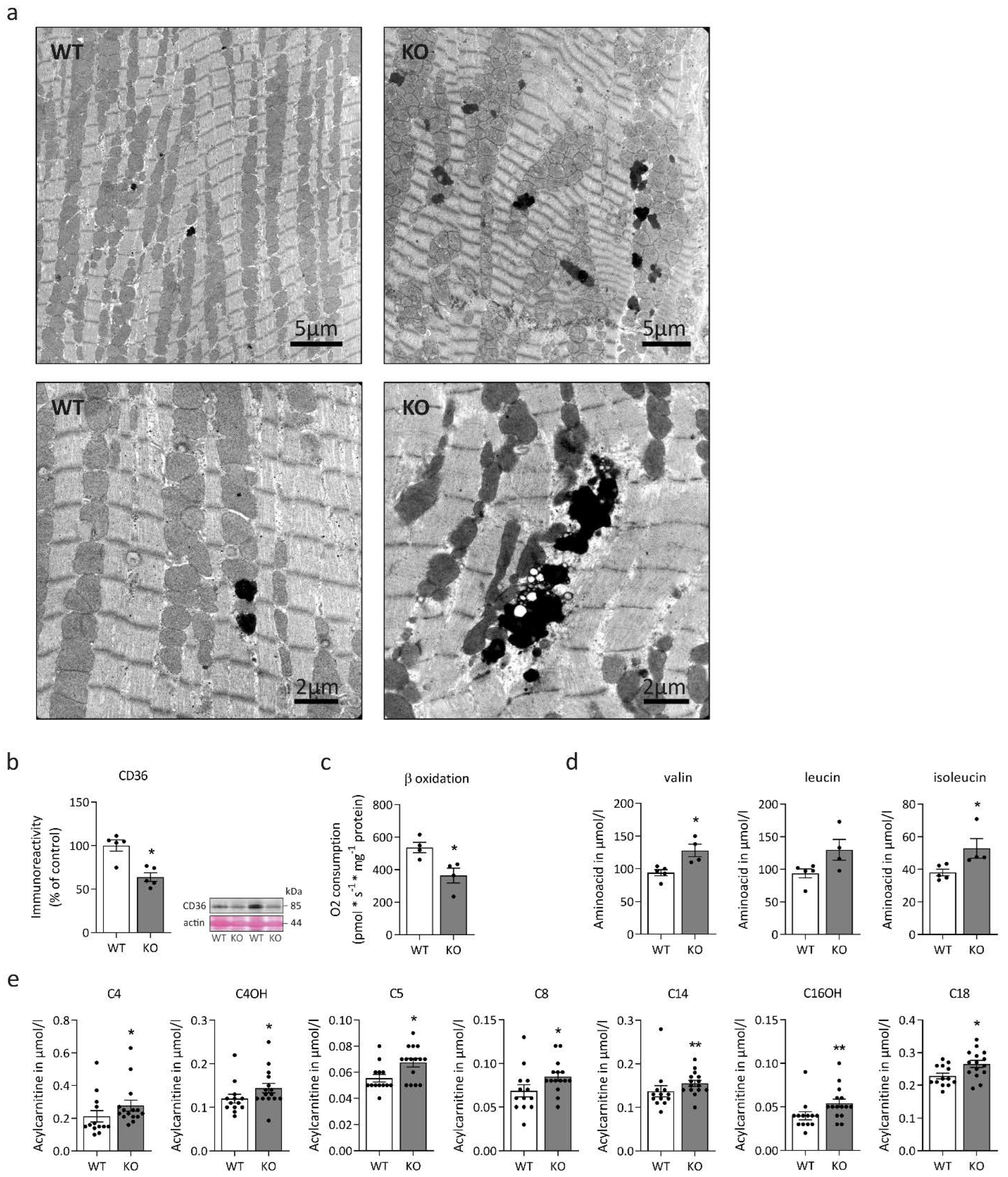
2.1. Desmin Deficiency Is Associated with Cardiac Mitochondrial Defects and Altered Fatty and Amino Acid Metabolism
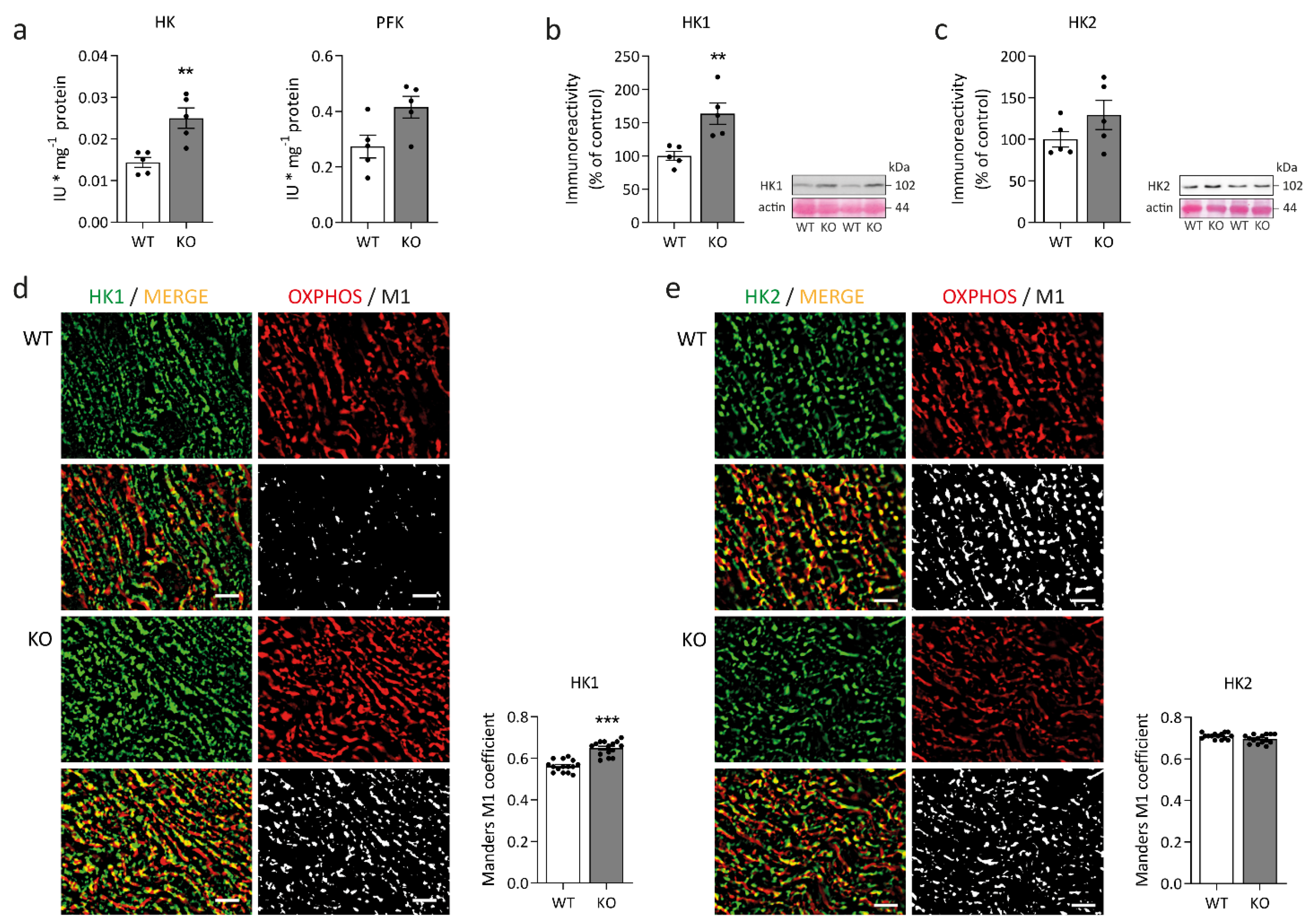
2.2. Concomitant Changes in Myocardial Glucose Metabolism
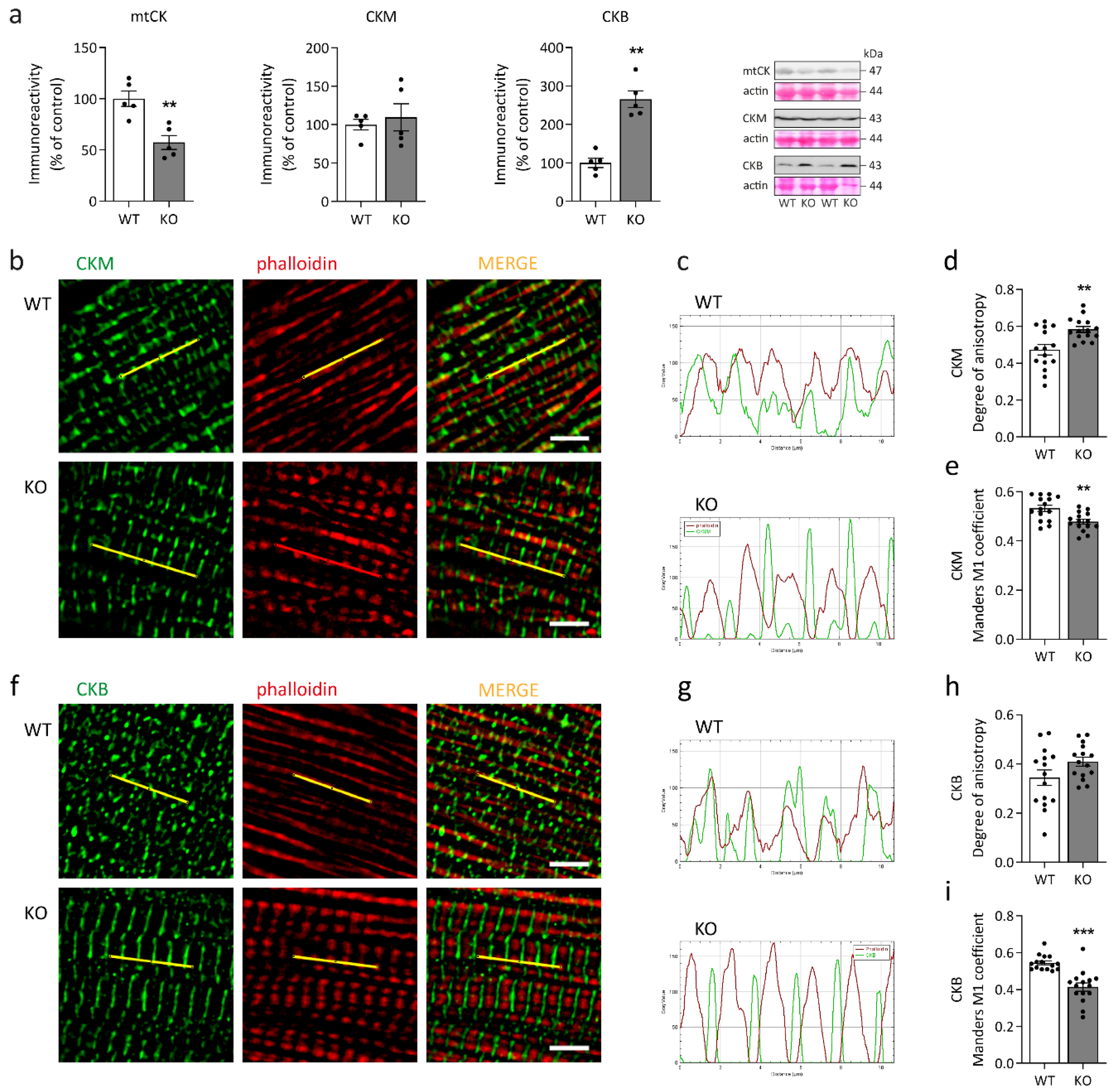
2.3. Imbalance in the Creatine Kinase System
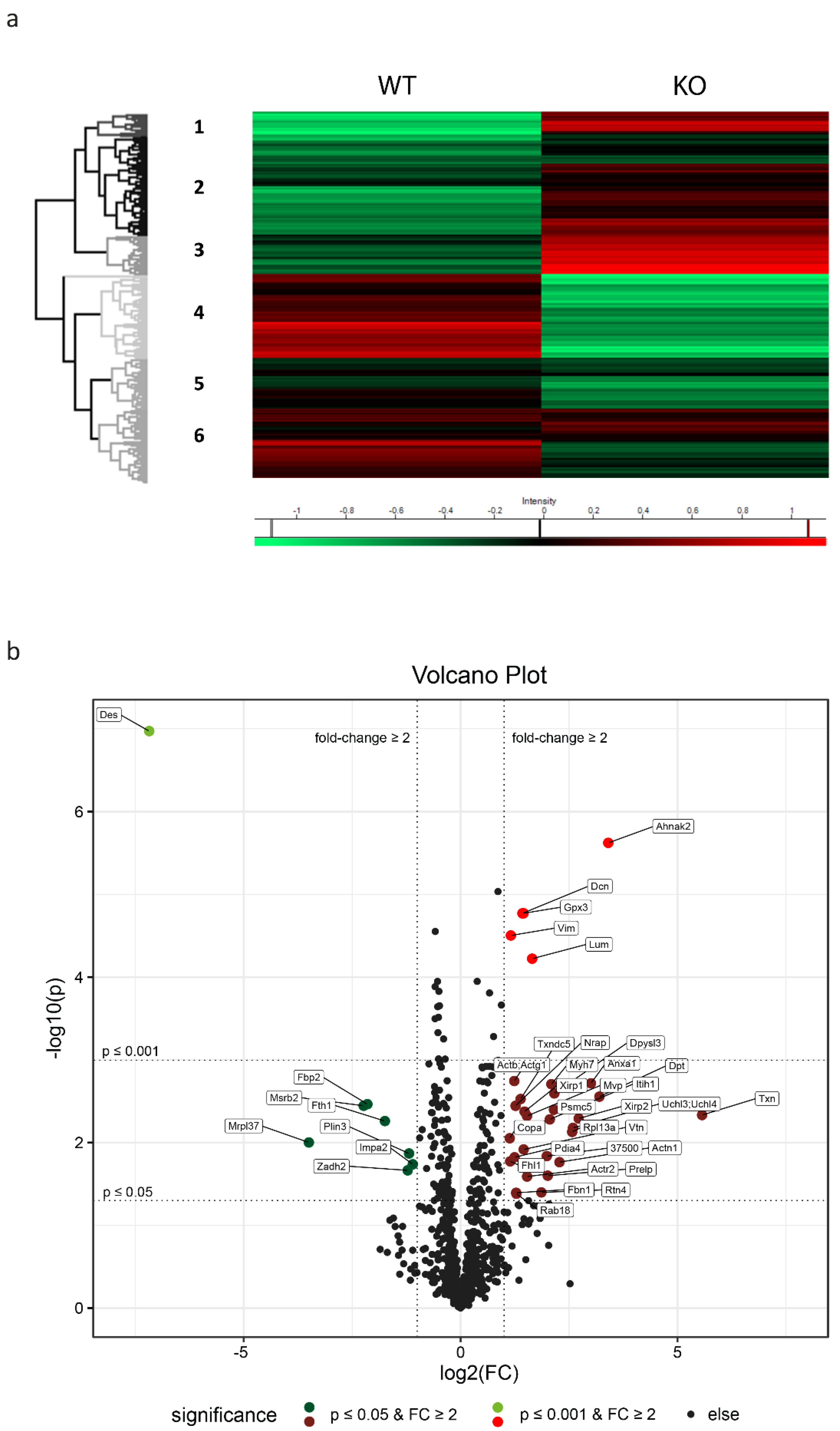
2.4. Proteome Analysis of Left Ventricular Cardiac Tissue Reveals Widespread Alterations Related to Subcellular Compartments and Metabolism
2.5. The Pattern of Significantly Regulated Proteins in Desmin Knock-Out Cardiac Tissue
2.6. A Closer Look at Metabolism-Related Proteins in Desmin Knock-Out Cardiac Tissue
3. Discussion
3.1. Desmin and Myocardial Mitochondria
3.2. Desmin and the Ultrastructure and Fragility of Mitochondria in Cardiac Muscle Tissue
3.3. From Aberrant Mitochondria to Changes in Fatty Acid Metabolism and Oxidative Phosphorylation
3.4. Metabolic Adaptations in Desmin Knock-Out Hearts
3.5. Desmin Deficiency and the Heart: A Combined Structural and Metabolic Disease
3.6. Translational Aspects Derived from the Analysis of Murine Desmin Knock-Out Hearts
4. Materials and Methods
4.1. Animals
4.2. Patients
4.3. Mass Spectrometric Analysis of Acylcarnitine and Amino Acid Levels in Blood
4.4. Cardiac Muscle Tissue Preparation
4.5. Enzyme Activity Measurements
4.6. High-Resolution Respirometry
4.7. SDS-PAGE and Immunoblotting
4.8. Immunofluorescence Staining, Imaging, and Image Analysis
4.9. Confocal Image Acquisition and Analysis
4.10. Ultrastructural Analysis
4.11. mtDNA Deletions Determination
4.12. mtDNA Copy Number Determination
4.13. Statistical Analysis
4.14. Proteomic Analysis: Sample Preparation, Mass Spectrometry, and Data Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Murphy, E.; Ardehali, H.; Balaban, R.S.; DiLisa, F.; Dorn, G.W., 2nd; Kitsis, R.N.; Otsu, K.; Ping, P.; Rizzuto, R.; Sack, M.N.; et al. Mitochondrial Function, Biology, and Role in Disease: A Scientific Statement from the American Heart Association. Circ. Res. 2016, 118, 1960–1991. [Google Scholar] [CrossRef] [PubMed]
- Karamanlidis, G.; Lee, C.F.; Garcia-Menendez, L.; Kolwicz, S.C., Jr.; Suthammarak, W.; Gong, G.; Sedensky, M.M.; Morgan, P.G.; Wang, W.; Tian, R. Mitochondrial complex I deficiency increases protein acetylation and accelerates heart failure. Cell Metab. 2013, 18, 239–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capetanaki, Y. Desmin cytoskeleton: A potential regulator of muscle mitochondrial behavior and function. Trends Cardiovasc. Med. 2002, 12, 339–348. [Google Scholar] [CrossRef]
- Fountoulakis, M.; Soumaka, E.; Rapti, K.; Mavroidis, M.; Tsangaris, G.; Maris, A.; Weisleder, N.; Capetanaki, Y. Alterations in the heart mitochondrial proteome in a desmin null heart failure model. J. Mol. Cell. Cardiol. 2005, 38, 461–474. [Google Scholar] [CrossRef]
- Lindén, M.; Li, Z.; Paulin, D.; Gotow, T.; Leterrier, J.F. Effects of desmin gene knockout on mice heart mitochondria. J. Bioenerg. Biomembr. 2001, 33, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Dalakas, M.C.; Park, K.Y.; Semino-Mora, C.; Lee, H.S.; Sivakumar, K.; Goldfarb, L.G. Desmin myopathy, a skeletal myopathy with cardiomyopathy caused by mutations in the desmin gene. N. Engl. J. Med. 2000, 342, 770–780. [Google Scholar] [CrossRef] [PubMed]
- Goldfarb, L.G.; Park, K.Y.; Cervenakova, L.; Gorokhova, S.; Lee, H.S.; Vasconcelos, O.; Nagle, J.W.; Semino-Mora, C.; Sivakumar, K.; Dalakas, M.C. Missense mutations in desmin associated with familial cardiac and skeletal myopathy. Nat. Genet. 1998, 19, 402–403. [Google Scholar] [CrossRef]
- Brodehl, A.; Gaertner-Rommel, A.; Milting, H. Molecular insights into cardiomyopathies associated with desmin (DES) mutations. Biophys. Rev. 2018, 10, 983–1006. [Google Scholar] [CrossRef]
- Clemen, C.S.; Herrmann, H.; Strelkov, S.V.; Schröder, R. Desminopathies: Pathology and mechanisms. Acta Neuropathol. 2013, 125, 47–75. [Google Scholar] [CrossRef] [Green Version]
- Van Spaendonck-Zwarts, K.; van Hessem, L.; Jongbloed, J.D.; de Walle, H.E.; Capetanaki, Y.; van der Kooi, A.J.; van Langen, I.M.; van den Berg, M.P.; van Tintelen, J.P. Desmin-related myopathy: A review and meta-analysis. Clin. Genet. 2010, 80, 354–366. [Google Scholar] [CrossRef]
- Clemen, C.S.; Stöckigt, F.; Strucksberg, K.H.; Chevessier, F.; Winter, L.; Schütz, J.; Bauer, R.; Thorweihe, J.M.; Wenzel, D.; Schlötzer-Schrehardt, U.; et al. The toxic effect of R350P mutant desmin in striated muscle of man and mouse. Acta Neuropathol. 2015, 129, 297–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrmann, H.; Cabet, E.; Chevalier, N.R.; Moosmann, J.; Schultheis, D.; Haas, J.; Schowalter, M.; Berwanger, C.; Weyerer, V.; Agaimy, A.; et al. Dual functional states of R406W-desmin assembly complexes cause cardiomyopathy with severe intercalated disc derangement in humans and in knock-in mice. Circulation 2020, 142, 2155–2171. [Google Scholar] [CrossRef] [PubMed]
- Carmignac, V.; Sharma, S.; Arbogast, S.; Fischer, D.; Serreri, C.; Serria, M.; Stoltenburg, G.; Maurage, C.A.; Herrmann, H.; Cuisset, J.M.; et al. A homozygous desmin deletion causes an Emery-Dreifuss like recessive myopathy with desmin depletion. Neuromuscul. Disord. 2009, 19, 600. [Google Scholar] [CrossRef]
- Durmus, H.; Ayhan, O.; Cirak, S.; Deymeer, F.; Parman, Y.; Franke, A.; Eiber, N.; Chevessier, F.; Schlötzer-Schrehardt, U.; Clemen, C.S.; et al. Neuromuscular endplate pathology in recessive desminopathies: Lessons from man and mice. Neurology 2016, 87, 799–805. [Google Scholar] [CrossRef] [PubMed]
- Henderson, M.; de Waele, L.; Hudson, J.; Eagle, M.; Sewry, C.; Marsh, J.; Charlton, R.; He, L.; Blakely, E.L.; Horrocks, I.; et al. Recessive desmin-null muscular dystrophy with central nuclei and mitochondrial abnormalities. Acta Neuropathol. 2013, 125, 917–919. [Google Scholar] [CrossRef]
- McLaughlin, H.M.; Kelly, M.A.; Hawley, P.P.; Darras, B.T.; Funke, B.; Picker, J. Compound heterozygosity of predicted loss-of-function DES variants in a family with recessive desminopathy. BMC Med. Genet. 2013, 14, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrmann, H.; Hesse, M.; Reichenzeller, M.; Aebi, U.; Magin, T.M. Functional complexity of intermediate filament cytoskeletons: From structure to assembly to gene ablation. Int. Rev. Cytol. 2003, 223, 83–175. [Google Scholar]
- Capetanaki, Y.; Bloch, R.J.; Kouloumenta, A.; Mavroidis, M.; Psarras, S. Muscle intermediate filaments and their links to membranes and membranous organelles. Exp. Cell Res. 2007, 313, 2063–2076. [Google Scholar] [CrossRef]
- Lazarides, E. Intermediate filaments as mechanical integrators of cellular space. Nature 1980, 283, 249–256. [Google Scholar] [CrossRef]
- Li, Z.; Colucci-Guyon, E.; Pincon-Raymond, M.; Mericskay, M.; Pournin, S.; Paulin, D.; Babinet, C. Cardiovascular lesions and skeletal myopathy in mice lacking desmin. Dev. Biol. 1996, 175, 362–366. [Google Scholar] [CrossRef] [Green Version]
- Milner, D.J.; Weitzer, G.; Tran, D.; Bradley, A.; Capetanaki, Y. Disruption of muscle architecture and myocardial degeneration in mice lacking desmin. J. Cell Biol. 1996, 134, 1255–1270. [Google Scholar] [CrossRef] [PubMed]
- Paulin, D.; Li, Z. Desmin: A major intermediate filament protein essential for the structural integrity and function of muscle. Exp. Cell Res. 2004, 301, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Diermeier, S.; Buttgereit, A.; Schürmann, S.; Winter, L.; Xu, H.; Murphy, R.M.; Clemen, C.S.; Schröder, R.; Friedrich, O. Preaged remodeling of myofibrillar cytoarchitecture in skeletal muscle expressing R349P mutant desmin. Neurobiol. Aging 2017, 58, 77–87. [Google Scholar] [CrossRef]
- Diermeier, S.; Iberl, J.; Vetter, K.; Haug, M.; Pollmann, C.; Reischl, B.; Buttgereit, A.; Schürmann, S.; Spörrer, M.; Goldmann, W.H.; et al. Early signs of architectural and biomechanical failure in isolated myofibers and immortalized myoblasts from desmin-mutant knock-in mice. Sci. Rep. 2017, 7, 1391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haug, M.; Reischl, B.; Prölß, G.; Pollmann, C.; Buckert, T.; Keidel, C.; Schürmann, S.; Hock, M.; Rupitsch, S.; Heckel, M.; et al. The MyoRobot: A novel automated biomechatronics system to assess voltage/Ca2+ biosensors and active/passive biomechanics in muscle and biomaterials. Biosens. Bioelectron. 2018, 102, 589–599. [Google Scholar] [CrossRef]
- Pollmann, C.; Haug, M.; Reischl, B.; Prölß, G.; Pöschel, T.; Rupitsch, S.J.; Clemen, C.S.; Schröder, R.; Friedrich, O. Growing Old Too Early: Skeletal Muscle Single Fiber Biomechanics in Ageing R349P Desmin Knock-in Mice Using the MyoRobot Technology. Int. J. Mol. Sci. 2020, 21, 5501. [Google Scholar] [CrossRef] [PubMed]
- Eiber, N.; Frob, F.; Schowalter, M.; Thiel, C.; Clemen, C.S.; Schröder, R.; Hashemolhosseini, S. Lack of Desmin in Mice Causes Structural and Functional Disorders of Neuromuscular Junctions. Front. Mol. Neurosci. 2020, 13, 567084. [Google Scholar] [CrossRef] [PubMed]
- Schröder, R.; Goudeau, B.; Simon, M.C.; Fischer, D.; Eggermann, T.; Clemen, C.S.; Li, Z.; Reimann, J.; Xue, Z.; Rudnik-Schöneborn, S.; et al. On noxious desmin: Functional effects of a novel heterozygous desmin insertion mutation on the extrasarcomeric desmin cytoskeleton and mitochondria. Hum. Mol. Genet. 2003, 12, 657–669, Erratum in Hum. Mol. Genet. 2007, 16, 2989–2990. [Google Scholar] [CrossRef]
- Winter, L.; Unger, A.; Berwanger, C.; Spörrer, M.; Türk, M.; Chevessier, F.; Strucksberg, K.H.; Schlötzer-Schrehardt, U.; Wittig, I.; Goldmann, W.H.; et al. Imbalances in protein homeostasis caused by mutant desmin. Neuropathol. Appl. Neurobiol. 2019, 45, 476–494. [Google Scholar] [CrossRef]
- Winter, L.; Wittig, I.; Peeva, V.; Eggers, B.; Heidler, J.; Chevessier, F.; Kley, R.A.; Barkovits, K.; Strecker, V.; Berwanger, C.; et al. Mutant desmin substantially perturbs mitochondrial morphology, function and maintenance in skeletal muscle tissue. Acta Neuropathol. 2016, 132, 453–473. [Google Scholar] [CrossRef] [Green Version]
- Milner, D.J.; Mavroidis, M.; Weisleder, N.; Capetanaki, Y. Desmin cytoskeleton linked to muscle mitochondrial distribution and respiratory function. J. Cell Biol. 2000, 150, 1283–1298. [Google Scholar] [CrossRef] [PubMed]
- Meyer, G.A.; Kiss, B.; Ward, S.R.; Morgan, D.L.; Kellermayer, M.S.; Lieber, R.L. Theoretical predictions of the effects of force transmission by desmin on intersarcomere dynamics. Biophys. J. 2010, 98, 258–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, S.B.; Davis, J.; Weisleder, N.; Kostavassili, I.; McCulloch, A.D.; Ralston, E.; Capetanaki, Y.; Lieber, R.L. Structural and functional roles of desmin in mouse skeletal muscle during passive deformation. Biophys. J. 2004, 86, 2993–3008. [Google Scholar] [CrossRef] [Green Version]
- Shah, S.B.; Su, F.C.; Jordan, K.; Milner, D.J.; Friden, J.; Capetanaki, Y.; Lieber, R.L. Evidence for increased myofibrillar mobility in desmin-null mouse skeletal muscle. J. Exp. Biol. 2002, 205 Pt 3, 321–325. [Google Scholar] [CrossRef] [PubMed]
- Agbulut, O.; Li, Z.; Perie, S.; Ludosky, M.A.; Paulin, D.; Cartaud, J.; Butler-Browne, G. Lack of desmin results in abortive muscle regeneration and modifications in synaptic structure. Cell. Motil. Cytoskelet. 2001, 49, 51–66. [Google Scholar] [CrossRef]
- Weisleder, N.; Taffet, G.E.; Capetanaki, Y. Bcl-2 overexpression corrects mitochondrial defects and ameliorates inherited desmin null cardiomyopathy. Proc. Natl. Acad. Sci. USA 2004, 101, 769–774. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Mericskay, M.; Agbulut, O.; Butler-Browne, G.; Carlsson, L.; Thornell, L.E.; Babinet, C.; Paulin, D. Desmin is essential for the tensile strength and integrity of myofibrils but not for myogenic commitment, differentiation, and fusion of skeletal muscle. J. Cell Biol. 1997, 139, 129–144. [Google Scholar] [CrossRef] [Green Version]
- Waskova-Arnostova, P.; Elsnicova, B.; Kasparova, D.; Hornikova, D.; Kolar, F.; Novotny, J.; Zurmanova, J. Cardioprotective adaptation of rats to intermittent hypobaric hypoxia is accompanied by the increased association of hexokinase with mitochondria. J. Appl. Physiol. 2015, 119, 1487–1493. [Google Scholar] [CrossRef] [Green Version]
- Waskova-Arnostova, P.; Kasparova, D.; Elsnicova, B.; Novotny, J.; Neckar, J.; Kolar, F.; Zurmanova, J. Chronic hypoxia enhances expression and activity of mitochondrial creatine kinase and hexokinase in the rat ventricular myocardium. Cell Physiol. Biochem. 2014, 33, 310–320. [Google Scholar] [CrossRef]
- Hornemann, T.; Stolz, M.; Wallimann, T. Isoenzyme-specific interaction of muscle-type creatine kinase with the sarcomeric M-line is mediated by NH2-terminal lysine charge-clamps. J. Cell Biol. 2000, 149, 1225–1234. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef] [PubMed]
- Leber, Y.; Ruparelia, A.A.; Kirfel, G.; van der Ven, P.F.; Hoffmann, B.; Merkel, R.; Bryson-Richardson, R.J.; Fürst, D.O. Filamin C is a highly dynamic protein associated with fast repair of myofibrillar microdamage. Hum. Mol. Genet. 2016, 25, 2776–2788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nilsson, M.I.; Nissar, A.A.; Al-Sajee, D.; Tarnopolsky, M.A.; Parise, G.; Lach, B.; Fürst, D.O.; van der Ven, P.F.M.; Kley, R.A.; Hawke, T.J. Xin is a marker of skeletal muscle damage severity in myopathies. Am. J. Pathol. 2013, 183, 1703–1709. [Google Scholar] [CrossRef] [PubMed]
- Orfanos, Z.; Godderz, M.P.; Soroka, E.; Godderz, T.; Rumyantseva, A.; van der Ven, P.F.; Hawke, T.J.; Fürst, D.O. Breaking sarcomeres by in vitro exercise. Sci. Rep. 2016, 6, 19614. [Google Scholar] [CrossRef] [Green Version]
- Molt, S.; Bührdel, J.B.; Yakovlev, S.; Schein, P.; Orfanos, Z.; Kirfel, G.; Winter, L.; Wiche, G.; van der Ven, P.F.; Rottbauer, W.; et al. Aciculin interacts with filamin C and Xin and is essential for myofibril assembly, remodeling and maintenance. J. Cell Sci. 2014, 127 Pt 16, 3578–3592. [Google Scholar] [CrossRef] [Green Version]
- Greenberg, C.C.; Connelly, P.S.; Daniels, M.P.; Horowits, R. Krp1 (Sarcosin) promotes lateral fusion of myofibril assembly intermediates in cultured mouse cardiomyocytes. Exp. Cell Res. 2008, 314, 1177–1191. [Google Scholar] [CrossRef] [Green Version]
- Dhume, A.; Lu, S.; Horowits, R. Targeted disruption of N-RAP gene function by RNA interference: A role for N-RAP in myofibril organization. Cell Motil. Cytoskelet. 2006, 63, 493–511. [Google Scholar] [CrossRef]
- Heckmann, M.B.; Bauer, R.; Jungmann, A.; Winter, L.; Rapti, K.; Strucksberg, K.H.; Clemen, C.S.; Li, Z.; Schröder, R.; Katus, H.A.; et al. AAV9-mediated gene transfer of desmin ameliorates cardiomyopathy in desmin-deficient mice. Gene Ther. 2016, 23, 673–679. [Google Scholar] [CrossRef] [Green Version]
- Ruppert, T.; Heckmann, M.B.; Rapti, K.; Schultheis, D.; Jungmann, A.; Katus, H.A.; Winter, L.; Frey, N.; Clemen, C.S.; Schröder, R.; et al. AAV-mediated cardiac gene transfer of wild-type desmin in mouse models for recessive desminopathies. Gene Ther. 2020, 27, 516–524. [Google Scholar] [CrossRef] [Green Version]
- Weisleder, N.; Soumaka, E.; Abbasi, S.; Taegtmeyer, H.; Capetanaki, Y. Cardiomyocyte-specific desmin rescue of desmin null cardiomyopathy excludes vascular involvement. J. Mol. Cell. Cardiol. 2004, 36, 121–128. [Google Scholar] [CrossRef]
- Carlsson, L.; Thornell, L.E. Desmin-related myopathies in mice and man. Acta Physiol. Scand. 2001, 171, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Kubanek, M.; Schimerova, T.; Piherova, L.; Brodehl, A.; Krebsova, A.; Ratnavadivel, S.; Stanasiuk, C.; Hansikova, H.; Zeman, J.; Palecek, T.; et al. Desminopathy: Novel Desmin Variants, a New Cardiac Phenotype, and Further Evidence for Secondary Mitochondrial Dysfunction. J. Clin. Med. 2020, 9, 937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winter, L.; Türk, M.; Harter, P.N.; Mittelbronn, M.; Kornblum, C.; Norwood, F.; Jungbluth, H.; Thiel, C.T.; Schlötzer-Schrehardt, U.; Schröder, R. Downstream effects of plectin mutations in epidermolysis bullosa simplex with muscular dystrophy. Acta Neuropathol. Commun. 2016, 4, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolling, M.C.; Pas, H.H.; de Visser, M.; Aronica, E.; Pfendner, E.G.; van den Berg, M.P.; Diercks, G.F.; Suurmeijer, A.J.; Jonkman, M.F. PLEC1 mutations underlie adult-onset dilated cardiomyopathy in epidermolysis bullosa simplex with muscular dystrophy. J. Investig. Dermatol. 2010, 130, 1178–1181. [Google Scholar] [CrossRef] [Green Version]
- Vicart, P.; Caron, A.; Guicheney, P.; Li, Z.; Prevost, M.C.; Faure, A.; Chateau, D.; Chapon, F.; Tome, F.; Dupret, J.M.; et al. A missense mutation in the αB-crystallin chaperone gene causes a desmin-related myopathy. Nat. Genet. 1998, 20, 92–95. [Google Scholar] [CrossRef]
- Brodehl, A.; Gaertner-Rommel, A.; Klauke, B.; Grewe, S.A.; Schirmer, I.; Peterschröder, A.; Faber, L.; Vorgerd, M.; Gummert, J.; Anselmetti, D.; et al. The novel αB-crystallin (CRYAB) mutation p.D109G causes restrictive cardiomyopathy. Hum. Mutat. 2017, 38, 947–952. [Google Scholar] [CrossRef]
- Alam, S.; Abdullah, C.S.; Aishwarya, R.; Morshed, M.; Nitu, S.S.; Miriyala, S.; Panchatcharam, M.; Kevil, C.G.; Orr, A.W.; Bhuiyan, M.S. Dysfunctional Mitochondrial Dynamic and Oxidative Phosphorylation Precedes Cardiac Dysfunction in R120G-αB-Crystallin-Induced Desmin-Related Cardiomyopathy. J. Am. Heart Assoc. 2020, 9, e017195. [Google Scholar] [CrossRef]
- Schröder, R.; Kunz, W.S.; Rouan, F.; Pfendner, E.; Tolksdorf, K.; Kappes-Horn, K.; Altenschmidt-Mehring, M.; Knoblich, R.; van der Ven, P.F.; Reimann, J.; et al. Disorganization of the desmin cytoskeleton and mitochondrial dysfunction in plectin-related epidermolysis bullosa simplex with muscular dystrophy. J. Neuropathol. Exp. Neurol. 2002, 61, 520–530. [Google Scholar] [CrossRef] [Green Version]
- Dayal, A.A.; Medvedeva, N.V.; Nekrasova, T.M.; Duhalin, S.D.; Surin, A.K.; Minin, A.A. Desmin Interacts Directly with Mitochondria. Int. J. Mol. Sci. 2020, 21, 8122. [Google Scholar] [CrossRef]
- Winter, L.; Kuznetsov, A.V.; Grimm, M.; Zeold, A.; Fischer, I.; Wiche, G. Plectin isoform P1b and P1d deficiencies differentially affect mitochondrial morphology and function in skeletal muscle. Hum. Mol. Genet. 2015, 24, 4530–4544. [Google Scholar] [CrossRef] [Green Version]
- Kuznetsov, A.V.; Javadov, S.; Grimm, M.; Margreiter, R.; Ausserlechner, M.J.; Hagenbuchner, J. Crosstalk between Mitochondria and Cytoskeleton in Cardiac Cells. Cells 2020, 9, 222. [Google Scholar] [CrossRef] [PubMed]
- Mado, K.; Chekulayev, V.; Shevchuk, I.; Puurand, M.; Tepp, K.; Kaambre, T. On the role of tubulin, plectin, desmin, and vimentin in the regulation of mitochondrial energy fluxes in muscle cells. Am. J. Physiol. Cell Physiol. 2019, 316, C657–C667. [Google Scholar] [CrossRef] [PubMed]
- Alam, S.; Abdullah, C.S.; Aishwarya, R.; Miriyala, S.; Panchatcharam, M.; Peretik, J.M.; Orr, A.W.; James, J.; Robbins, J.; Bhuiyan, M.S. Aberrant Mitochondrial Fission Is Maladaptive in Desmin Mutation-Induced Cardiac Proteotoxicity. J. Am. Heart Assoc. 2018, 7, e009289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Y.; Chiang, W.C.; Sumpter, R., Jr.; Mishra, P.; Levine, B. Prohibitin 2 Is an Inner Mitochondrial Membrane Mitophagy Receptor. Cell 2017, 168, 224–238.e210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thornell, L.; Carlsson, L.; Li, Z.; Mericskay, M.; Paulin, D. Null mutation in the desmin gene gives rise to a cardiomyopathy. J. Mol. Cell. Cardiol. 1997, 29, 2107–2124. [Google Scholar] [CrossRef]
- Lopaschuk, G.D.; Ussher, J.R.; Folmes, C.D.; Jaswal, J.S.; Stanley, W.C. Myocardial fatty acid metabolism in health and disease. Physiol. Rev. 2010, 90, 207–258. [Google Scholar] [CrossRef]
- Zaarour, N.; Berenguer, M.; le Marchand-Brustel, Y.; Govers, R. Deciphering the role of GLUT4 N-glycosylation in adipocyte and muscle cell models. Biochem. J. 2012, 445, 265–273. [Google Scholar] [CrossRef] [Green Version]
- Sadler, J.B.; Bryant, N.J.; Gould, G.W.; Welburn, C.R. Posttranslational modifications of GLUT4 affect its subcellular localization and translocation. Int. J. Mol. Sci. 2013, 14, 9963–9978. [Google Scholar] [CrossRef] [Green Version]
- Shao, D.; Tian, R. Glucose Transporters in Cardiac Metabolism and Hypertrophy. Compr. Physiol. 2015, 6, 331–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montessuit, C.; Thorburn, A. Transcriptional activation of the glucose transporter GLUT1 in ventricular cardiac myocytes by hypertrophic agonists. J. Biol. Chem. 1999, 274, 9006–9012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quest, A.F.; Soldati, T.; Hemmer, W.; Perriard, J.C.; Eppenberger, H.M.; Wallimann, T. Phosphorylation of chicken brain-type creatine kinase affects a physiologically important kinetic parameter and gives rise to protein microheterogeneity in vivo. FEBS Lett. 1990, 269, 457–464. [Google Scholar] [CrossRef]
- Zurmanova, J.; Difato, F.; Malacova, D.; Mejsnar, J.; Stefl, B.; Zahradnik, I. Creatine kinase binds more firmly to the M-band of rabbit skeletal muscle myofibrils in the presence of its substrates. Mol. Cell. Biochem. 2007, 305, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Gregor, M.; Janovska, A.; Stefl, B.; Zurmanova, J.; Mejsnar, J. Substrate channelling in a creatine kinase system of rat skeletal muscle under various pH conditions. Exp. Physiol. 2003, 88, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Schlattner, U.; Tokarska-Schlattner, M.; Wallimann, T. Mitochondrial creatine kinase in human health and disease. Biochim. Biophys. Acta 2006, 1762, 164–180. [Google Scholar] [CrossRef] [PubMed]
- Hernando-Rodriguez, B.; Artal-Sanz, M. Mitochondrial Quality Control Mechanisms and the PHB (Prohibitin) Complex. Cells 2018, 7, 238. [Google Scholar] [CrossRef] [Green Version]
- Merkwirth, C.; Langer, T. Prohibitin function within mitochondria: Essential roles for cell proliferation and cristae morphogenesis. Biochim. Biophys. Acta 2009, 1793, 27–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niederwieser, A.; Steinmann, B.; Exner, U.; Neuheiser, F.; Redweik, U.; Wang, M.; Rampini, S.; Wendel, U. Multiple acyl-Co A dehydrogenation deficiency (MADD) in a boy with nonketotic hypoglycemia, hepatomegaly, muscle hypotonia and cardiomyopathy. Detection of N-isovalerylglutamic acid and its monoamide. Helv. Paediatr. Acta 1983, 38, 9–26. [Google Scholar]
- Singla, M.; Guzman, G.; Griffin, A.J.; Bharati, S. Cardiomyopathy in multiple Acyl-CoA dehydrogenase deficiency: A clinico-pathological correlation and review of literature. Pediatr. Cardiol. 2008, 29, 446–451. [Google Scholar] [CrossRef]
- Couce, M.L.; Sanchez-Pintos, P.; Diogo, L.; Leao-Teles, E.; Martins, E.; Santos, H.; Bueno, M.A.; Delgado-Pecellin, C.; Castineiras, D.E.; Cocho, J.A.; et al. Newborn screening for medium-chain acyl-CoA dehydrogenase deficiency: Regional experience and high incidence of carnitine deficiency. Orphanet J. Rare Dis. 2013, 8, 102. [Google Scholar] [CrossRef] [Green Version]
- Houten, S.M.; Denis, S.; Brinke, H.T.; Jongejan, A.; van Kampen, A.H.; Bradley, E.J.; Baas, F.; Hennekam, R.C.; Millington, D.S.; Young, S.P.; et al. Mitochondrial NADP(H) deficiency due to a mutation in NADK2 causes dienoyl-CoA reductase deficiency with hyperlysinemia. Hum. Mol. Genet. 2014, 23, 5009–5016. [Google Scholar] [CrossRef] [Green Version]
- Ranjbarvaziri, S.; Kooiker, K.B.; Ellenberger, M.; Fajardo, G.; Zhao, M.; Roest, A.S.V.; Woldeyes, R.A.; Koyano, T.T.; Fong, R.; Ma, N.; et al. Altered Cardiac Energetics and Mitochondrial Dysfunction in Hypertrophic Cardiomyopathy. Circulation 2021, 144, 1714–1731. [Google Scholar] [CrossRef] [PubMed]
- Koopman, W.J.; Willems, P.H.; Smeitink, J.A. Monogenic mitochondrial disorders. N. Engl. J. Med. 2012, 366, 1132–1141. [Google Scholar] [CrossRef] [PubMed]
- Rapti, K.; Diokmetzidou, A.; Kloukina, I.; Milner, D.J.; Varela, A.; Davos, C.H.; Capetanaki, Y. Opposite effects of catalase and MnSOD ectopic expression on stress induced defects and mortality in the desmin deficient cardiomyopathy model. Free Radic. Biol. Med. 2017, 110, 206–218. [Google Scholar] [CrossRef] [PubMed]
- Diokmetzidou, A.; Soumaka, E.; Kloukina, I.; Tsikitis, M.; Makridakis, M.; Varela, A.; Davos, C.H.; Georgopoulos, S.; Anesti, V.; Vlahou, A.; et al. Desmin and αB-crystallin interplay in the maintenance of mitochondrial homeostasis and cardiomyocyte survival. J. Cell Sci. 2016, 129, 3705–3720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olsen, R.K.; Olpin, S.E.; Andresen, B.S.; Miedzybrodzka, Z.H.; Pourfarzam, M.; Merinero, B.; Frerman, F.E.; Beresford, M.W.; Dean, J.C.; Cornelius, N.; et al. ETFDH mutations as a major cause of riboflavin-responsive multiple acyl-CoA dehydrogenation deficiency. Brain 2007, 130 Pt 8, 2045–2054. [Google Scholar] [CrossRef] [Green Version]
- Mosegaard, S.; Dipace, G.; Bross, P.; Carlsen, J.; Gregersen, N.; Olsen, R.K.J. Riboflavin Deficiency-Implications for General Human Health and Inborn Errors of Metabolism. Int. J. Mol. Sci. 2020, 21, 3847. [Google Scholar] [CrossRef]
- Gempel, K.; Topaloglu, H.; Talim, B.; Schneiderat, P.; Schoser, B.G.; Hans, V.H.; Palmafy, B.; Kale, G.; Tokatli, A.; Quinzii, C.; et al. The myopathic form of coenzyme Q10 deficiency is caused by mutations in the electron-transferring-flavoprotein dehydrogenase (ETFDH) gene. Brain 2007, 130 Pt 8, 2037–2044. [Google Scholar] [CrossRef]
- Liang, W.C.; Ohkuma, A.; Hayashi, Y.K.; Lopez, L.C.; Hirano, M.; Nonaka, I.; Noguchi, S.; Chen, L.H.; Jong, Y.J.; Nishino, I. ETFDH mutations, CoQ10 levels, and respiratory chain activities in patients with riboflavin-responsive multiple acyl-CoA dehydrogenase deficiency. Neuromuscul. Disord. 2009, 19, 212–216. [Google Scholar] [CrossRef]
- Strnadová, K.A.; Holub, M.; Mühl, A.; Heinze, G.; Ratschmann, R.; Mascher, H.; Stöckler-Ipsiroglu, S.; Waldhauser, F.; Votava, F.; Lebl, J.; et al. Long-term stability of amino acids and acylcarnitines in dried blood spots. Clin. Chem. 2007, 53, 717–722. [Google Scholar] [CrossRef] [Green Version]
- Neckar, J.; Svatonova, A.; Weissova, R.; Drahota, Z.; Zajickova, P.; Brabcova, I.; Kolar, D.; Alanova, P.; Vasinova, J.; Silhavy, J.; et al. Selective replacement of mitochondrial DNA increases the cardioprotective effect of chronic continuous hypoxia in spontaneously hypertensive rats. Clin. Sci. 2017, 131, 865–881. [Google Scholar] [CrossRef]
- Gnaiger, E.; Kuznetsov, A.V. Mitochondrial respiration at low levels of oxygen and cytochrome c. Biochem. Soc. Trans. 2002, 30, 252–258. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsov, A.V.; Schneeberger, S.; Seiler, R.; Brandacher, G.; Mark, W.; Steurer, W.; Saks, V.; Usson, Y.; Margreiter, R.; Gnaiger, E. Mitochondrial defects and heterogeneous cytochrome c release after cardiac cold ischemia and reperfusion. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H1633–H1641. [Google Scholar] [CrossRef] [PubMed]
- Kohutova, J.; Elsnicova, B.; Holzerova, K.; Neckar, J.; Sebesta, O.; Jezkova, J.; Vecka, M.; Vebr, P.; Hornikova, D.; Bacova, B.S.; et al. Anti-arrhythmic Cardiac Phenotype Elicited by Chronic Intermittent Hypoxia Is Associated with Alterations in Connexin-43 Expression, Phosphorylation, and Distribution. Front. Endocrinol. 2018, 9, 789. [Google Scholar] [CrossRef] [PubMed]
- Manders, E.M.M.; Verbeek, F.J.; Aten, J.A. Measurement of co-localization of objects in dual-colour confocal images. J. Microsc. 1993, 169, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Zuiderveld, K. Contrast limited adaptive histogram equalization. In Graphics Gems IV; Academic Press Professional, Inc.: Cambridge, MA, USA, 1994; pp. 474–485. [Google Scholar]
- Bankhead, P. Analyzing Fluorescence Microscopy Images with ImageJ; Queen’s University Belfast: Belfast, UK, 2014. [Google Scholar]
- Doube, M.; Klosowski, M.M.; Arganda-Carreras, I.; Cordelieres, F.P.; Dougherty, R.P.; Jackson, J.S.; Schmid, B.; Hutchinson, J.R.; Shefelbine, S.J. BoneJ: Free and extensible bone image analysis in ImageJ. Bone 2010, 47, 1076–1079. [Google Scholar] [CrossRef] [Green Version]
- Schmid, B.; Tripal, P.; Fraaß, T.; Kersten, C.; Ruder, B.; Grüneboom, A.; Huisken, J.; Palmisano, R. 3Dscript: Animating 3D/4D microscopy data using a natural-language-based syntax. Nat. Methods 2019, 16, 278–280. [Google Scholar] [CrossRef]
- Zhao, S.; Fernald, R.D. Comprehensive algorithm for quantitative real-time polymerase chain reaction. J. Comput. Biol. 2005, 12, 1047–1064. [Google Scholar] [CrossRef] [Green Version]
- Zsurka, G.; Baron, M.; Stewart, J.D.; Kornblum, C.; Bos, M.; Sassen, R.; Taylor, R.W.; Elger, C.E.; Chinnery, P.F.; Kunz, W.S. Clonally expanded mitochondrial DNA mutations in epileptic individuals with mutated DNA polymerase gamma. J. Neuropathol. Exp. Neurol. 2008, 67, 857–866. [Google Scholar] [CrossRef] [Green Version]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE guidelines: Minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef] [Green Version]
- Oertzen-Hagemann, V.; Kirmse, M.; Eggers, B.; Pfeiffer, K.; Marcus, K.; de Marees, M.; Platen, P. Effects of 12 Weeks of Hypertrophy Resistance Exercise Training Combined with Collagen Peptide Supplementation on the Skeletal Muscle Proteome in Recreationally Active Men. Nutrients 2019, 11, 1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plum, S.; Helling, S.; Theiss, C.; Leite, R.E.P.; May, C.; Jacob-Filho, W.; Eisenacher, M.; Kuhlmann, K.; Meyer, H.E.; Riederer, P.; et al. Combined enrichment of neuromelanin granules and synaptosomes from human substantia nigra pars compacta tissue for proteomic analysis. J. Proteom. 2013, 94, 202–206. [Google Scholar] [CrossRef] [PubMed]
- Plum, S.; Eggers, B.; Helling, S.; Stepath, M.; Theiss, C.; Leite, R.E.P.; Molina, M.; Grinberg, L.T.; Riederer, P.; Gerlach, M.; et al. Proteomic Characterization of Synaptosomes from Human Substantia Nigra Indicates Altered Mitochondrial Translation in Parkinson’s Disease. Cells 2020, 9, 2580. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 2016, 11, 2301–2319. [Google Scholar] [CrossRef] [PubMed]
- UniProt Consortum. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Schwanhäusser, B.; Busse, D.; Li, N.; Dittmar, G.; Schuchhardt, J.; Wolf, J.; Chen, W.; Selbach, M. Global quantification of mammalian gene expression control. Nature 2011, 473, 337–342. [Google Scholar] [CrossRef] [Green Version]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Acylcarnitines in µmol/L | Desmin Knock-out (Mean ± SEM; n) | Wild-Type Littermate (Mean ± SEM; n) | p-Value (Two-Tailed) (Mann–Whitney Test) |
|---|---|---|---|
| Carnitine (C0) | 17.52 ± 0.54; 15 | 16.79 ± 0.63; 13 | 0.434 |
| Acetyl-carnitine (C2) | 15.09 ± 0.84; 15 | 13.02 ± 0.38; 13 | 0.140 |
| Propionyl-carnitine (C3) | 0.51 ± 0.02; 15 | 0.45 ± 0.02; 13 | 0.059 |
| Malonyl-carnitine (C3DC) | 0.14 ± 0.01; 14 | 0.14 ± 0.02; 11 | 0.298 |
| Butyryl-carnitine (C4) | 0.28 ± 0.03; 15 | 0.21 ± 0.04; 13 | 0.036 |
| Methylmalonyl-carnitine (C4DC) | undetectable | undetectable | ― |
| 3OH-Butyryl-carnitine (C4OH) | 0.14 ± 0.01; 15 | 0.12 ± 0.01; 13 | 0.027 |
| Isovaleryl-carnitine (C5) | 0.07 ± 0.00; 15 | 0.06 ± 0.00; 13 | 0.023 |
| Tiglyl-carnitine (C5:1) | undetectable | undetectable | ― |
| Glutaryl-carnitine (C5DC) | 0.07 ±0.01; 15 | 0.07 ± 0.01; 11 | 0.938 |
| 3OH-Isovaleryl-carnitine (C5OH) | undetectable | undetectable | ― |
| Hexanoyl-carnitine (C6) | 0.07 ± 0.00; 15 | 0.07 ± 0.01; 13 | 0.645 |
| Octanoyl-carnitine (C8) | 0.08 ± 0.00; 15 | 0.07 ± 0.01; 13 | 0.030 |
| Octenoyl-carnitine (C8:1) | undetectable | undetectable | ― |
| Decanoyl-carnitine (C10) | 0.04 ± 0.00; 15 | 0.04 ± 0.00; 13 | 0.447 |
| Cis4-Decanoyl-carnitine (C10:1) | 0.03 ± 0.00; 10 | 0.02 ± 0.00; 11 | 0.139 |
| Dodecanoyl-carnitine (C12) | 0.21 ± 0.01; 15 | 0.19 ± 0.02; 13 | 0.214 |
| Tetradecanoyl-carnitine (C14) | 0.16 ± 0.01; 15 | 0.14 ± 0.01; 13 | 0.010 |
| Tetradecenoyl-carnitine (C14:1) | 0.07 ± 0.00; 15 | 0.07 ± 0.00; 13 | 0.369 |
| Tetradecadienyl-carnitine (C14:2) | 0.03 ± 0.00; 15 | 0.02 ± 0.00; 13 | 0.475 |
| 3OH-Tetradecanoyl-carnitine (C14OH) | 0.02 ± 0.00; 15 | 0.01 ± 0.00; 13 | 0.174 |
| Palmitoyl-carnitine (C16) | 1.00 ± 0.04; 15 | 0.92 ± 0.04; 13 | 0.088 |
| Palmitoleyl-carnitine (C16:1) | 0.08 ± 0.00; 15 | 0.08 ± 0.01; 13 | 0.279 |
| 3OH-Palmitoyl-carnitine (C16OH) | 0.05 ± 0.00; 15 | 0.04 ± 0.00; 13 | 0.012 |
| 3OH-Hexadecenoyl-carnitine (C16:1OH) | 0.032 ± 0.002; 15 | 0.026 ± 0.003; 13 | 0.076 |
| Octadecanoyl-carnitine (C18) | 0.27 ± 0.01; 15 | 0.23 ± 0.01; 13 | 0.025 |
| Oleoyl-carnitine (C18:1) | 0.34 ± 0.02; 15 | 0.30 ± 0.02; 13 | 0.065 |
| 3OH-Stearoyl-carnitine (C18OH) | 0.01 ± 0.00; 14 | 0.01 ± 0.00; 11 | 0.584 |
| 3OH-Oleoyl-carnitine (C18:1OH) | 0.02 ± 0.00; 15 | 0.02 ± 0.00; 13 | 0.645 |
| 3OH-Linolyl-carnitine (C18:2OH) | 0.03 ± 0.00; 15 | 0.03 ± 0.00; 13 | 0.333 |
| Acylcarnitine ratios | Desmin knock-out (mean ± SEM; n) | Wild-type littermate (mean ± SEM; n) | p-Value (one-tailed) (Mann–Whitney test) |
| C3/C0 | 0.03 ± 0.00; 15 | 0.03 ± 0.00; 13 | 0.099 |
| C3/C2 | 0.03 ± 0.00; 15 | 0.03 ± 0.00; 13 | 0.418 |
| C5/C2 | 0.00 ± 0.00; 15 | 0.00 ± 0.00; 13 | 0.238 |
| C8/C2 | 0.01 ± 0.00; 15 | 0.01 ± 0.00; 13 | 0.111 |
| C8/C10 | 2.12 ± 0.11; 15 | 1.85 ± 0.12; 13 | 0.049 |
| C8/C12 | 0.41 ± 0.02; 15 | 0.40 ± 0.06; 13 | 0.191 |
| C14:1/C14 | 0.48 ± 0.02; 15 | 0.52 ± 0.04; 13 | 0.282 |
| C14:1/C16 | 0.08 ± 0.00; 15 | 0.07 ± 0.00; 13 | 0.356 |
| Amino acids in µmol/l | Desmin knock-out (mean ± SEM; n) | Wild-type littermate (mean ± SEM; n) | p-Value (two-tailed) (Mann–Whitney test) |
| Alanine | 303 ± 51; 4 | 256 ± 31; 5 | 0.806 |
| Allo-Isoleucine | <2.0; 4 | <2.0; 5 | ― |
| Arginine | 25.1 ± 2.4; 4 | 25.4 ± 2.3; 5 | 0.624 |
| Citrulline | 13.6 ± 1.3; 4 | 14.9 ± 1.8; 5 | 1.000 |
| Glutamine | 107.2 ± 16.7; 4 | 94.6 ± 18.0; 5 | 0.462 |
| Glycine | 64.9 ± 6.0; 4 | 53.3 ± 5.5; 5 | 0.221 |
| Histidine | 3.54 ± 0.83; 4 | 2.51 ± 0.30; 5 | 0.327 |
| Isoleucine | 52.9 ± 6.0; 4 | 38.0 ± 2.0; 5 | 0.014 |
| Leucine | 129.9 ± 16.1; 4 | 93.7 ± 6.9; 5 | 0.086 |
| Lysine | 20.1 ± 1.8; 4 | 19.1 ± 2.2; 5 | 1.000 |
| Methionine | 12.9 ± 1.0; 4 | 11.8 ± 1.4; 5 | 1.000 |
| 3-O-Methyldopa | 0.063 ± 0.003; 4 | 0.056 ± 0.002; 5 | 0.178 |
| Ornithine | 2.6 ± 0.6; 4 | <2.0; 5 | ― |
| Phenylalanine | 59.0 ± 12.2; 4 | 44.6 ± 8.1; 5 | 0.086 |
| Proline | 93.3 ± 16.0; 4 | 73.9 ± 12.1; 5 | 0.327 |
| Serine | 83.5 ± 10.4; 4 | 62.6 ± 9.6; 5 | 0.221 |
| Threonine | 52.9 ± 1.5; 4 | 44.7 ± 4.9; 5 | 0.221 |
| Tryptophan | 3.47 ± 0.44; 4 | 2.45 ± 0.23 | 0.086 |
| Tyrosine | 59.1 ± 12.7; 4 | 50.6 ± 9.5; 5 | 0.327 |
| Valine | 128.0 ± 9.6; 4 | 94.1 ± 4.7; 5 | 0.014 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elsnicova, B.; Hornikova, D.; Tibenska, V.; Kolar, D.; Tlapakova, T.; Schmid, B.; Mallek, M.; Eggers, B.; Schlötzer-Schrehardt, U.; Peeva, V.; et al. Desmin Knock-Out Cardiomyopathy: A Heart on the Verge of Metabolic Crisis. Int. J. Mol. Sci. 2022, 23, 12020. https://doi.org/10.3390/ijms231912020
Elsnicova B, Hornikova D, Tibenska V, Kolar D, Tlapakova T, Schmid B, Mallek M, Eggers B, Schlötzer-Schrehardt U, Peeva V, et al. Desmin Knock-Out Cardiomyopathy: A Heart on the Verge of Metabolic Crisis. International Journal of Molecular Sciences. 2022; 23(19):12020. https://doi.org/10.3390/ijms231912020
Chicago/Turabian StyleElsnicova, Barbara, Daniela Hornikova, Veronika Tibenska, David Kolar, Tereza Tlapakova, Benjamin Schmid, Markus Mallek, Britta Eggers, Ursula Schlötzer-Schrehardt, Viktoriya Peeva, and et al. 2022. "Desmin Knock-Out Cardiomyopathy: A Heart on the Verge of Metabolic Crisis" International Journal of Molecular Sciences 23, no. 19: 12020. https://doi.org/10.3390/ijms231912020