
Non-Covalent Interactions in the Crystal Structures of Perbrominated Sulfonium Derivatives of the closo-Decaborate Anion
 , , , , and
, , , , and
Abstract
:1. Introduction
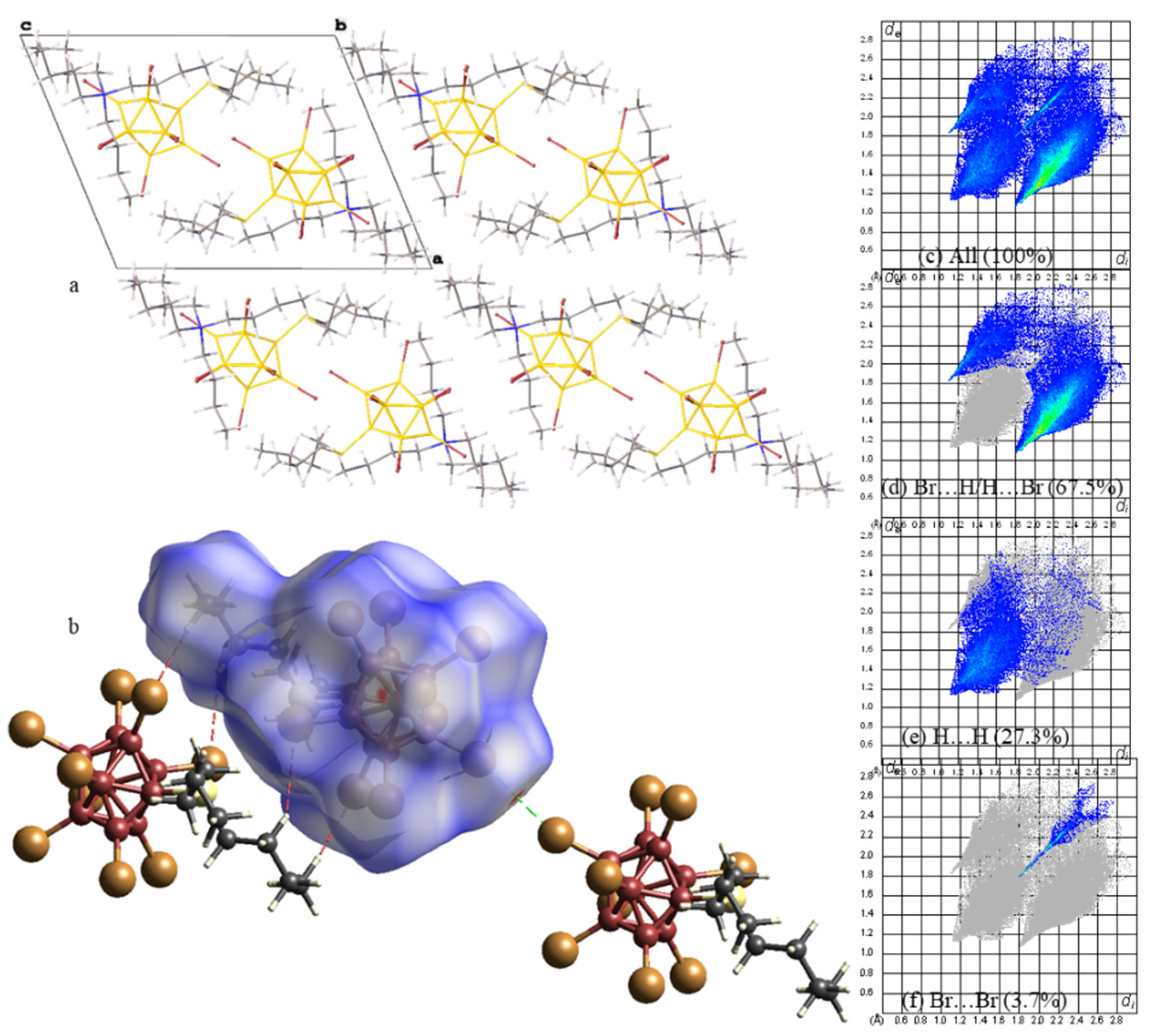
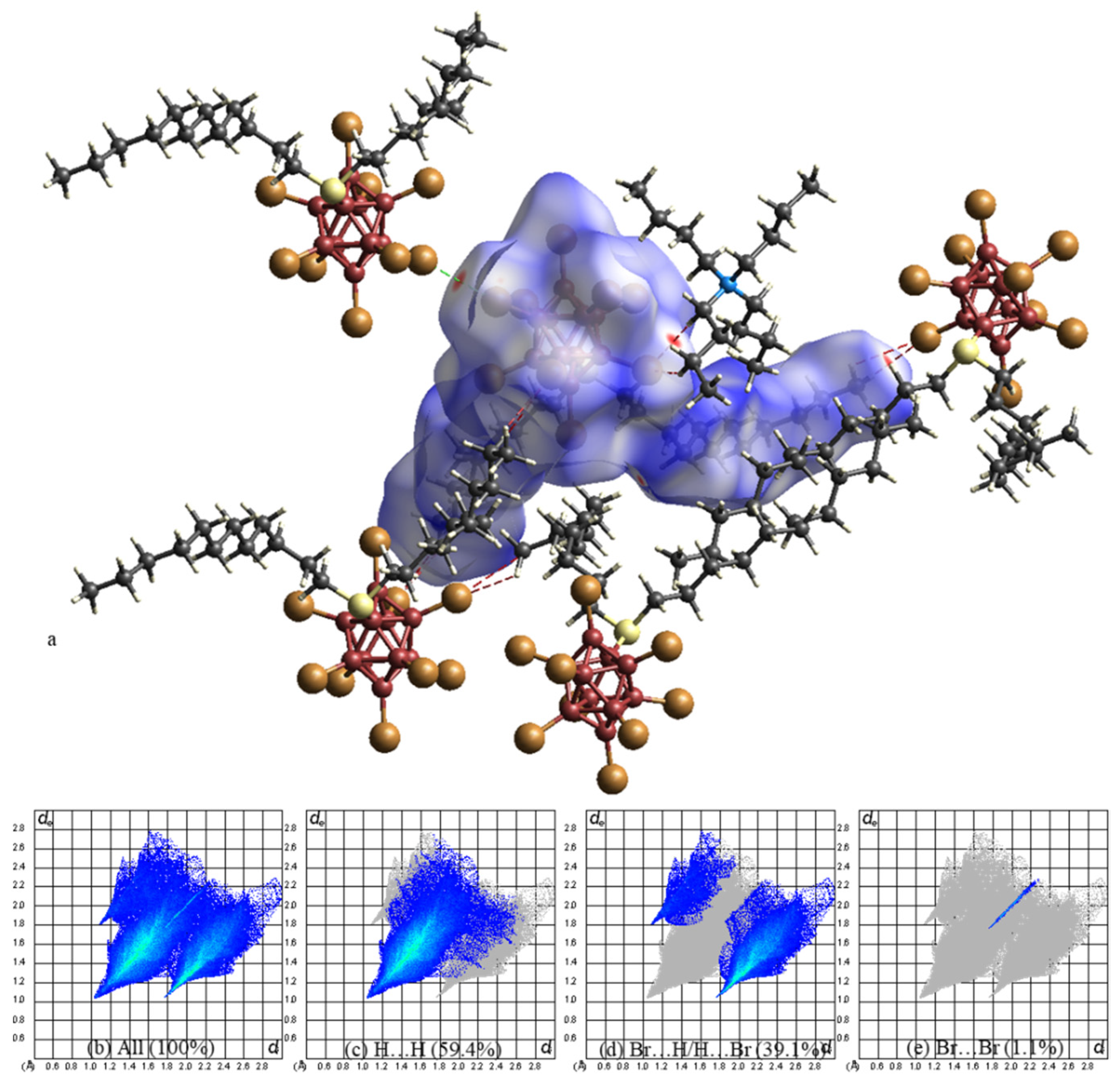
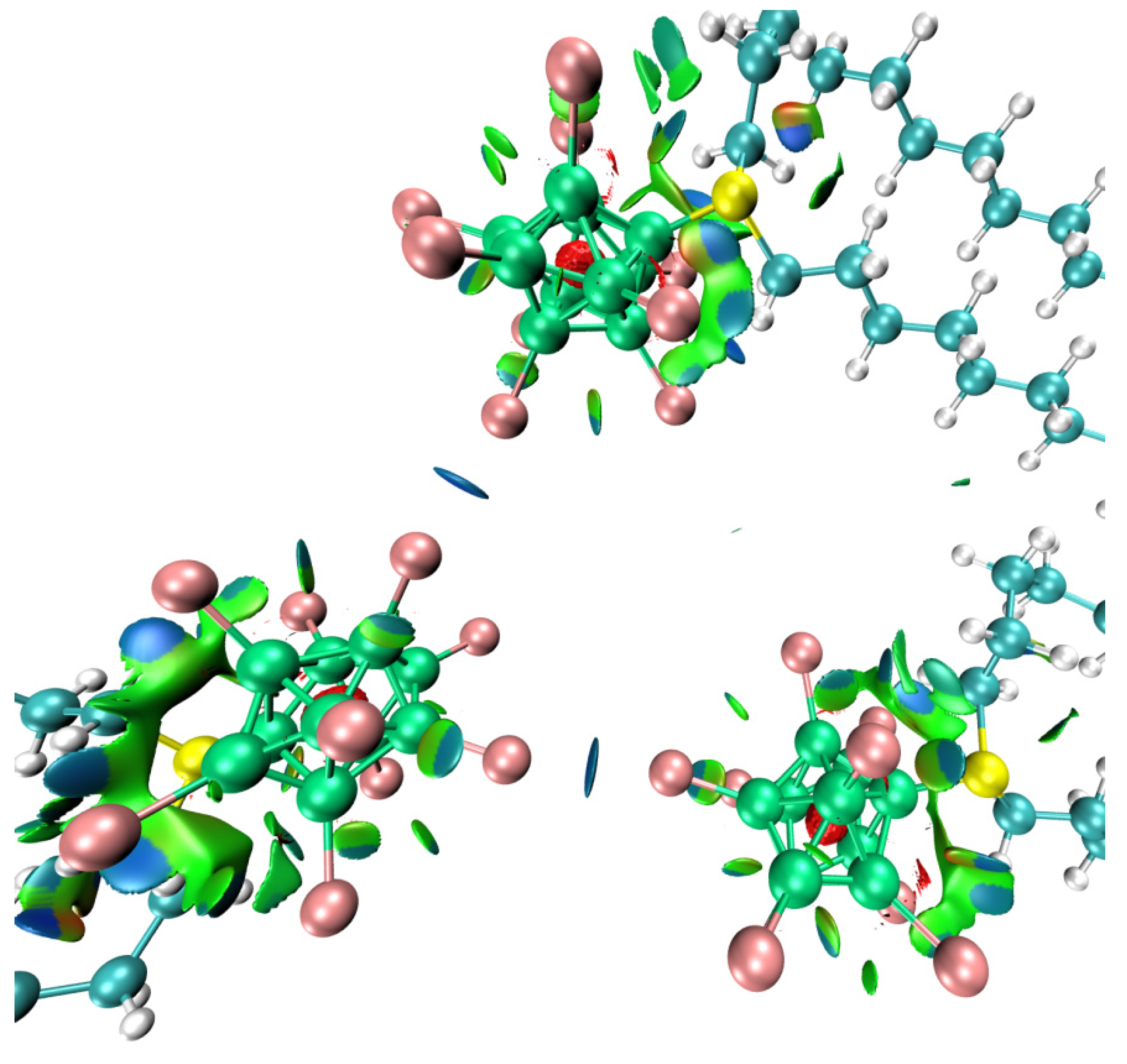
2. Results
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brammer, L.; Espallargas, G.M.; Libri, S. Combining Metals with Halogen Bonds. CrystEngComm 2008, 10, 1712–1727. [Google Scholar] [CrossRef]
- Bulfield, D.; Engelage, E.; Mancheski, L.; Stoesser, J.; Huber, S.M. Crystal Engineering with Multipoint Halogen Bonding: Double Two-Point Donors and Acceptors at Work. Chem. A Eur. J. 2020, 26, 1567–1575. [Google Scholar] [CrossRef] [Green Version]
- Rissanen, K. Halogen Bonded Supramolecular Complexes and Networks. CrystEngComm 2008, 10, 1107–1113. [Google Scholar] [CrossRef]
- Von Der Heiden, D.; Detmar, E.; Kuchta, R.; Breugst, M. Activation of Michael Acceptors by Halogen-Bond Donors. Synlett 2018, 29, 1307–1313. [Google Scholar] [CrossRef] [Green Version]
- Virovets, A.V.; Vakulenko, N.N.; Volkov, V.V.; Podberezskaya, N.V. Crystal Structure of Di (1-n-Dodecylpyridine) Decahydro-Closo-Decaborate(2-) (C5H5N-C12H25)2[B10H10]. J. Struct. Chem. 1995, 35, 339–344. [Google Scholar] [CrossRef]
- Songkram, C.; Ohta, K.; Yamaguchi, K.; Pichierri, F.; Endo, Y. Conformational Control of Benzyl-o-Carboranylbenzene Derivatives and Molecular Encapsulation of Acetone in the Dynamically Formed Space of 1,3,5-Tris(2-Benzyl-o-Carboran-1-Yl)Benzene. Inorg. Chem. 2010, 49, 11174–11183. [Google Scholar] [CrossRef] [PubMed]
- Štíbr, B.; Bakardjiev, M.; Holub, J.; Růžička, A.; Padělková, Z.; Štěpnička, P. Additive Character of Electron Donation by Methyl Substituents within a Complete Series of Polymethylated [1-(H6-MenC6H6-n)-Closo-1,2,3-FeC2B9H11] Complexes. Linear Correlations of the NMR Parameters and Fe II/III Redox Potentials with the Number of Are. Inorg. Chem. 2011, 50, 3097–3102. [Google Scholar] [CrossRef]
- Wang, Z.; Zhao, J.; Muddassir, M.; Guan, R.; Tao, S. Recovering the Thermally Activated Delayed Fluorescence in Aggregation-Induced Emitters of Carborane. Inorg. Chem. 2021, 60, 4705–4716. [Google Scholar] [CrossRef]
- Sivaev, I.B.; Prikaznov, A.V.; Naoufal, D. Fifty Years of the Closo-Decaborate Anion Chemistry. Collect. Czechoslov. Chem. Commun. 2010, 75, 1149–1199. [Google Scholar] [CrossRef]
- Zhizhin, K.Y.; Zhdanov, A.P.; Kuznetsov, N.T. Derivatives of Closo-Decaborate Anion [B10H10]2− with Exo-Polyhedral Substituents. Russ. J. Inorg. Chem. 2010, 55, 2089–2127. [Google Scholar] [CrossRef]
- Kubasov, A.S.; Turishev, E.S.; Polyakova, I.N.; Matveev, E.Y.; Zhizhin, K.Y.; Kuznetsov, N.T. The Method for Synthesis of 2-Sulfanyl Closo-Decaborate Anion and Its S-Alkyl and S-Acyl Derivatives. J. Organomet. Chem. 2017, 828, 106–115. [Google Scholar] [CrossRef]
- Kaszynski, P.; Huang, J.; Jenkins, G.S.; Bairamov, K.A.; Lipiak, D. Boron Clusters in Liquid Crystals. Mol. Cryst. Liq. Cryst. Sci. Technol. Sect. A Mol. Cryst. Liq. Cryst. 1995, 260, 315–332. [Google Scholar] [CrossRef]
- Ringstrand, B.; Kaszynski, P. Anion for the Construction of New Classes of Liquid Crystals. Acc. Chem. Res. 2013, 46, 214–225. [Google Scholar] [CrossRef] [PubMed]
- Ringstrand, B.; Jankowiak, A.; Johnson, L.E.; Kaszynski, P.; Pociecha, D.; Górecka, E. Anion-Driven Mesogenicity: A Comparative Study of Ionic Liquid Crystals Based on the [Closo-1-CB9H10]− and [Closo-1-CB11H12]− Clusters. J. Mater. Chem. 2012, 22, 4874–4880. [Google Scholar] [CrossRef]
- Turyshev, E.S.; Kopytin, A.V.; Zhizhin, K.Y.; Kubasov, A.S.; Shpigun, L.K.; Kuznetsov, N.T. Potentiometric Quantitation of General Local Anesthetics with a New Highly Sensitive Membrane Sensor. Talanta 2022, 241, 123239. [Google Scholar] [CrossRef] [PubMed]
- Jankowiak, A.; Baliński, A.; Harvey, J.E.; Mason, K.; Januszko, A.; Kaszyński, P.; Young, V.G.; Persoons, A. [Closo-B10H10]2− as a Structural Element for Quadrupolar Liquid Crystals: A New Class of Liquid Crystalline NLO Chromophores. J. Mater. Chem. C 2013, 1, 1144–1159. [Google Scholar] [CrossRef]
- Kaszyński, P.; Ringstrand, B. Functionalization of Closo-Borates via Iodonium Zwitterions. Angew. Chem. Int. Ed. 2015, 54, 6576–6581. [Google Scholar] [CrossRef] [PubMed]
- Kessler, M.; Knapp, C.; Sagawe, V.; Scherer, H.; Uzun, R. Synthesis, Characterization, and Crystal Structures of Silylium Compounds of the Weakly Coordinating Dianion [B12Cl12]2−. Inorg. Chem. 2010, 49, 5223–5230. [Google Scholar] [CrossRef]
- Kim, K.C.; Reed, C.A.; Long, G.S.; Sen, A. Et2Al+ Alumenium Ion-like Chemistry. Synthesis and Reactivity toward Alkenes and Alkene Oxides. J. Am. Chem. Soc. 2002, 124, 7662–7663. [Google Scholar] [CrossRef] [Green Version]
- Saleh, M.; Powell, D.R.; Wehmschulte, R.J. Chlorination of 1-Carba-Closo-Dodecaborate and 1-Ammonio-Closo-Dodecaborate Anions. Inorg. Chem. 2016, 55, 10617–10627. [Google Scholar] [CrossRef]
- Justus, E.; Rischka, K.; Wishart, J.F.; Werner, K.; Gabel, D. Trialkylammoniododecaborates: Anions for Ionic Liquids with Potassium, Lithium and Protons as Cations. Chem. A Eur. J. 2008, 14, 1918–1923. [Google Scholar] [CrossRef] [PubMed]
- Dymon, J.; Wibby, R.; Kleingardner, J.; Tanski, J.M.; Guzei, I.A.; Holbrey, J.D.; Larsen, A.S. Designing Ionic Liquids with Boron Cluster Anions: Alkylpyridinium and Imidazolium [Nido-C2B9H11] and [Closo-CB11H12] Carborane Salts. Dalt. Trans. 2008, 22, 2999–3006. [Google Scholar] [CrossRef] [PubMed]
- Zhou, N.; Zhao, G.; Dong, K.; Sun, J.; Shao, H. Investigations on a Series of Novel Ionic Liquids Containing the [Closo-B12Cl12]2− Dianion. RSC Adv. 2012, 2, 9830. [Google Scholar] [CrossRef]
- Jenne, C.; Kirsch, C. Alkoxy Substituted Halogenated Closo-Dodecaborates as Anions for Ionic Liquids. Dalt. Trans. 2015, 44, 13119–13124. [Google Scholar] [CrossRef] [Green Version]
- Holub, J.; El Anwar, S.; Jelínek, T.; Fojt, L.; Růžičková, Z.; Šolínová, V.; Kašička, V.; Gabel, D.; Grüner, B. Polyhalogenated Decaborate and 1-Ammoniododecaborate Ions: An Improved Synthesis with Elemental Halogens, and Physicochemical and Chemical Properties. Eur. J. Inorg. Chem. 2017, 2017, 4499–4509. [Google Scholar] [CrossRef] [Green Version]
- El Anwar, S.; Holub, J.; Tok, O.; Jelínek, T.; Růžičková, Z.; Fojt, L.; Šolínová, V.; Kašička, V.; Grüner, B. Synthesis and Selected Properties of Nonahalogenated 2-Ammonio-Decaborate Anions and Their Derivatives Substituted at N-Centre. J. Organomet. Chem. 2018, 865, 189–199. [Google Scholar] [CrossRef]
- Golubev, A.V.; Kubasov, A.S.; Bykov, A.Y.; Zhizhin, K.Y.; Kravchenko, E.A.; Gippius, A.A.; Zhurenko, S.V.; Semenova, V.A.; Korlyukov, A.A.; Kuznetsov, N.T. Synthesis of Perchlorinated Sulfonium Derivatives of Closo-Decaborate Anion [2-B10Cl9SR2]− (R = i-C3H7, n-C3H7, n-C4H9, n-C8H17, n-C12H25, n-C18H37, CH2Ph, and Cyclo-S(CH2)4). Inorg. Chem. 2021, 60, 8592–8604. [Google Scholar] [CrossRef]
- Kubasov, A.S.; Golubev, A.V.; Bykov, A.Y.; Matveev, E.Y.; Zhizhin, K.Y.; Kuznetsov, N.T. Synthesis, Structures, DFT Calculations, and Hirshfeld Surface Analysis of Sulfonium Derivatives of the Closo-Decaborate Anion [B10X9-Cyclo-S(CH2)4]− and [B10X9-Cyclo-S(CH2CH2)2O]− (X = H, Cl, Br). J. Mol. Struct. 2021, 1241, 130591. [Google Scholar] [CrossRef]
- Golubev, A.V.; Kubasov, A.S.; Turyshev, E.S.; Bykov, A.Y.; Zhizhin, K.Y.; Kuznetsov, N.T. Perbrominated Sulfonium-Substituted Closo-Decaborates with Exo-Polyhedral Amino Groups [2-B10Br9S((CH2)nNH2)2]− (n = 1–3). Russ. J. Inorg. Chem. 2020, 65, 1333–1342. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Clarendon Press: Oxford, UK, 1990; ISBN 0198558651. [Google Scholar]
- Espinosa, E.; Molins, E.; Lecomte, C. Hydrogen Bond Strengths Revealed by Topological Analyses of Experimentally Observed Electron Densities. Chem. Phys. Lett. 1998, 285, 170–173. [Google Scholar] [CrossRef]
- Espinosa, E.; Lecomte, C.; Molins, E. Experimental Electron Density Overlapping in Hydrogen Bonds: Topology vs. Energetics. Chem. Phys. Lett. 1999, 300, 745–748. [Google Scholar] [CrossRef]
- Kravchenko, E.A.; Gippius, A.A.; Korlyukov, A.A.; Vologzhanina, A.V.; Avdeeva, V.V.; Malinina, E.A.; Ulitin, E.O.; Kuznetsov, N.T. Secondary Interactions in Decachloro-Closo-Decaborates R2[B10Cl10] (R = Et3NH+, Ph4P+, and [Ag(NH3)2]+): 35Cl NQR, PW-DFT, and X-Ray Studies. Inorg. Chim. Acta 2016, 447, 22–31. [Google Scholar] [CrossRef]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [Green Version]
- Gu, W.; Ozerov, O.V. Exhaustive Chlorination of [B12H12]2− without Chlorine Gas and the Use of [B12Cl12]2− as a Supporting Anion in Catalytic Hydrodefluorination of Aliphatic C-F Bonds. Inorg. Chem. 2011, 50, 2726–2728. [Google Scholar] [CrossRef] [PubMed]
- Justus, E.; Vöge, A.; Gabel, D. N-Alkylation of Ammonioundecahydro-Closo-Dodecaborate(1-) for the Preparation of Anions for Ionic Liquids. Eur. J. Inorg. Chem. 2008, 2008, 5245–5250. [Google Scholar] [CrossRef]
- Kubasov, A.S.; Matveev, E.Y.; Turyshev, E.S.; Polyakova, I.N.; Nichugovskiy, A.I.; Zhizhin, K.Y.; Kuznetsov, N.T. Synthesis and Stability Studies of Derivatives of the 2-Sulfanyl-Closo-Decaborate Anion [2-B10H9SH]2−. Inorg. Chim. Acta 2018, 477, 277–283. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Spackman, P.R.; Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer: A Program for Hirshfeld Surface Analysis, Visualization and Quantitative Analysis of Molecular Crystals. J. Appl. Crystallogr. 2021, 54, 1006–1011. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector Augmented-Wave Method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method. Phys. Rev. B Condens. Matter Mater. Phys. 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimme, S. Semiempirical GGA-Type Density Functional Constructed with a Long-Range Dispersion Correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Hafner, J. Ab Initio Molecular-Dynamics Simulation of the Liquid-Metalamorphous- Semiconductor Transition in Germanium. Phys. Rev. B 1994, 49, 14251–14269. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Hafner, J. Ab Initio Molecular Dynamics for Liquid Metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of Ab-Initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmuller, J. Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Gonze, X.; Beuken, J.M.; Caracas, R.; Detraux, F.; Fuchs, M.; Rignanese, G.M.; Sindic, L.; Verstraete, M.; Zerah, G.; Jollet, F.; et al. First-Principles Computation of Material Properties: The ABINIT Software Project. Comput. Mater. Sci. 2002, 25, 478–492. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | 1 | 2 | 3 | 5 | 6 |
|---|---|---|---|---|---|
| B–Brap(av.) | 1.935(5) | 1.954(8) | 1.939(6) | 1.931(6) | 1.941(6) |
| B–Breq(av.) | 1.951(5) | 1.951(8) | 1.959(6) | 1.955(6) | 1.947(5) |
| B–S | 1.889(5) | 1.912(8) | 1.887(6) | 1.896(6) | 1.893(6) |
| S–C(av.) | 1.815(5) | 1.831(15) | 1.824(6) | 1.833(5) | 1.821(6) |
| C–S–C | 104.2(3) | 104.4(5) * | 102.8(3) | 102.9(3) | 102.0(2) |
| C–S–B(av.) | 103.8(2) | 107.0(5) | 104.0(3) | 102.9(3) | 102.8(2) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Golubev, A.V.; Kubasov, A.S.; Bykov, A.Y.; Zhdanov, A.P.; Buzanov, G.A.; Korlyukov, A.A.; Zhizhin, K.Y.; Kuznetsov, N.T. Non-Covalent Interactions in the Crystal Structures of Perbrominated Sulfonium Derivatives of the closo-Decaborate Anion. Int. J. Mol. Sci. 2022, 23, 12022. https://doi.org/10.3390/ijms231912022
Golubev AV, Kubasov AS, Bykov AY, Zhdanov AP, Buzanov GA, Korlyukov AA, Zhizhin KY, Kuznetsov NT. Non-Covalent Interactions in the Crystal Structures of Perbrominated Sulfonium Derivatives of the closo-Decaborate Anion. International Journal of Molecular Sciences. 2022; 23(19):12022. https://doi.org/10.3390/ijms231912022
Chicago/Turabian StyleGolubev, Aleksei V., Alexey S. Kubasov, Alexander Yu. Bykov, Andrey P. Zhdanov, Grigorii A. Buzanov, Alexander A. Korlyukov, Konstantin Yu. Zhizhin, and Nikolay T. Kuznetsov. 2022. "Non-Covalent Interactions in the Crystal Structures of Perbrominated Sulfonium Derivatives of the closo-Decaborate Anion" International Journal of Molecular Sciences 23, no. 19: 12022. https://doi.org/10.3390/ijms231912022