
Identification of Candidate mRNA Isoforms for Prostate Cancer-Risk SNPs Utilizing Iso-eQTL and sQTL Methods
 , ,
, ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
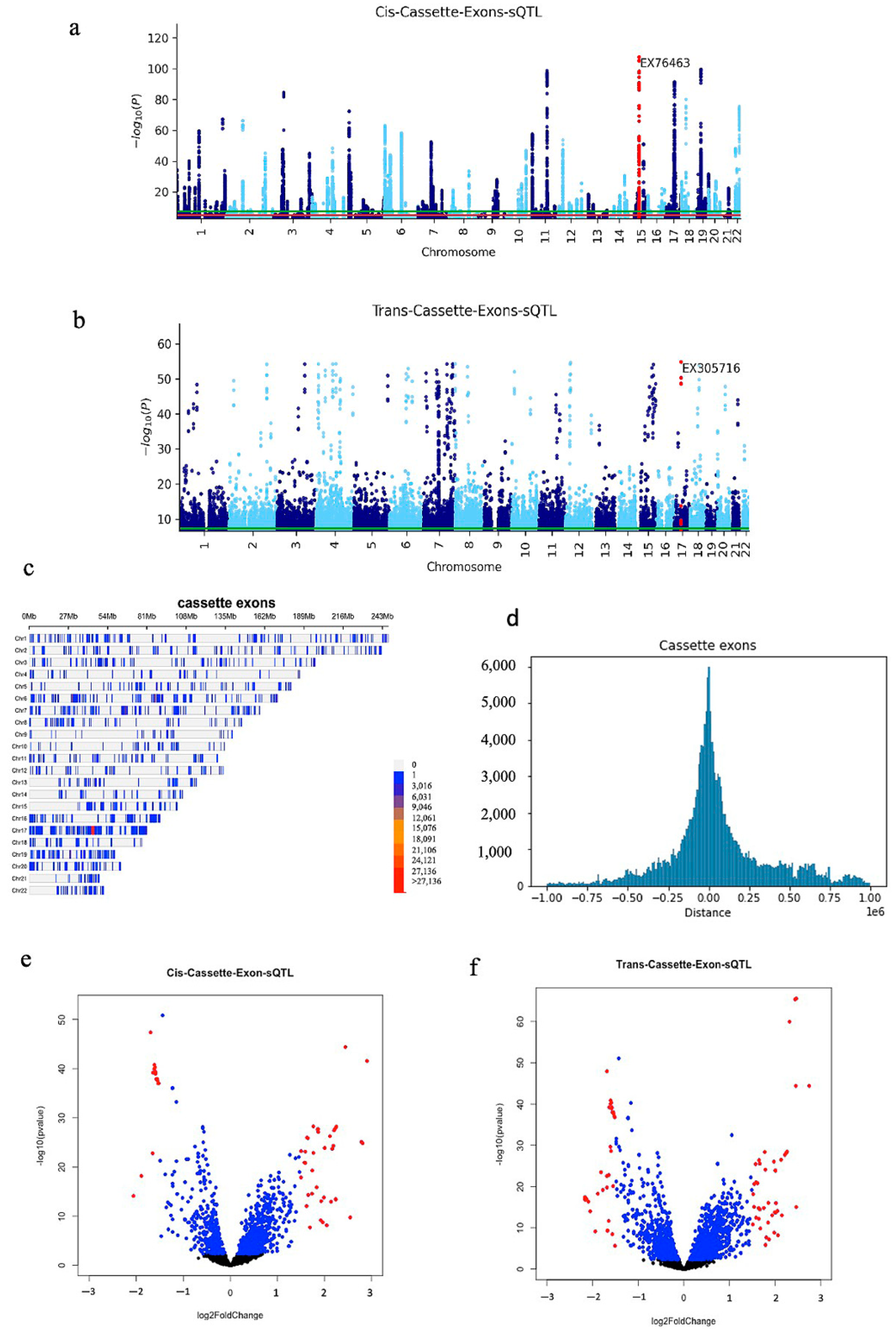
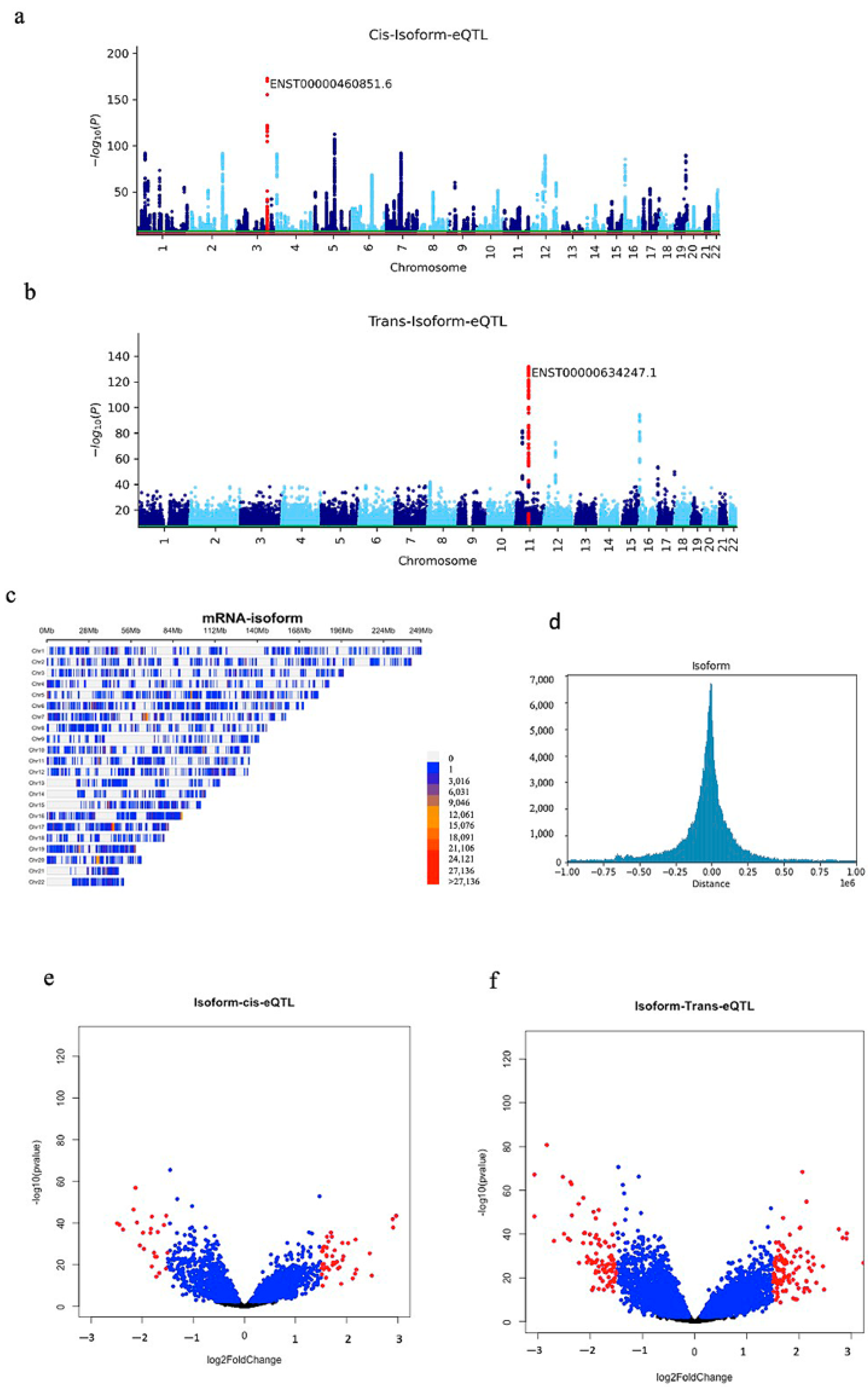
2.1. Identifying Iso-eQTLs and sQTL
2.2. Isoform-Structure Prediction
3. Discussion
4. Materials and Methods
4.1. Data Collection, Genotype Data pre-Processing and Imputation
4.2. Isoform Expression Analysis
4.3. Splicing Event Data
4.4. Iso-eQTL and sQTL Analysis
4.5. GWAS Related Iso-eQTL and sQTL
4.6. Isoform-Structure Prediction
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Xue, L.L.; Wang, F.; Xiong, L.L.; Du, R.L.; Zhou, H.L.; Zou, Y.; Wu, M.X.; Yang, M.A.; Dai, J.; He, M.X.; et al. A single-nucleotide polymorphism induced alternative splicing in Tacr3 involves in hypoxic-ischemic brain damage. Brain. Res. Bull. 2020, 154, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Monlong, J.; Calvo, M.; Ferreira, P.G.; Guigo, R. Identification of genetic variants associated with alternative splicing using sQTLseekeR. Nat. Commun. 2014, 5, 4698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernando, A.; Liyanage, C.; Moradi, A.; Janaththani, P.; Batra, J. Identification and Characterization of Alternatively Spliced Transcript Isoforms of IRX4 in Prostate Cancer. Genes 2021, 12, 615. [Google Scholar] [CrossRef] [PubMed]
- Herzel, L.; Ottoz, D.S.M.; Alpert, T.; Neugebauer, K.M. Splicing and transcription touch base: Co-transcriptional spliceosome assembly and function. Nat. Rev. Mol. Cell. Biol. 2017, 18, 637–650. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Martin, D.; Borsari, B.; Calvo, M.; Reverter, F.; Guigo, R. Identification and analysis of splicing quantitative trait loci across multiple tissues in the human genome. Nat. Commun. 2021, 12, 727. [Google Scholar] [CrossRef]
- Ellegren, H. Microsatellites: Simple sequences with complex evolution. Nat. Rev. Genet. 2004, 5, 435–445. [Google Scholar] [CrossRef]
- Brouwer, J.R.; Huguet, A.; Nicole, A.; Munnich, A.; Gourdon, G. Transcriptionally Repressive Chromatin Remodelling and CpG Methylation in the Presence of Expanded CTG-Repeats at the DM1 Locus. J. Nucleic Acids 2013, 2013, 567435. [Google Scholar] [CrossRef]
- Moradi, A.; Srinivasan, S.; Clements, J.; Batra, J. Beyond the biomarker role: Prostate-specific antigen (PSA) in the prostate cancer microenvironment. Cancer Metastasis Rev. 2019, 38, 333–346. [Google Scholar] [CrossRef] [PubMed]
- Hu, R.; Dunn, T.A.; Wei, S.; Isharwal, S.; Veltri, R.W.; Humphreys, E.; Han, M.; Partin, A.W.; Vessella, R.L.; Isaacs, W.B.; et al. Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res. 2009, 69, 16–22. [Google Scholar] [CrossRef] [Green Version]
- Hu, R.; Isaacs, W.B.; Luo, J. A snapshot of the expression signature of androgen receptor splicing variants and their distinctive transcriptional activities. Prostate 2011, 71, 1656–1667. [Google Scholar] [CrossRef]
- Yuan, X.; Cai, C.; Chen, S.; Chen, S.; Yu, Z.; Balk, S.P. Androgen receptor functions in castration-resistant prostate cancer and mechanisms of resistance to new agents targeting the androgen axis. Oncogene 2014, 33, 2815–2825. [Google Scholar] [CrossRef] [Green Version]
- Mashhadi, R.; Pourmand, G.; Kosari, F.; Mehrsai, A.; Salem, S.; Pourmand, M.R.; Alatab, S.; Khonsari, M.; Heydari, F.; Beladi, L.; et al. Role of steroid hormone receptors in formation and progression of bladder carcinoma: A case-control study. Urol. J. 2014, 11, 1968–1973. [Google Scholar]
- Tuygun, C.; Kankaya, D.; Imamoglu, A.; Sertcelik, A.; Zengin, K.; Oktay, M.; Sertcelik, N. Sex-specific hormone receptors in urothelial carcinomas of the human urinary bladder: A comparative analysis of clinicopathological features and survival outcomes according to receptor expression. Urol. Oncol. 2011, 29, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Van der Steen, T.; Tindall, D.J. Are androgen receptor variants a substitute for the full-length receptor? Nat. Rev. Urol. 2015, 12, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Antonarakis, E.S.; Lu, C.; Wang, H.; Luber, B.; Nakazawa, M.; Roeser, J.C.; Chen, Y.; Mohammad, T.A.; Chen, Y.; Fedor, H.L.; et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N. Engl. J. Med. 2014, 371, 1028–1038. [Google Scholar] [CrossRef] [Green Version]
- Farashi, S.; Kryza, T.; Clements, J.; Batra, J. Post-GWAS in prostate cancer: From genetic association to biological contribution. Nat. Rev. Cancer 2019, 19, 46–59. [Google Scholar] [CrossRef] [Green Version]
- Tian, J.; Wang, Z.; Mei, S.; Yang, N.; Yang, Y.; Ke, J.; Zhu, Y.; Gong, Y.; Zou, D.; Peng, X.; et al. CancerSplicingQTL: A database for genome-wide identification of splicing QTLs in human cancer. Nucleic Acids Res. 2019, 47, D909–D916. [Google Scholar] [CrossRef]
- Noble, J.D.; Balmant, K.M.; Dervinis, C.; de Los Campos, G.; Resende, M.F.R., Jr.; Kirst, M.; Barbazuk, W.B. The Genetic Regulation of Alternative Splicing in Populus deltoides. Front. Plant Sci. 2020, 11, 590. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Hu, Y.; Li, J.; Zhang, X. ulfasQTL: An ultra-fast method of composite splicing QTL analysis. BMC Genom. 2017, 18, 963. [Google Scholar] [CrossRef] [Green Version]
- Saferali, A.; Yun, J.H.; Parker, M.M.; Sakornsakolpat, P.; Chase, R.P.; Lamb, A.; Hobbs, B.D.; Boezen, M.H.; Dai, X.; de Jong, K.; et al. Analysis of genetically driven alternative splicing identifies FBXO38 as a novel COPD susceptibility gene. PLoS Genet. 2019, 15, e1008229. [Google Scholar] [CrossRef] [Green Version]
- Gibson, G.; Powell, J.E.; Marigorta, U.M. Expression quantitative trait locus analysis for translational medicine. Genome Med. 2015, 7, 60. [Google Scholar] [CrossRef] [PubMed]
- Shabalin, A.A. Matrix eQTL: Ultra fast eQTL analysis via large matrix operations. Bioinformatics 2012, 28, 1353–1358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chrifi, I.; Hermkens, D.; Brandt, M.M.; van Dijk, C.G.M.; Burgisser, P.E.; Haasdijk, R.; Pei, J.; van de Kamp, E.H.M.; Zhu, C.; Blonden, L.; et al. Cgnl1, an endothelial junction complex protein, regulates GTPase mediated angiogenesis. Cardiovasc. Res. 2017, 113, 1776–1788. [Google Scholar] [CrossRef] [Green Version]
- Nowsheen, S.; Aziz, K.; Luo, K.; Deng, M.; Qin, B.; Yuan, J.; Jeganathan, K.B.; Yu, J.; Zhang, H.; Ding, W.; et al. ZNF506-dependent positive feedback loop regulates H2AX signaling after DNA damage. Nat. Commun. 2018, 9, 2736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Guo, M.; Xia, X.; Zhang, C.; Zeng, Y.; Wu, S. XRRA1 Targets ATM/CHK1/2-Mediated DNA Repair in Colorectal Cancer. Biomed. Res. Int. 2017, 2017, 5718968, Erratum in Biomed. Res. Int. 2021, 2021, 3030267. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Forman, L.W.; Williams, R.M.; Faller, D.V. Protein kinase C-delta inactivation inhibits the proliferation and survival of cancer stem cells in culture and in vivo. BMC Cancer 2014, 14, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkat, M.; Bast, H.; Drees, R.; Dunser, J.; Mahr, A.; Azoitei, N.; Marienfeld, R.; Frank, F.; Brhel, M.; Ushmorov, A.; et al. Adenosine receptor 2B activity promotes autonomous growth, migration as well as vascularization of head and neck squamous cell carcinoma cells. Int. J. Cancer 2020, 147, 202–217. [Google Scholar] [CrossRef] [Green Version]
- Xue, Y.; Li, P.D.; Tang, X.M.; Yan, Z.H.; Xia, S.S.; Tian, H.P.; Liu, Z.L.; Zhou, T.; Tang, X.G.; Zhang, G.J. Cytochrome C Oxidase Assembly Factor 1 Homolog Predicts Poor Prognosis and Promotes Cell Proliferation in Colorectal Cancer by Regulating PI3K/AKT Signaling. Onco. Targets Ther. 2020, 13, 11505–11516. [Google Scholar] [CrossRef]
- Chunli, W.; Jiajie, H.; Lifei, W.; Beiqing, P.; Xin, X.; Yan, C.; Mingrong, W.; Xuemei, J. IGHMBP2 overexpression promotes cell migration and invasion in esophageal squamous carcinoma. Yi Chuan 2015, 37, 360–366. [Google Scholar] [CrossRef]
- Pan, Q.; Shai, O.; Lee, L.J.; Frey, B.J.; Blencowe, B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008, 40, 1413–1415. [Google Scholar] [CrossRef]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef]
- Wang, G.S.; Cooper, T.A. Splicing in disease: Disruption of the splicing code and the decoding machinery. Nat. Rev. Genet. 2007, 8, 749–761. [Google Scholar] [CrossRef] [PubMed]
- Caceres, J.F.; Kornblihtt, A.R. Alternative splicing: Multiple control mechanisms and involvement in human disease. Trends Genet. 2002, 18, 186–193. [Google Scholar] [CrossRef]
- Pickrell, J.K.; Marioni, J.C.; Pai, A.A.; Degner, J.F.; Engelhardt, B.E.; Nkadori, E.; Veyrieras, J.B.; Stephens, M.; Gilad, Y.; Pritchard, J.K. Understanding mechanisms underlying human gene expression variation with RNA sequencing. Nature 2010, 464, 768–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adamopoulos, P.G.; Kontos, C.K.; Scorilas, A. Discovery of novel transcripts of the human tissue kallikrein (KLK1) and kallikrein-related peptidase 2 (KLK2) in human cancer cells, exploiting Next-Generation Sequencing technology. Genomics 2019, 111, 642–652. [Google Scholar] [CrossRef] [PubMed]
- Marees, A.T.; de Kluiver, H.; Stringer, S.; Vorspan, F.; Curis, E.; Marie-Claire, C.; Derks, E.M. A tutorial on conducting genome-wide association studies: Quality control and statistical analysis. Int. J. Methods Psychiatr. Res. 2018, 27, e1608. [Google Scholar] [CrossRef] [Green Version]
- Das, S.; Forer, L.; Schonherr, S.; Sidore, C.; Locke, A.E.; Kwong, A.; Vrieze, S.I.; Chew, E.Y.; Levy, S.; McGue, M.; et al. Next-generation genotype imputation service and methods. Nat. Genet. 2016, 48, 1284–1287. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Jonassen, I. RASflow: An RNA-Seq analysis workflow with Snakemake. BMC Bioinform. 2020, 21, 110. [Google Scholar] [CrossRef] [Green Version]
- Tabrez, S.S.; Sharma, R.D.; Jain, V.; Siddiqui, A.A.; Mukhopadhyay, A. Differential alternative splicing coupled to nonsense-mediated decay of mRNA ensures dietary restriction-induced longevity. Nat. Commun. 2017, 8, 306. [Google Scholar] [CrossRef] [Green Version]
- Buniello, A.; MacArthur, J.A.L.; Cerezo, M.; Harris, L.W.; Hayhurst, J.; Malangone, C.; McMahon, A.; Morales, J.; Mountjoy, E.; Sollis, E.; et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019, 47, D1005–D1012. [Google Scholar] [CrossRef] [Green Version]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moradi, A.; Sharma, H.; Sharma, R.D.; Fernando, A.; Barrero, R.A.; Batra, J. Identification of Candidate mRNA Isoforms for Prostate Cancer-Risk SNPs Utilizing Iso-eQTL and sQTL Methods. Int. J. Mol. Sci. 2022, 23, 12406. https://doi.org/10.3390/ijms232012406
Moradi A, Sharma H, Sharma RD, Fernando A, Barrero RA, Batra J. Identification of Candidate mRNA Isoforms for Prostate Cancer-Risk SNPs Utilizing Iso-eQTL and sQTL Methods. International Journal of Molecular Sciences. 2022; 23(20):12406. https://doi.org/10.3390/ijms232012406
Chicago/Turabian StyleMoradi, Afshin, Harsh Sharma, Ravi Datta Sharma, Achala Fernando, Roberto A. Barrero, and Jyotsna Batra. 2022. "Identification of Candidate mRNA Isoforms for Prostate Cancer-Risk SNPs Utilizing Iso-eQTL and sQTL Methods" International Journal of Molecular Sciences 23, no. 20: 12406. https://doi.org/10.3390/ijms232012406