
In Vitro/In Vivo Translation of Synergistic Combination of MDM2 and MEK Inhibitors in Melanoma Using PBPK/PD Modelling: Part I

Abstract
:1. Introduction
2. Results
2.1. In Vitro Cytotoxicity
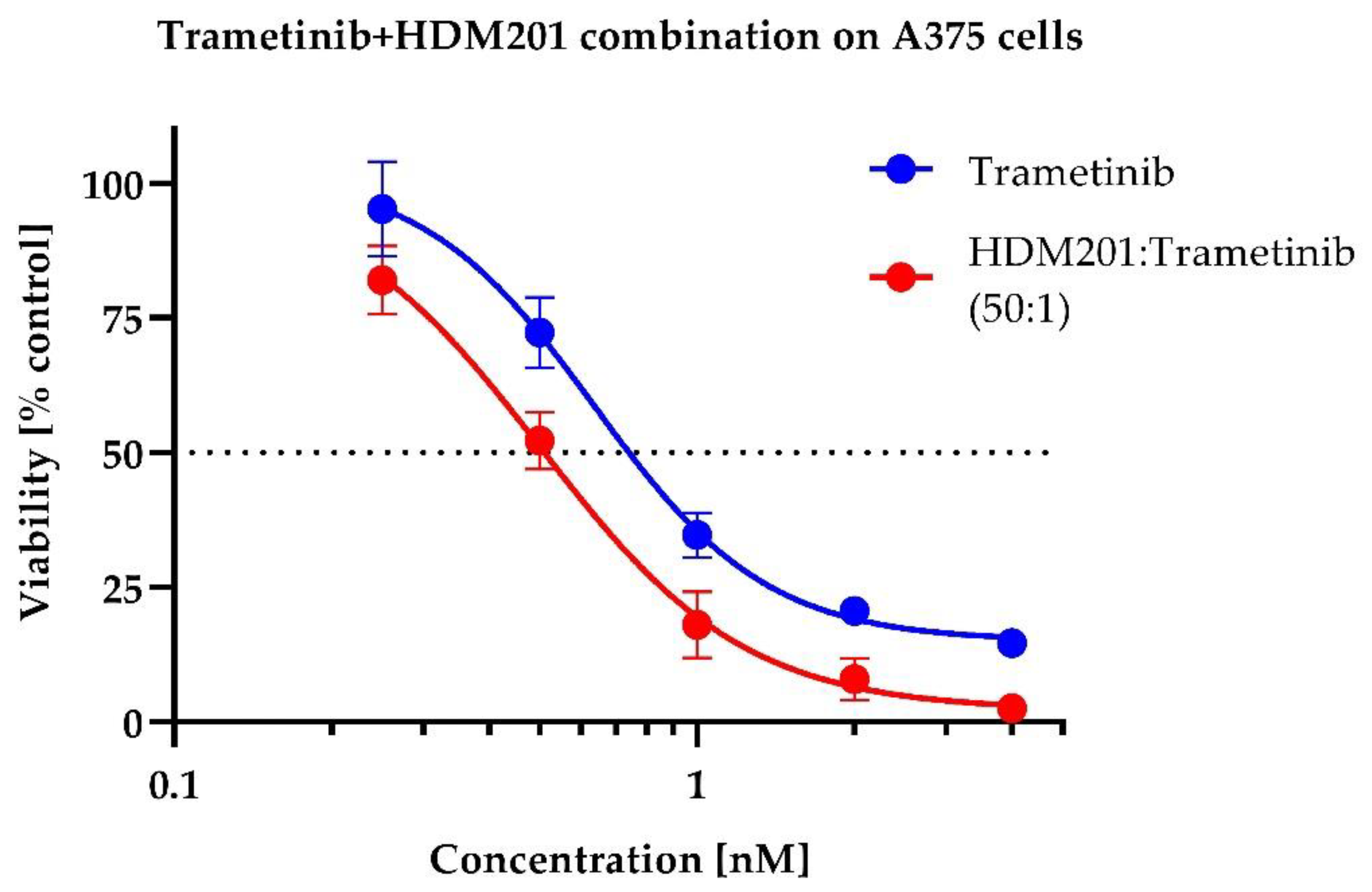
2.2. In Vitro Drug Combination Analysis
2.3. Siremadlin and Trametinib Pharmacokinetics (PK)
2.4. Siremadlin and Trametinib Pharmacodynamics (PD)
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Software
4.3. In Vitro Drug Combination Studies
4.4. Drug Combination Interaction Analysis
4.5. Studies Involving Animals
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Eroglu, Z.; Ribas, A. Combination Therapy with BRAF and MEK Inhibitors for Melanoma: Latest Evidence and Place in Therapy. Adv. Med. Oncol. 2016, 8, 48–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saiki, A.Y.; Caenepeel, S.; Yu, D.; Lofgren, J.A.; Osgood, T.; Robertson, R.; Canon, J.; Su, C.; Jones, A.; Zhao, X.; et al. MDM2 Antagonists Synergize Broadly and Robustly with Compounds Targeting Fundamental Oncogenic Signaling Pathways. Oncotarget 2014, 5, 2030–2043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.-E.; Koay, T.S.; Esfandiari, A.; Ho, Y.-H.; Lovat, P.; Lunec, J. ATM Dependent DUSP6 Modulation of P53 Involved in Synergistic Targeting of MAPK and P53 Pathways with Trametinib and MDM2 Inhibitors in Cutaneous Melanoma. Cancers 2018, 11, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caenepeel, S.; CANON, J.; Hughes, P.; Oliner, J.D.; Rickles, R.J.; Saiki, A.Y. Combination Therapy Including an MDM2 Inhibitor and One or More Additional Pharmaceutically Active Agents for the Treatment of Cancers. U.S. Patent 10,881,648, 5 January 2021. [Google Scholar]
- Cinatl, J.; Speidel, D.; Hardcastle, I.; Michaelis, M. Resistance Acquisition to MDM2 Inhibitors. Biochem. Soc. Trans. 2014, 42, 752–757. [Google Scholar] [CrossRef] [PubMed]
- Hoffman-Luca, C.G.; Yang, C.-Y.; Lu, J.; Ziazadeh, D.; McEachern, D.; Debussche, L.; Wang, S. Significant Differences in the Development of Acquired Resistance to the MDM2 Inhibitor SAR405838 between In Vitro and In Vivo Drug Treatment. PLoS ONE 2015, 10, e0128807. [Google Scholar] [CrossRef] [Green Version]
- Tripathi, R.; Liu, Z.; Jain, A.; Lyon, A.; Meeks, C.; Richards, D.; Liu, J.; He, D.; Wang, C.; Nespi, M.; et al. Combating Acquired Resistance to MAPK Inhibitors in Melanoma by Targeting Abl1/2-Mediated Reactivation of MEK/ERK/MYC Signaling. Nat. Commun. 2020, 11, 5463. [Google Scholar] [CrossRef]
- Berberich, A.; Kessler, T.; Thomé, C.M.; Pusch, S.; Hielscher, T.; Sahm, F.; Oezen, I.; Schmitt, L.-M.; Ciprut, S.; Hucke, N.; et al. Targeting Resistance against the MDM2 Inhibitor RG7388 in Glioblastoma Cells by the MEK Inhibitor Trametinib. Clin. Cancer Res. 2019, 25, 253–265. [Google Scholar] [CrossRef] [Green Version]
- Shangary, S.; Qin, D.; McEachern, D.; Liu, M.; Miller, R.S.; Qiu, S.; Nikolovska-Coleska, Z.; Ding, K.; Wang, G.; Chen, J.; et al. Temporal Activation of P53 by a Specific MDM2 Inhibitor Is Selectively Toxic to Tumours and Leads to Complete Tumour Growth Inhibition. Proc. Natl. Acad. Sci. USA 2008, 105, 3933–3938. [Google Scholar] [CrossRef] [Green Version]
- De la Puente, P.; Muz, B.; Jin, A.; Azab, F.; Luderer, M.; Salama, N.N.; Azab, A.K. MEK Inhibitor, TAK-733 Reduces Proliferation, Affects Cell Cycle and Apoptosis, and Synergizes with Other Targeted Therapies in Multiple Myeloma. Blood Cancer J. 2016, 6, e399. [Google Scholar] [CrossRef] [Green Version]
- Moschos, S.J.; Sandhu, S.K.; Lewis, K.D.; Sullivan, R.J.; Johnson, D.B.; Zhang, Y.; Rasmussen, E.; Henary, H.A.; Long, G.V. Phase 1 Study of the P53-MDM2 Inhibitor AMG 232 Combined with Trametinib plus Dabrafenib or Trametinib in Patients (Pts) with TP53 Wild Type (TP53WT) Metastatic Cutaneous Melanoma (MCM). J. Clin. Oncol. 2017, 35, 2575. [Google Scholar] [CrossRef]
- Greco, W.; Unkelbach, H.-D.; Pöch, G.; Sühnel, J.; Kundi, M.; Bödeker, W. Consensus on Concepts and Terminology for Combined-Action Assessment: The Saariselkä Agreement. Arch. Complex Environ. Stud. 1992, 4, 65–69. [Google Scholar]
- Roell, K.R.; Reif, D.M.; Motsinger-Reif, A.A. An Introduction to Terminology and Methodology of Chemical Synergy—Perspectives from Across Disciplines. Front. Pharm. 2017, 8, 158. [Google Scholar] [CrossRef] [Green Version]
- Loewe, S. The Problem of Synergism and Antagonism of Combined Drugs. Arzneimittelforschung 1953, 3, 285–290. [Google Scholar]
- Bliss, C.I. The Toxicity of Poisons Applied Jointly1. Ann. Appl. Biol. 1939, 26, 585–615. [Google Scholar] [CrossRef]
- Berenbaum, M.C. What Is Synergy? Pharm. Rev. 1989, 41, 93–141. [Google Scholar]
- Yadav, B.; Wennerberg, K.; Aittokallio, T.; Tang, J. Searching for Drug Synergy in Complex Dose–Response Landscapes Using an Interaction Potency Model. Comput. Struct. Biotechnol. J. 2015, 13, 504–513. [Google Scholar] [CrossRef] [Green Version]
- Meyer, C.T.; Wooten, D.J.; Paudel, B.B.; Bauer, J.; Hardeman, K.N.; Westover, D.; Lovly, C.M.; Harris, L.A.; Tyson, D.R.; Quaranta, V. Quantifying Drug Combination Synergy along Potency and Efficacy Axes. Cell Syst. 2019, 8, 97–108.e16. [Google Scholar] [CrossRef] [Green Version]
- Tang, J.; Wennerberg, K.; Aittokallio, T. What Is Synergy? The Saariselkä Agreement Revisited. Front. Pharm. 2015, 6, 181. [Google Scholar] [CrossRef]
- Cokol, M.; Chua, H.N.; Tasan, M.; Mutlu, B.; Weinstein, Z.B.; Suzuki, Y.; Nergiz, M.E.; Costanzo, M.; Baryshnikova, A.; Giaever, G.; et al. Systematic Exploration of Synergistic Drug Pairs. Mol. Syst. Biol. 2011, 7, 544. [Google Scholar] [CrossRef]
- Sen, P.; Saha, A.; Dixit, N.M. You Cannot Have Your Synergy and Efficacy Too. Trends Pharmacol. Sci. 2019, 40, 811–817. [Google Scholar] [CrossRef] [Green Version]
- Ianevski, A.; He, L.; Aittokallio, T.; Tang, J. SynergyFinder: A Web Application for Analyzing Drug Combination Dose–Response Matrix Data. Bioinformatics 2017, 33, 2413–2415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wooten, D.J.; Albert, R. Synergy: A Python Library for Calculating, Analyzing and Visualizing Drug Combination Synergy. Bioinformatics 2021, 37, 1473–1474. [Google Scholar] [CrossRef] [PubMed]
- Di Veroli, G.Y.; Fornari, C.; Wang, D.; Mollard, S.; Bramhall, J.L.; Richards, F.M.; Jodrell, D.I. Combenefit: An Interactive Platform for the Analysis and Visualization of Drug Combinations. Bioinformatics 2016, 32, 2866–2868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehàr, J.; Krueger, A.S.; Avery, W.; Heilbut, A.M.; Johansen, L.M.; Price, E.R.; Rickles, R.J.; Short, G.F.; Staunton, J.E.; Jin, X.; et al. Synergistic Drug Combinations Improve Therapeutic Selectivity. Nat. Biotechnol. 2009, 27, 659–666. [Google Scholar] [CrossRef] [PubMed]
- Malyutina, A.; Majumder, M.M.; Wang, W.; Pessia, A.; Heckman, C.A.; Tang, J. Drug Combination Sensitivity Scoring Facilitates the Discovery of Synergistic and Efficacious Drug Combinations in Cancer. PLoS Comput. Biol. 2019, 15, e1006752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wooten, D.J.; Meyer, C.T.; Lubbock, A.L.R.; Quaranta, V.; Lopez, C.F. MuSyC Is a Consensus Framework That Unifies Multi-Drug Synergy Metrics for Combinatorial Drug Discovery. Nat. Commun. 2021, 12, 4607. [Google Scholar] [CrossRef]
- Wooten, D. MuSyC Model Description (Github). Available online: https://github.com/djwooten/synergy/blob/b13e2ac961f8738d58c01f79e5b93ec2dd78f555/src/synergy/combination/musyc.py (accessed on 27 May 2022).
- Meyer, C.T.; Wooten, D.J.; Lopez, C.F.; Quaranta, V. Charting the Fragmented Landscape of Drug Synergy. Trends Pharmacol. Sci. 2020, 41, 266–280. [Google Scholar] [CrossRef]
- Day, D.; Siu, L.L. Approaches to Modernize the Combination Drug Development Paradigm. Genome Med. 2016, 8, 115. [Google Scholar] [CrossRef] [Green Version]
- Zagidullin, B.; Aldahdooh, J.; Zheng, S.; Wang, W.; Wang, Y.; Saad, J.; Malyutina, A.; Jafari, M.; Tanoli, Z.; Pessia, A.; et al. DrugComb: An Integrative Cancer Drug Combination Data Portal. Nucleic Acids Res. 2019, 47, W43–W51. [Google Scholar] [CrossRef] [Green Version]
- Jeay, S.; Ferretti, S.; Holzer, P.; Fuchs, J.; Chapeau, E.A.; Wartmann, M.; Sterker, D.; Romanet, V.; Murakami, M.; Kerr, G.; et al. Dose and Schedule Determine Distinct Molecular Mechanisms Underlying the Efficacy of the P53–MDM2 Inhibitor HDM201. Cancer Res. 2018, 78, 6257–6267. [Google Scholar] [CrossRef] [Green Version]
- Jing, J.; Greshock, J.; Holbrook, J.D.; Gilmartin, A.; Zhang, X.; McNeil, E.; Conway, T.; Moy, C.; Laquerre, S.; Bachman, K.; et al. Comprehensive Predictive Biomarker Analysis for MEK Inhibitor GSK1120212. Mol. Cancer Ther. 2012, 11, 720–729. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Zhu, Q.; Chen, M.; Huang, Q.; Wang, W.; Li, Q.; Huang, Y.; Di, W. The Changing 50% Inhibitory Concentration (IC 50) of Cisplatin: A Pilot Study on the Artifacts of the MTT Assay and the Precise Measurement of Density-Dependent Chemoresistance in Ovarian Cancer. Oncotarget 2016, 7, 70803–70821. [Google Scholar] [CrossRef] [Green Version]
- Howard, G.R.; Johnson, K.E.; Rodriguez Ayala, A.; Yankeelov, T.E.; Brock, A. A Multi-State Model of Chemoresistance to Characterize Phenotypic Dynamics in Breast Cancer. Sci. Rep. 2018, 8, 12058. [Google Scholar] [CrossRef]
- Yoon, N.; Vander Velde, R.; Marusyk, A.; Scott, J.G. Optimal Therapy Scheduling Based on a Pair of Collaterally Sensitive Drugs. Bull. Math. Biol. 2018, 80, 1776–1809. [Google Scholar] [CrossRef]
- Drummond, C.J.; Esfandiari, A.; Liu, J.; Lu, X.; Hutton, C.; Jackson, J.; Bennaceur, K.; Xu, Q.; Makimanejavali, A.R.; Bello, F.D.; et al. TP53 Mutant MDM2-Amplified Cell Lines Selected for Resistance to MDM2-P53 Binding Antagonists Retain Sensitivity to Ionizing Radiation. Oncotarget 2016, 7, 46203–46218. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.-E.; Koay, T.S.; Ho, Y.-H.; Lovat, P.; Lunec, J. Abstract 3034: TP53 Mutant Cell Lines Selected for Resistance to MDM2 Inhibitors Retain Growth Inhibition by MAPK Pathway Inhibitors but a Reduced Apoptotic Response. Cancer Res. 2019, 79, 3034. [Google Scholar] [CrossRef]
- Deben, C.; Boullosa, L.F.; Domen, A.; Wouters, A.; Cuypers, B.; Laukens, K.; Lardon, F.; Pauwels, P. Characterization of Acquired Nutlin-3 Resistant Non-Small Cell Lung Cancer Cells. Cancer Drug Resist. 2021, 4, 233–243. [Google Scholar] [CrossRef]
- Michaelis, M.; Rothweiler, F.; Schneider, C.; Rothenburger, T.; Mernberger, M.; Nist, A.; Stiewe, T.; Cinatl, J.; Nevels, M. Long-Term Cultivation Using Ineffective MDM2 Inhibitor Concentrations Alters the Drug Sensitivity Profiles of PL21 Leukaemia Cells. Exp. Results 2020, 1, e5. [Google Scholar] [CrossRef] [Green Version]
- Poulikakos, P.I.; Persaud, Y.; Janakiraman, M.; Kong, X.; Ng, C.; Moriceau, G.; Shi, H.; Atefi, M.; Titz, B.; Gabay, M.T.; et al. RAF Inhibitor Resistance Is Mediated by Dimerization of Aberrantly Spliced BRAF(V600E). Nature 2011, 480, 387–390. [Google Scholar] [CrossRef] [Green Version]
- Basile, K.J.; Abel, E.V.; Dadpey, N.; Hartsough, E.J.; Fortina, P.; Aplin, A.E. In Vivo MAPK Reporting Reveals the Heterogeneity in Tumoural Selection of Resistance to RAF Inhibitors. Cancer Res. 2013, 73, 7101–7110. [Google Scholar] [CrossRef] [Green Version]
- Witkowski, J.; Polak, S.; Rogulski, Z.; Pawelec, D. In Vitro/In Vivo Translation of Synergistic Combination of MDM2 and MEK Inhibitors in Melanoma Using PBPK/PD Modelling: Part II. Int. J. Mol. Sci. 2022, 23, 11939. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Tang, J. Synergyfinder Package Documentation. Available online: https://bioconductor.org/packages/release/bioc/manuals/synergyfinder/man/synergyfinder.pdf (accessed on 13 June 2022).
- Curry, S.H.; Whelpton, R. Appendix 1: Mathematical Concepts and the Trapezoidal Method. In Introduction to Drug Disposition and Pharmacokinetics; John Wiley & Sons: Hoboken, NJ, USA, 2016; pp. 293–299. ISBN 978-1-119-26108-7. [Google Scholar]
- Jensen, M.M.; Jørgensen, J.T.; Binderup, T.; Kjær, A. Tumour Volume in Subcutaneous Mouse Xenografts Measured by MicroCT Is More Accurate and Reproducible than Determined by 18F-FDG-MicroPET or External Caliper. BMC Med. Imaging 2008, 8, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hather, G.; Liu, R.; Bandi, S.; Mettetal, J.; Manfredi, M.; Shyu, W.-C.; Donelan, J.; Chakravarty, A. Growth Rate Analysis and Efficient Experimental Design for Tumour Xenograft Studies. Cancer Inf. 2014, 13, 65–72. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Description | δ Score Value |
|---|---|
| Antagonism | ≤−5 |
| Additivity | (−5; 5) |
| Synergism | ≥5 |
| Description | α12/α21 Score Value | β Score Value | γ12/γ21 Score Value |
|---|---|---|---|
| Antagonism | <1 | <0 | <1 |
| Additivity | 1 | 0 | 1 |
| Synergism | >1 | >0 | >1 |
| Compound | MTS IC50 ± SD (nM) | RealTime-Glo IC50 ± SD (nM) | Literature IC50 (nM) |
|---|---|---|---|
| Siremadlin (HDM201) | 65.7 ± 4.7 | 260.1 ± 170.5 | 764.1 [32] 1 |
| Trametinib | 0.58 ± 0.03 | 0.8 ± 0.4 | 1.0 [33] 2 |
| Assay | Timepoints (h) | ZIP δ ± SD | Loewe δ ± SD | HSA δ ± SD | Bliss δ ± SD | Mean across Models δ ± SD |
|---|---|---|---|---|---|---|
| MTS | 72 | 5.353 ± 2.613 | 5.111 ± 1.926 | 12.394 ± 2.085 | 4.881 ± 3.117 | 6.935 ± 2.420 |
| RealTime-Glo | 28–80 | 4.858 ± 1.346 | 7.113 ± 4.355 | 13.513 ± 3.111 | 5.540 ± 1.957 | 7.756 ± 1.614 |
| Mean | - | 5.023 ± 1.768 | 6.446 ± 3.546 | 13.140 ± 2.769 | 5.321 ± 2.344 | 7.482 ± 1.883 |
| Assay | Timepoints (h) | α12/α21 ± SD | β ± SD | γ12/γ21 ± SD |
|---|---|---|---|---|
| MTS | 72 | 2.229 ± 1.065/ 1.498 ± 0.351 | 0.217 ± 0.045 | 0.402 ± 0.102/ 0.710 ± 0.286 |
| RealTime-Glo | 28–80 | 2.095 ± 0.780/ 12,507 ± 26,999 | 0.244 ± 0.050 | 0.901 ± 0.136/ 6878 ± 21,748 |
| Mean | 2.162 ± 0.923/ 6254 ± 13,500 | 0.231 ± 0.048 | 0.652 ± 0.119/ 3440 ± 10,874 |
| Conditions | Tissue | AUC0–24h ± SD (nM × h) | Cmax ± SD (nM) | Tmax ± SD (h) |
|---|---|---|---|---|
| HDM201 without Trametinib | Plasma | 95,092.97 ± 34,215.83 | 9777.67 ± 2976.84 | 1.50 ± 1.44 |
| HDM201 with Trametinib | Plasma | 107,993.98 ± 26,303.00 | 14,559.95 ± 7433.26 | 1.50 ± 1.44 |
| Trametinib without HDM201 | Plasma | 5580.83 ± 566.66 | 567.02 ± 49.38 | 4.00 ± 1.44 |
| Trametinib with HDM201 | Plasma | 4484.99 ± 1171.06 | 353.65 ± 105.55 | 4.00 ± 1.44 |
| HDM201 without Trametinib | A375 tumour | 179,026.48 ± 65,901.61 | 16,214.30 ± 5459.78 | 1.50 ± 1.44 |
| HDM201 with Trametinib | A375 tumour | 218,677.07 ± 91,168.31 | 28,613.74 ± 16,751.20 | 1.50 ± 1.44 |
| Trametinib without HDM201 | A375 tumour | 9131.17 ± 1296.84 | 587.25 ± 66.35 | 4.00 ± 0.00 |
| Trametinib with HDM201 | A375 tumour | 9656.67 ± 1393.80 | 714.53 ± 197.48 | 4.00 ± 0.00 |
| Group | Max TGI (%) ± SEM |
|---|---|
| HDM201 40 mg/kg qdx3 | 33.39 ± 13.90 |
| HDM201 100 mg/kg qdx3 | 76.94 ± 5.38 |
| Trametinib 0.3 mg/kg qdx6 | 65.47 ± 21.29 |
| Trametinib 1 mg/kg qdx6 | 90.05 ± 1.13 |
| HDM201 + Trametinib 40 + 0.3 mg/kg qdx3/qdx6 | 91.83 ± 1.37 |
| HDM201 + Trametinib 40 + 1 mg/kg qdx3/qdx6 | 93.68 ± 1.63 |
| HDM201 + Trametinib 100 + 0.3 mg/kg qdx3/qdx6 | 94.56 ± 1.77 |
| HDM201 + Trametinib 100 + 1 mg/kg qdx3/qdx6 | 95.99 ± 0.84 |
| Compound | Initial Tumour Volume (mm3) | Doses (mg/kg) | Dose Schedule | N | Comments |
|---|---|---|---|---|---|
| Vehicle | ~162 | - | qdx6 | 11 | Efficacy |
| Siremadlin | ~163–172 | 40/100 | qdx3 | 6 | Efficacy |
| Trametinib | ~167–180 | 0.3/1 | qdx6 | 6 | Efficacy |
| Siremadlin+ Trametinib | ~165–169 | 40 + 0.3/40 + 1/ 100 + 0.3/100 + 1 | qdx3/qdx6 | 6 | Efficacy |
| Siremadlin | ~300 | 100 | qdx1 | 12 | PK |
| Trametinib | ~300 | 1 | qdx1 | 12 | PK |
| Siremadlin+ Trametinib | ~300 | 100 + 1 | qdx1 | 12 | PK |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Witkowski, J.; Polak, S.; Rogulski, Z.; Pawelec, D. In Vitro/In Vivo Translation of Synergistic Combination of MDM2 and MEK Inhibitors in Melanoma Using PBPK/PD Modelling: Part I. Int. J. Mol. Sci. 2022, 23, 12984. https://doi.org/10.3390/ijms232112984
Witkowski J, Polak S, Rogulski Z, Pawelec D. In Vitro/In Vivo Translation of Synergistic Combination of MDM2 and MEK Inhibitors in Melanoma Using PBPK/PD Modelling: Part I. International Journal of Molecular Sciences. 2022; 23(21):12984. https://doi.org/10.3390/ijms232112984
Chicago/Turabian StyleWitkowski, Jakub, Sebastian Polak, Zbigniew Rogulski, and Dariusz Pawelec. 2022. "In Vitro/In Vivo Translation of Synergistic Combination of MDM2 and MEK Inhibitors in Melanoma Using PBPK/PD Modelling: Part I" International Journal of Molecular Sciences 23, no. 21: 12984. https://doi.org/10.3390/ijms232112984