
Targeted Therapies for Hepatocellular Carcinoma Treatment: A New Era Ahead—A Systematic Review
 ,
,  , , , , ,
, , , , ,
Abstract
:1. Introduction
1.1. Liver Lobule: The Organization of Hepatic Parenchyma
1.2. Staging Systems
1.3. Expression of Growth Factors
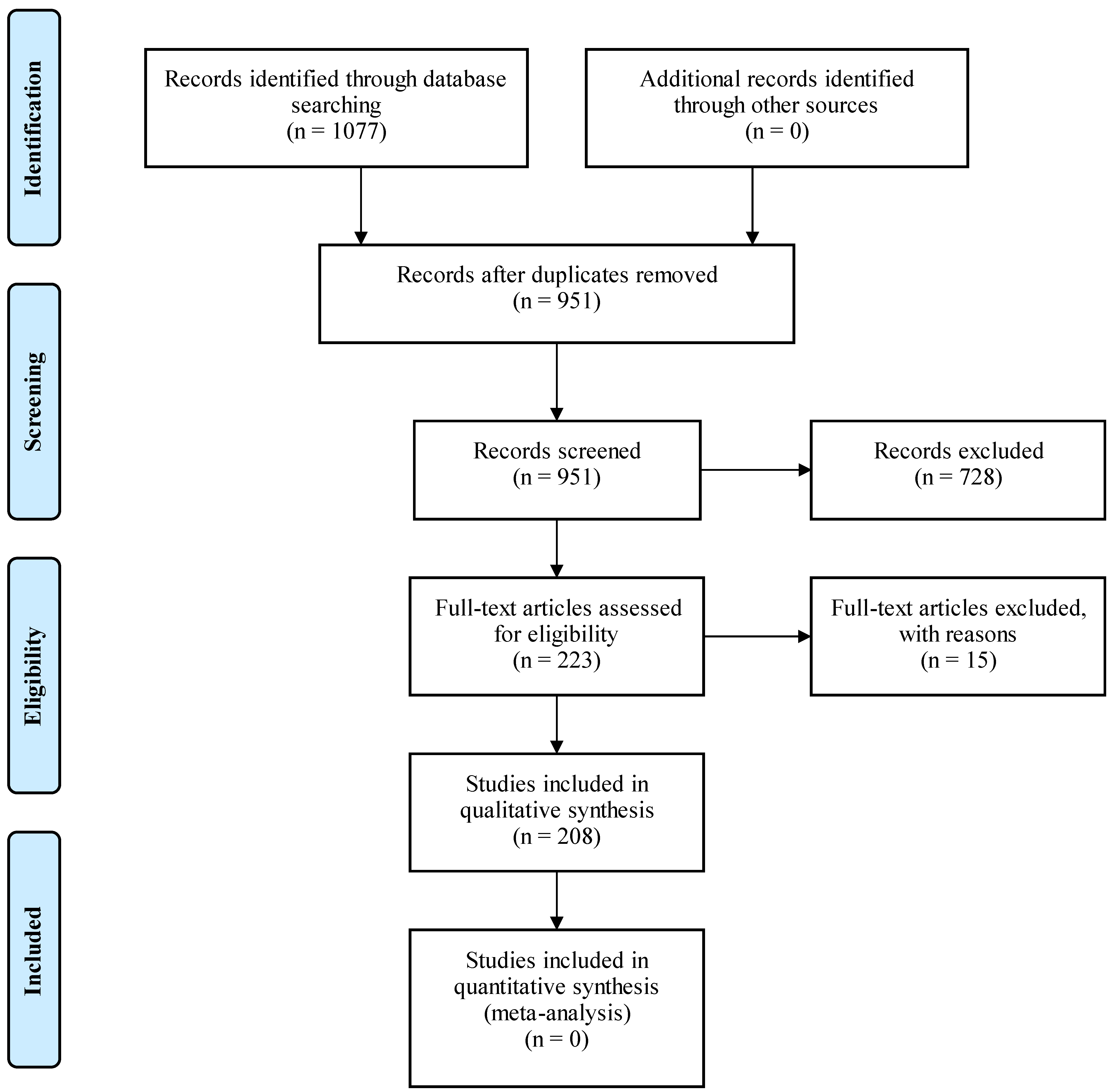
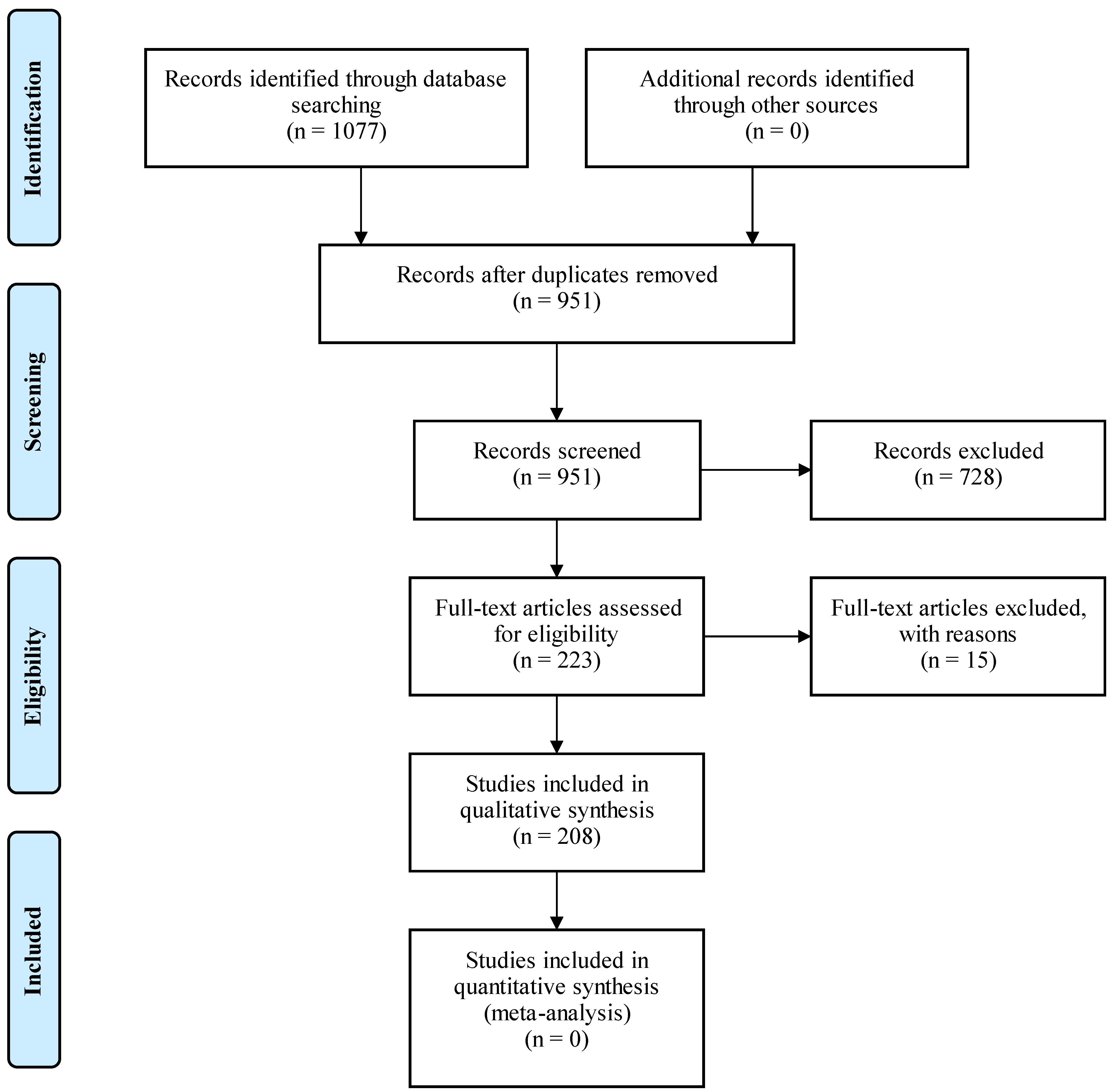
2. Materials and Methods
3. Results
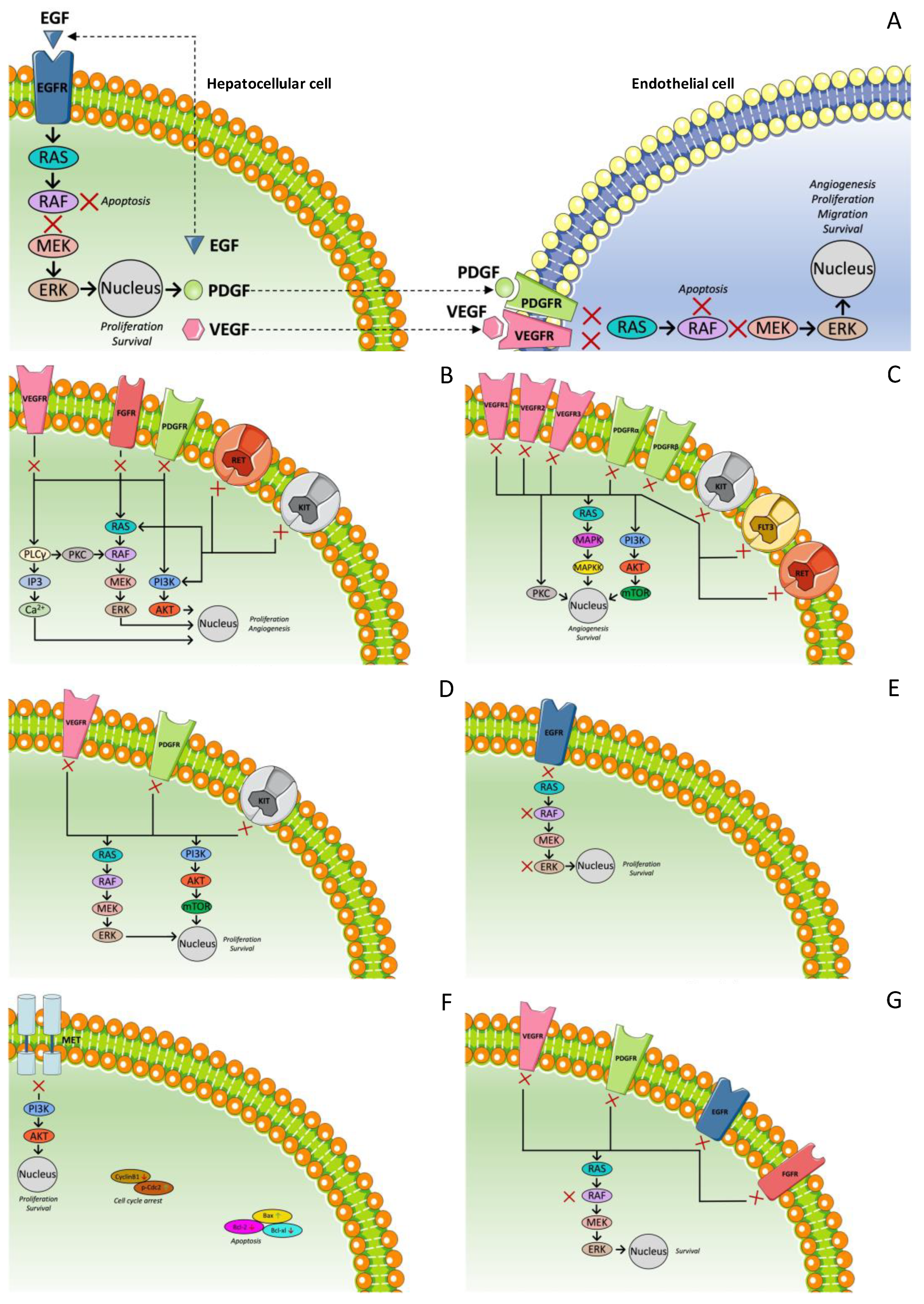
3.1. Multi-Targeted Tyrosine Kinase Inhibitors: First-Line Treatment
3.1.1. Sorafenib
3.1.2. Lenvatinib
3.1.3. Sunitinib
3.1.4. Linifanib
3.1.5. Erlotinib
3.1.6. Foretinib
3.1.7. Donafenib

3.2. Multi-Targeted Tyrosine Kinase Inhibitors: Second-Line Treatment
3.2.1. Regorafenib
3.2.2. Cabozantinib
3.2.3. Tivantinib
3.2.4. Axitinib
3.2.5. Anlotinib
3.2.6. Tepotinib

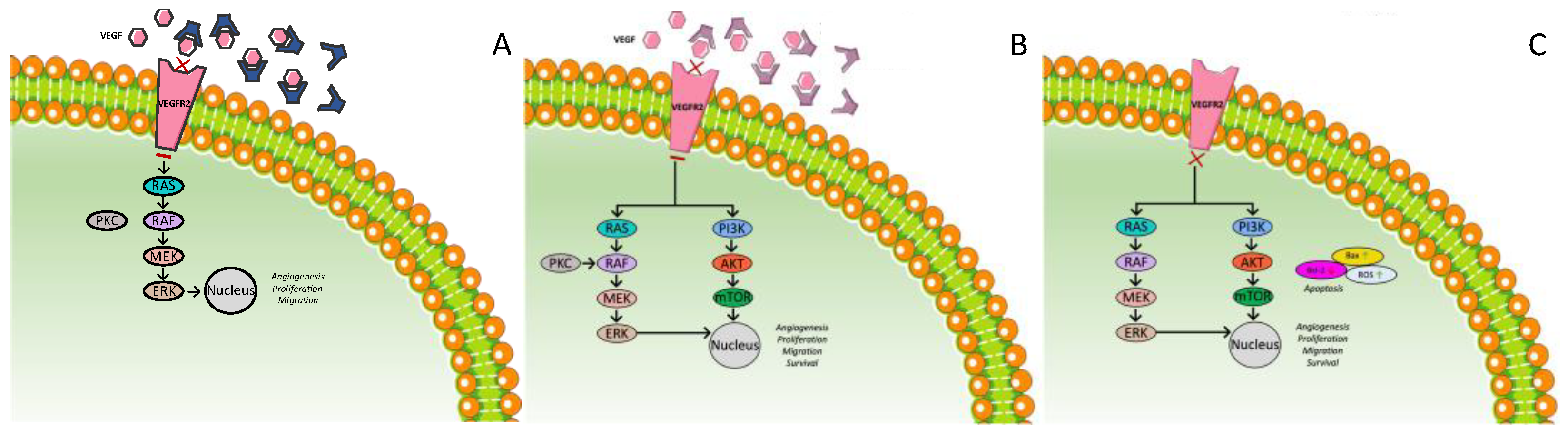
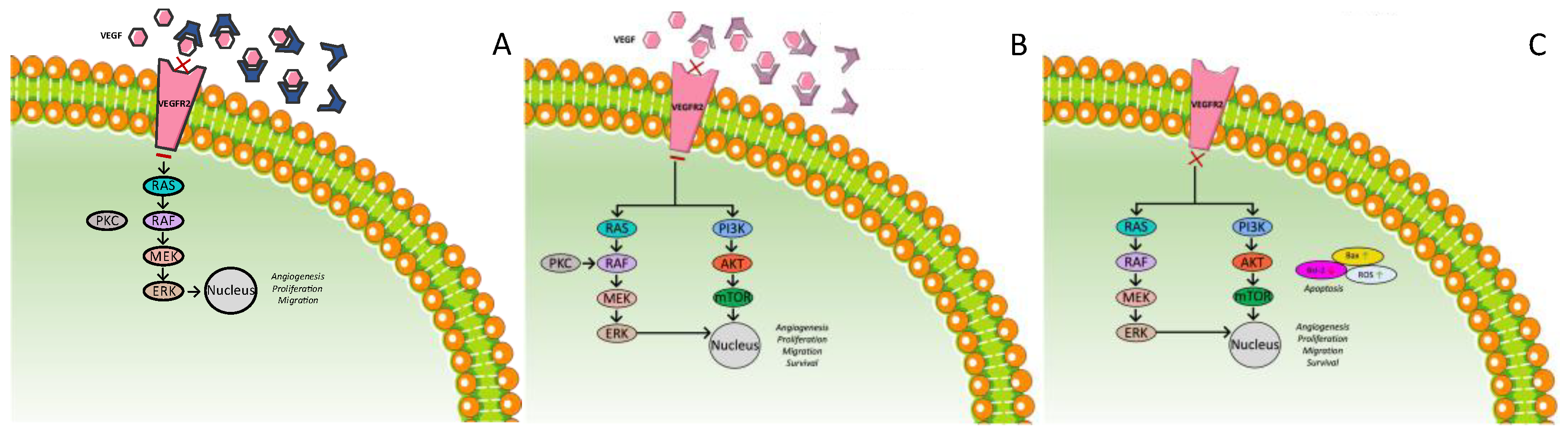
3.3. Anti-VEGF Therapies
3.3.1. Ramucirumab
3.3.2. Bevacizumab
3.3.3. Apatinib
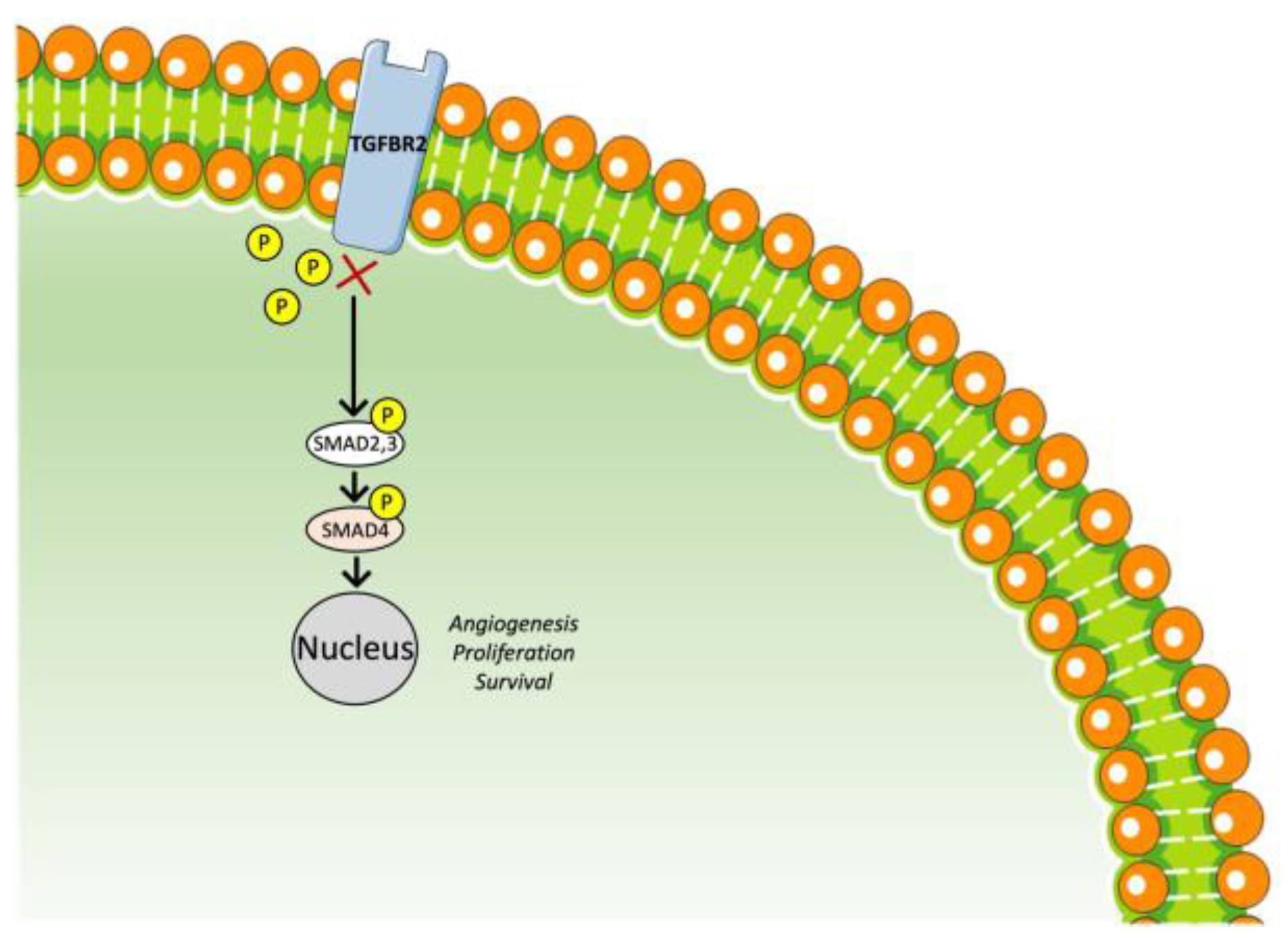
3.4. TGF-β Receptor Inhibitor
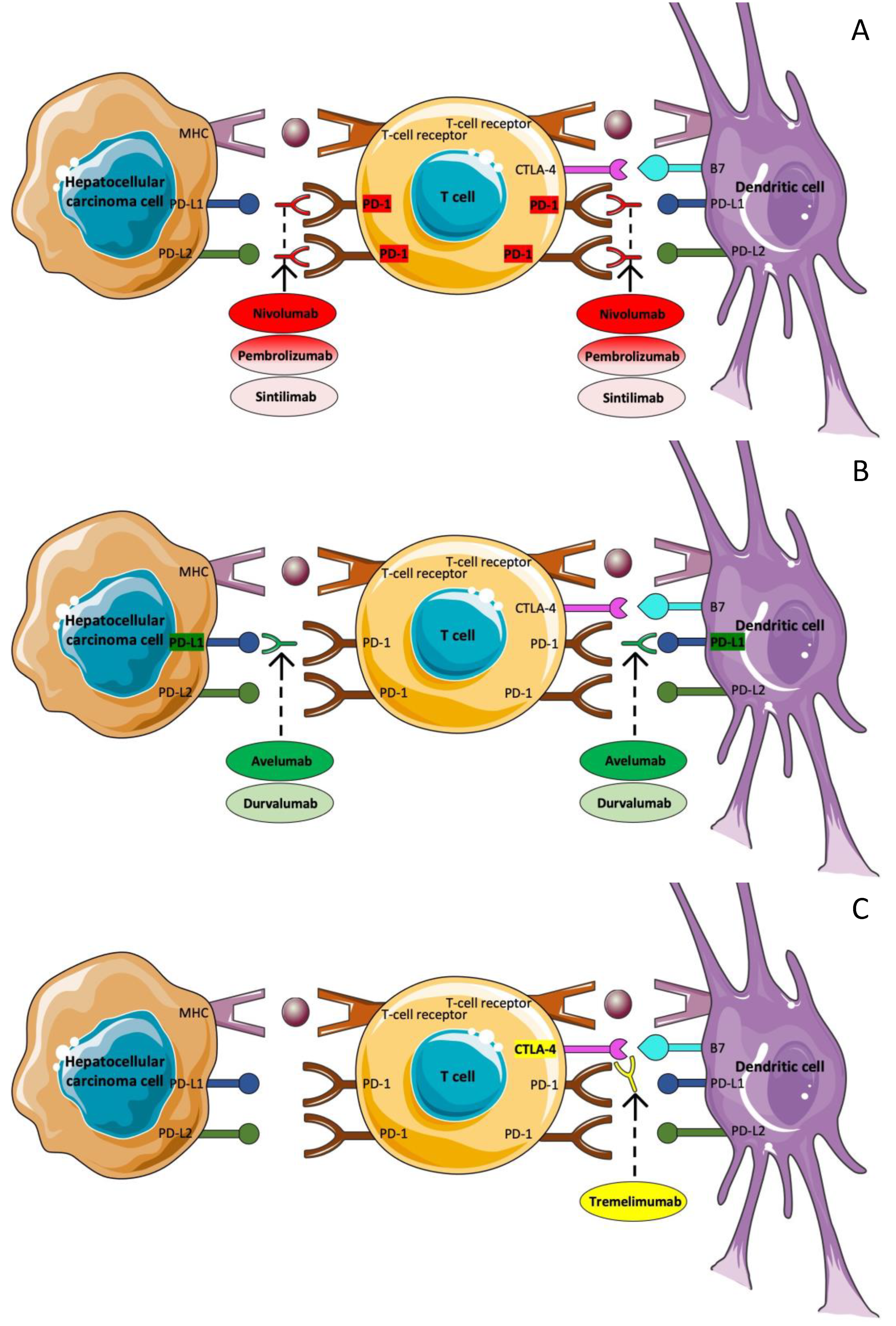
3.5. Immunotherapy
3.5.1. Nivolumab
3.5.2. Pembrolizumab
3.5.3. Avelumab
3.5.4. Tremelimumab plus Durvalumab
3.5.5. Sintilimab

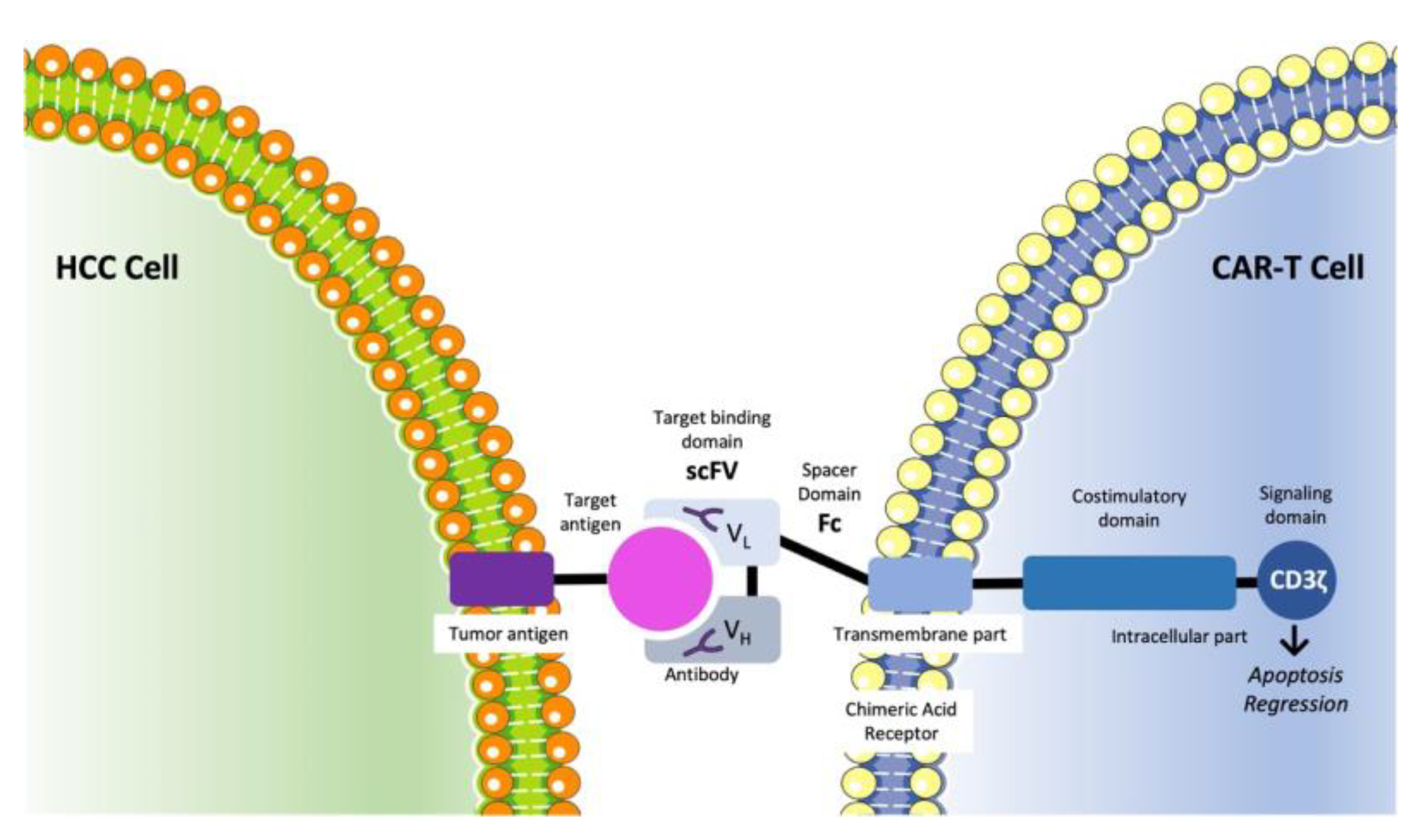
3.6. CAR-T Cell Therapy
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Dhanasekaran, R.; Limaye, A.; Cabrera, R. Hepatocellular carcinoma: Current trends in worldwide epidemiology, risk factors, diagnosis, and therapeutics. Hepat. Med. 2012, 8, 19–37. [Google Scholar]
- McGlynn, K.A.; London, W.T. The global epidemiology of hepatocellular carcinoma: Present and future. Clin. Liver. Dis. 2011, 15, 223–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, P.; Xie, S.H.; Hu, S.; Cheng, X.; Gao, T.; Zhang, C.; Song, Z. Age-specific sex difference in the incidence of hepatocellular carcinoma in the United States. Oncotarget 2017, 8, 68131–68137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dimitroulis, D.; Damaskos, C.; Valsami, S.; Davakis, S.; Garmpis, N.; Spartalis, E.; Athanasiou, A.; Moris, D.; Sakellariou, S.; Kykalos, S.; et al. From diagnosis to treatment of hepatocellular carcinoma: An epidemic problem for both developed and developing world. World J. Gastroenterol. 2017, 23, 5282–5294. [Google Scholar] [CrossRef] [PubMed]
- Damaskos, C.; Kaskantamis, A.; Garmpis, N.; Dimitroulis, D.; Mantas, D.; Garmpi, A.; Sakellariou, S.; Angelou, A.; Syllaios, A.; Kostakis, A.; et al. Intensive care unit outcomes following orthotopic liver transplantation: Single-center experience and review of the literature. G. Chir. 2019, 40, 463–480. [Google Scholar] [PubMed]
- Harris, P.S.; Hansen, R.M.; Gray, M.E.; Massoud, O.I.; McGuire, B.M.; Shoreibah, M.G. Hepatocellular carcinoma surveillance: An evidence-based approach. World J. Gastroenterol. 2019, 25, 1550–1559. [Google Scholar] [CrossRef]
- Zakharia, K.; Luther, C.A.; Alsabbak, H.; Roberts, L.R. Hepatocellular carcinoma: Epidemiology, pathogenesis and surveillance—Implications for sub-Saharan Africa. S. Afr. Med. J. 2018, 108, 35–40. [Google Scholar]
- Colli, A.; Fraquelli, M.; Casazza, G. Accuracy of ultrasonography, spiral CT, magnetic resonance, and alpha-fetoprotein in diagnosing hepatocellular carcinoma: A systematic review. Am. J. Gastroenterol. 2006, 101, 513–523. [Google Scholar] [CrossRef]
- Cicalese, L. Hepatocellular Carcinoma Workup Group. Available online: https://emedicine.medscape.com/article/197319-workup (accessed on 2 March 2022).
- NCCN Guidelines, Version 2.2019, Hepatocellular Carcinoma; NCCN: Plymouth Meeting, PA, USA, 2019.
- White, D.L.; Li, D.; Nurgalieva, Z.; El-Serag, H.B. Genetic variants of glutathione S-transferase as possible risk factors for hepatocellular carcinoma: A HuGE systematic review and meta-analysis. Am. J. Epidemiol. 2008, 167, 377–389. [Google Scholar] [CrossRef] [Green Version]
- Feitelson, M.A.; Pan, J.; Lian, Z. Early molecular and genetic determinants of primary liver malignancy. Surg. Clin. N. Am. 2004, 84, 339–354. [Google Scholar] [CrossRef]
- Martins-Filho, S.N.; Paiva, C.; Azevedo, R.S.; Alves, V.A.F. Histological grading of hepatocellular carcinoma-A systematic review of literature. Front. Med. 2017, 4, 193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiba, T.; Zheng, Y.W.; Kita, K.; Yokosuka, O.; Saisho, H.; Onodera, M.; Miyoshi, H.; Nakano, M.; Zen, Y.; Nakanuma, Y.; et al. Enhanced self-renewal capability in hepatic stem/progenitor cells drives cancer initiation. Gastroenterology 2007, 133, 937–950. [Google Scholar] [CrossRef] [PubMed]
- Avila, M.A.; Berasain Sangro, C.B.; Prieto, J. New therapies for hepatocellular carcinoma. Oncogene 2006, 25, 3866–3884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, L.R.; Gores, G.J. Hepatocellular carcinoma: Molecular pathways and new therapeutic targets. Semin. Liver Dis. 2005, 25, 212–225. [Google Scholar] [CrossRef] [Green Version]
- Abu Rmilah, A.; Zhou, W.; Nelson, E.; Lin, L.; Amiot, B.; Nyberg, S.L. Understanding the marvels behind liver regeneration. Wiley Interdiscip. Rev. Dev. Biol. 2019, 8, e340. [Google Scholar] [CrossRef]
- Petersen, B.E.; Bowen, W.C.; Patrene, K.D.; Mars, W.M.; Sullivan, A.K.; Murase, N.; Boggs, S.S.; Greenberger, J.S.; Goff, J.P. Bone marrow as a potential source of hepatic oval cells. Science 1999, 284, 1168–1170. [Google Scholar] [CrossRef]
- Best, D.H.; Coleman, W.B. Liver regeneration by small hepatocyte-like progenitor cells after necrotic injury by carbon tetrachloride in retrorsine-exposed rats. Exp. Mol. Pathol. 2010, 89, 92–98. [Google Scholar] [CrossRef]
- Cogliati, B.; Aloia, T.P.A.; Bosch, R.V.; Alves, V.A.F.; Hernandez-Blazquez, F.J.; Dagli, M.L.Z. Identification of hepatic stem/progenitor cells in canine hepatocellular and cholangiocellular carcinoma. Vet. Comp. Oncol. 2010, 8, 112–121. [Google Scholar] [CrossRef]
- Kietzmann, T. Metabolic zonation of the liver: The oxygen gradient revisited. Redox Biol. 2017, 11, 622–630. [Google Scholar] [CrossRef]
- Mafra, K.; Nakagaki, B.N.; Castro Oliveira, H.M.; Rezende, R.M.; Antunes, M.M.; Menezes, G.B. The liver as a nursery for leukocytes. J. Leukoc. Biol. 2019, 106, 687–693. [Google Scholar] [CrossRef]
- Severi, T.; van Malenstein, H.; Verslype, C.; van Pelt, J.F. Tumor initiation and progression in hepatocellular carcinoma: Risk factors, classification, and therapeutic targets. Acta Pharmacol. Sin. 2010, 31, 1409–1420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, B.; Ren, J.; Jiang, H.F.; Jia, J. Antitumor activities against hepatocellular carcinoma induced by bone marrow mesenchymal stem cells pulsed with tumor-derived exosomes. Beijing Da Xue Xue Bao Yi Xue Ban 2008, 40, 494–499. [Google Scholar] [PubMed]
- Roskams, T.; Kojiro, M. Pathology of early hepatocellular carcinoma: Conventional and molecular diagnosis. Semin. Liver Dis. 2010, 30, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Klonisch, T.; Wiechec, E.; Hombach-Klonisch, S.; Ande, S.R.; Wesselborg, S.; Schulze-Osthoff, K.; Los, M. Cancer stem cell markers in common cancers—Therapeutic implications. Trends Mol. Med. 2008, 14, 450–460. [Google Scholar] [CrossRef] [Green Version]
- Shah, S.; Shukla, A.; Paunipagar, B. Radiological features of hepatocellular carcinoma. J. Clin. Exp. Hepatol. 2014, 4, S63–S66. [Google Scholar] [CrossRef] [Green Version]
- Greten, T.F.; Papendorf, F.; Bleck, J.S.; Kirchhoff, T.; Wohlberedt, T.; Kubicka, S.; Klempnauer, J.; Galanski, M.; Manns, M.P. Survival rate in patients with hepatocellular carcinoma: A retrospective analysis of 389 patients. Br. J. Cancer 2005, 92, 1862–1868. [Google Scholar] [CrossRef]
- Llovet, J.M.; Brú, C.; Bruix, J. Prognosis of hepatocellular carcinoma: The BCLC staging classification. Semin. Liver Dis. 1999, 19, 329–338. [Google Scholar] [CrossRef]
- Forner, A.; Llovet, J.M.; Bruix, J. Hepatocellular carcinoma. Lancet 2012, 379, 1245–1255. [Google Scholar] [CrossRef]
- Marrero, J.A.; Kudo, M.; Bronowicki, J.P. The challenge of prognosis and staging for hepatocellular carcinoma. Oncologist 2010, 15, 23–33. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Wang, Z.G.; Fu, S.Y.; Li, A.J.; Pan, Z.Y.; Zhou, W.P.; Lau, W.Y.; Wu, M.C. Randomized clinical trial of chemoembolization plus radiofrequency ablation versus partial hepatectomy for hepatocellular carcinoma within the Milan criteria. Br. J. Surg. 2016, 103, 348–356. [Google Scholar] [CrossRef] [Green Version]
- Yin, L.; Li, H.; Li, A.J.; Lau, W.Y.; Pan, Z.Y.; Lai, E.C.; Wu, M.C.; Zhou, W.P. Partial hepatectomy vs. transcatheter arterial chemoembolization for resectable multiple hepatocellular carcinoma beyond Milan criteria: A RCT. J. Hepatol. 2014, 1, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Ou, T.M.; Hsu, C.W.; Horng, C.T.; Lee, C.C.; Tsai, Y.Y.; Tsai, C.C.; Liou, Y.S.; Yang, C.C.; Hsueh, C.W.; et al. Current systemic treatment of hepatocellular carcinoma: A review of the literature. World J. Hepatol. 2015, 7, 1412–1420. [Google Scholar] [CrossRef] [PubMed]
- Yau, T.C.C.; Lencioni, R.; Sukeepaisarnjaroen, W.; Chao, Y.; Yen, C.J.; Lausoontornsiri, W.; Chen, P.J.; Sanpajit, T.; Camp, A.; Cox, D.S.; et al. A phase I/II multicenter study of single-agent foretinib as first-line therapy in patients with advanced hepatocellular carcinoma. Clin. Cancer Res. 2017, 23, 2405–2413. [Google Scholar] [CrossRef] [Green Version]
- Garmpis, N.; Damaskos, C.; Garmpi, A.; Georgakopoulou, V.E.; Sarantis, P.; Antoniou, E.A.; Karamouzis, M.V.; Nonni, A.; Schizas, D.; Diamantis, E.; et al. Histone deacetylase inhibitors in the treatment of hepatocellular carcinoma: Current evidence and future opportunities. J. Pers. Med. 2021, 11, 223. [Google Scholar] [CrossRef]
- Carmeliet, P.; Jain, R.K. Angiogenesis in cancer and other diseases. Nature 2000, 14, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Xu, B.; Fu, L.; Hao, X.S. Correlation of four vascular specific growth factors with carcinogenesis and portal vein tumor thrombus formation in human hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2006, 25, 403–409. [Google Scholar] [PubMed]
- Stock, P.; Monga, D.; Tan, X.; Micsenyi, A.; Loizos, N.; Monga, S.P.S. Platelet-derived growth factor receptor-α: A novel therapeutic target in human hepatocellular cancer. Mol. Cancer Ther. 2007, 6, 1932–1941. [Google Scholar] [CrossRef] [Green Version]
- El-Assal, O.N.; Yamanoi, A.; Ono, T.; Kohno, H.; Nagasue, N. The clinicopathological significance of heparanase and basic fibroblast growth factor expressions in hepatocellular carcinoma. Clin. Cancer Res. 2001, 7, 1299–1305. [Google Scholar]
- Krause, D.S.; van Etten, R.A. Tyrosine kinases as targets for cancer therapy. N. Engl. J. Med. 2005, 353, 172–187. [Google Scholar] [CrossRef] [Green Version]
- Madhusudan, S.; Ganesan, T.S. Tyrosine kinase inhibitors in cancer therapy. Clin. Biochem. 2004, 37, 618–635. [Google Scholar] [CrossRef]
- Kontomanolis, E.N.; Koutras, A.; Syllaios, A.; Schizas, D.; Mastoraki, A.; Garmpis, N.; Diakosavvas, M.; Angelou, K.; Tsatsaris, G.; Pagkalos, A.; et al. Role of oncogenes and tumor-suppressor genes in carcinogenesis: A review. Anticancer Res. 2020, 40, 6009–6015. [Google Scholar] [CrossRef] [PubMed]
- Downward, J. Targeting RAS signaling pathways in cancer therapy. Nat. Rev. Cancer 2003, 3, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Cheng, H.; Roberts, T.M.; Zhao, J.J. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat. Rev. Drug Discov. 2009, 8, 627–644. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Decaens, T.; Raoul, J.L.; Boucher, E.; Kudo, M.; Chang, C.; Kang, Y.K.; Assenat, E.; Lim, H.Y.; Boige, V.; et al. Brivanib in patients with advanced hepatocellular carcinoma who were intolerant to sorafenib or for whom sorafenib failed: Results from the randomized phase III BRISK-PS study. J. Clin. Oncol. 2013, 31, 3509–3516. [Google Scholar] [CrossRef]
- Huynh, H.; Nguyen, T.T.; Chow, K.H.; Tan, P.H.; Soo, K.C.; Tran, E. Over-expression of the mitogen-activated protein kinase (MAPK) kinase (MEK)-MAPK in hepatocellular carcinoma: Its role in tumor progression and apoptosis. BMC Gastroenterol. 2003, 3, 19. [Google Scholar] [CrossRef] [Green Version]
- Iyer, R.; Bharthuar, A. A review of erlotinib--An oral, selective epidermal growth factor receptor tyrosine kinase inhibitor. Expert Opin. Pharmacother. 2010, 11, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Shi, Y.; Jiang, C.Y.; Wei, L.X.; Wang, Y.L.; Dai, G.H. Expression and prognostic role of pan-Ras, Raf-1, pMEK1 and pERK1/2 in patients with hepatocellular carcinoma. Eur. J. Surg. Oncol. 2011, 37, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Siddiqui, A. Hepatitis B virus X protein stimulates the mitochondrial translocation of Raf-1 via oxidative stress. J. Virol. 2007, 81, 6757–6760. [Google Scholar] [CrossRef] [Green Version]
- Ezzoukhry, Z.; Louandre, C.; Trécherel, E.; Godin, C.; Chauffert, B.; Dupont, S.; Diouf, M.; Barbare, J.C.; Mazière, J.C.; Galmiche, A. EGFR activation is a potential determinant of primary resistance of hepatocellular carcinoma cells to sorafenib. Int. J. Cancer 2012, 131, 2961–2969. [Google Scholar] [CrossRef]
- Zhu, A.X.; Rosmorduc, O.; Evans, T.R.; Ross, P.J.; Santoro, A.; Carrilho, F.J.; Bruix, J.; Qin, S.; Thuluvath, P.J.; Llovet, J.M.; et al. Search: A phase III, randomized, double-blind, placebo-controlled trial of sorafenib plus erlotinib in patients with advanced hepatocellular carcinoma. J. Clin. Oncol. 2015, 33, 559–566. [Google Scholar] [CrossRef]
- Schiffer, E.; Housset, C.; Cacheux, C.W.; Wendum, D.; Desbois-Mouthon, C.; Rey, C.; Clergue, F.; Poupon, R.; Barbu, V.; Rosmorduc, O. Gefitinib, an EGFR inhibitor, prevents hepatocellular carcinoma development in the rat liver with cirrhosis. Hepatology 2005, 41, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Sǎftoiu, A.; Ciurea, T.; Baniţǎ, M.; Georgescu, C.; Comǎnescu, V.; Rogoveanu, I.; Gorunescu, F.; Georgescu, I. Immunohistochemical assessment of angiogenesis in primary hepatocellular carcinoma. Rom. J. Gastroenterol. 2004, 13, 3–8. [Google Scholar] [PubMed]
- Yakes, F.M.; Chen, J.; Tan, J.; Yamaguchi, K.; Shi, Y.; Yu, P.; Qian, F.; Chu, F.; Bentzien, F.; Cancilla, B.; et al. Cabozantinib (XL184), a novel MET and VEGFR2 inhibitor, simultaneously suppresses metastasis, angiogenesis, and tumor growth. Mol. Cancer Ther. 2011, 10, 2298–2308. [Google Scholar] [CrossRef] [PubMed]
- Buitrago-Molina, L.E.; Pothiraju, D.; Lamlé, J.; Marhenke, S.; Kossatz, U.; Breuhahn, K.; Manns, M.P.; Malek, N.; Vogel, A. Rapamycin delays tumor development in murine livers by inhibiting proliferation of hepatocytes with DNA damage. Hepatology 2009, 50, 500–509. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A.X.; Abrams, T.A.; Miksad, R.; Blaszkowsky, L.S.; Meyerhardt, J.A.; Zheng, H.; Muzikansky, A.; Clark, J.W.; Kwak, E.L.; Schrag, D.; et al. Phase 1/2 study of everolimus in advanced hepatocellular carcinoma. Cancer 2011, 117, 5094–5102. [Google Scholar] [CrossRef] [Green Version]
- Shiah, H.S.; Chen, C.Y.; Dai, C.Y.; Hsiao, C.F.; Lin, Y.J.; Su, W.C.; Chang, J.Y.; Whang-Peng, J.; Lin, P.W.; Huang, J.D.; et al. Randomised clinical trial: Comparison of two everolimus dosing schedules in patients with advanced hepatocellular carcinoma. Aliment Pharmacol. Ther. 2013, 37, 62–73. [Google Scholar] [CrossRef] [Green Version]
- Grothey, A.; van Cutsem, E.; Sobrero, A.; Siena, S.; Falcon, A.; Ychou, M.; Humblet, Y.; Bouché, O.; Mineur, L.; Barone, C.; et al. Correct Study Group Regorafenib monotherapy for previously treated metastatic colorectal cancer (Correct): An international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 2013, 381, 303–312. [Google Scholar] [CrossRef]
- Chen, Y.L.; Law, P.Y.; Loh, H.H. Inhibition of P13K/Akt signaling: An emerging paradigm for targeted cancer therapy. Curr. Med. Chem. Anticancer Agents 2005, 5, 575–589. [Google Scholar] [CrossRef]
- Chen, K.F.; Chen, H.L.; Tai, W.T.; Feng, W.C.; Hsu, C.H.; Chen, P.J.; Cheng, A.L. Activation of phosphatidylinositol 3-kinase/Akt signaling pathway mediates acquired resistance to sorafenib in hepatocellular carcinoma cells. J. Pharmacol. Exp. Ther. 2011, 337, 155–161. [Google Scholar] [CrossRef] [Green Version]
- Hu, T.H.; Huang, C.C.; Lin, P.R.; Chang, H.W.; Ger, L.P.; Lin, Y.W.; Changchien, C.S.; Lee, C.M.; Tai, M.H. Expression and prognostic role of tumor suppressor gene PTEN/MMAC1/TEP1 in hepatocellular carcinoma. Cancer 2003, 97, 1929–1940. [Google Scholar] [CrossRef]
- Wilhelm, S.M.; Dumas, J.; Adnane, L.; Lynch, M.; Carter, C.A.; Schütz, G.; Thierauch, K.H.; Zopf, D. Regorafenib (BAY 73-4506): A new oral multikinase inhibitor of angiogenic, stromal and oncogenic receptor tyrosine kinases with potent preclinical antitumor activity. Int. J. Cancer 2011, 129, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Abou-Elkacem, L.; Arns, S.; Brix, G.; Gremse, F.; Zopf, D.; Kiessling, F.; Lederle, W. Regorafenib inhibits growth, angiogenesis, and metastasis in a highly aggressive, orthotopic colon cancer model. Mol. Cancer Ther. 2013, 12, 1322–1331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mross, K.; Frost, A.; Steinbild, S.; Hedbom, S.; Büchert, M.; Fasol, U.; Unger, C.; Krätzschmar, J.; Heinig, R.; Boix, O.; et al. A phase I dose-escalation study of regorafenib (BAY 73-4506), an inhibitor of oncogenic, angiogenic, and stromal kinases, in patients with advanced solid tumors. Clin. Cancer Res. 2012, 18, 2658–2667. [Google Scholar] [CrossRef] [PubMed]
- Bruix, J.; Qin, S.; Merle, P.; Granito, A.; Huang, Y.H.; Bodoky, G.; Pracht, M.; Yokosuka, O.; Rosmorduc, O.; Breder, V.; et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 389, 56–66. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Goh, B.C.; Albert, D.H.; Chen, C.S. ABT-869, a promising multi-targeted tyrosine kinase inhibitor: From bench to bedside. J. Hematol. Oncol. 2009, 2, 33. [Google Scholar] [CrossRef] [Green Version]
- Cainap, C.; Qin, S.; Huang, W.T.; Chung, I.J.; Pan, H.; Cheng, Y.; Kudo, M.; Kang, Y.K.; Chen, P.J.; Toh, H.C.; et al. Linifanib versus sorafenib in patients with advanced hepatocellular carcinoma: Results of a randomized phase III trial. J. Clin. Oncol. 2015, 33, 172–179. [Google Scholar] [CrossRef]
- Cai, Z.W.; Zhang, Y.; Borzilleri, R.M.; Qian, L.; Barbosa, S.; Wei, D.; Zheng, X.; Wu, L.; Fan, J.; Shi, Z.; et al. Discovery of brivanib alaninate((S)-((R)-1-(4-(4-fluoro-2-methyl-1H-indol5-yloxy)-5-methylpyrrolo [2,1-f][1,2,4]triazin-6-yloxy)propan-2-yl)2 aminopropanoate), a novel prodrug of dual vascular endothelial growth factor receptor-2 and fibroblast growth factor receptor-1 kinase inhibitor (BMS540215). J. Med. Chem. 2008, 51, 1976–1980. [Google Scholar]
- Johnson, P.J.; Qin, S.; Park, J.W.; Poon, R.T.; Raoul, J.L.; Philip, P.A.; Hsu, C.H.; Hu, T.H.; Heo, J.; Xu, J.; et al. Brivanib versus sorafenib as first-line therapy in patients with unresectable, advanced hepatocellular carcinoma: Results from the randomized phase III BRISK-FL study. J. Clin. Oncol. 2013, 3, 3517–3524. [Google Scholar] [CrossRef] [Green Version]
- Ozanne, B.W.; Spence, H.J.; McGarry, L.C.; Hennigan, R.F. Transcription factors control invasion: AP-1 the first among equals. Oncogene 2007, 26, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Shimotohno, K.; Watashi, K.; Tsuchihara, K.; Fukuda, K.; Marusawa, H.; Hijikata, M. Hepatitis C virus and its roles in cell proliferation. J. Gastroenterol. 2002, 37, 50–54. [Google Scholar] [CrossRef]
- Sananbenesi, F.; Fischer, A.; Schrick, C.; Spiess, J.; Radulovic, J. Phosphorylation of hippocampal Erk-1/2, Elk-1, and p90-Rsk-1 during contextual fear conditioning: Interactions between Erk-1/2 and Elk-1. Mol. Cell. Neurosci. 2002, 21, 463–476. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, K.J.; Wohlschlaeger, J.; Lang, H.; Sotiropoulos, G.C.; Malago, M.; Steveling, K.; Reis, H.; Cicinnati, V.R.; Schmid, K.W.; Baba, H.A. Activation of the ERK and AKT signalling pathway predicts poor prognosis in hepatocellular carcinoma and ERK activation in cancer tissue is associated with hepatitis C virus infection. J. Hepatol. 2008, 48, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Gerber, H.P.; LeCouter, J. The biology of VEGF and its receptors. Nat. Med. 2003, 9, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Ishigami, S.I.; Arii, S.; Furutani, M.; Niwano, M.; Harada, T.; Mizumoto, M.; Mori, A.; Onodera, H.; Imamura, M. Predictive value of vascular endothelial growth factor (VEGF) in metastasis and prognosis of human colorectal cancer. Br. J. Cancer 1998, 78, 1379–1384. [Google Scholar] [CrossRef] [PubMed]
- Maeda, K.; Chung, Y.S.; Ogawa, Y. Prognostic value of vascular endothelial growth factor expression in gastric carcinoma. Cancer 1996, 77, 858–863. [Google Scholar] [CrossRef]
- Inoue, K.; Ozeki, Y.; Suganuma, T.; Sugiura, Y.; Tanaka, S. Vascular endothelial growth factor expression in primary esophageal squamous cell carcinoma. Association with angiogenesis and tumor progression. Cancer 1997, 79, 206–213. [Google Scholar] [CrossRef]
- Grothey, A.; Galanis, E. Targeting angiogenesis: Progress with anti-VEGF treatment with large molecules. Nat. Rev. Clin. Oncol. 2009, 6, 507–518. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhou, X.; Shen, H.; Wang, D.; Wang, Y. Phosphorylated ERK is a potential predictor of sensitivity to sorafenib when treating hepatocellular carcinoma: Evidence from an in vitro study. BMC Med. 2009, 7, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Roberts, L.R.; Gores, G.J. Emerging drugs for hepatocellular carcinoma. Expert Opin. Emerg. Drugs 2006, 11, 469–487. [Google Scholar] [CrossRef]
- Antoniou, E.A.; Koutsounas, I.; Damaskos, C.; Koutsounas, S. Remission of psoriasis in a patient with hepatocellular carcinoma treated with sorafenib. In Vivo 2016, 30, 677–680. [Google Scholar]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; Cosme de Oliveira, A.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sharp Investigators Study Group. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, A.L.; Kang, Y.K.; Chen, Z.; Tsao, C.J.; Qin, S.; Kim, J.S.; Luo, R.; Feng, J.; Ye, S.; Yang, T.S.; et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: A phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009, 10, 25–34. [Google Scholar] [CrossRef]
- Printz, C. Clinical trials of note. Sorafenib as adjuvant treatment in the prevention of disease recurrence in patients with hepatocellular carcinoma (HCC) (STORM). Cancer 2009, 115, 4646. [Google Scholar] [CrossRef] [PubMed]
- Takezawa, K.; Okamoto, I.; Yonesaka, K.; Hatashita, E.; Yamada, Y.; Fukuoka, M.; Nakagawa, K. Sorafenib inhibits non-small cell lung cancer cell growth by targeting B-RAF in KRAS wild-type cells and C-RAF in KRAS mutant cells. Cancer Res. 2009, 69, 6515–6521. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.K.; Abou-Alfa, G.K. An update on clinical trials in the treatment of advanced hepatocellular carcinoma. J. Clin. Gastroenterol. 2013, 47, S16–S19. [Google Scholar] [CrossRef]
- Wilhelm, S.M.; Carter, C.; Tang, L.; Wilkie, D.; McNabola, A.; Rong, H.; Chen, C.; Zhang, X.; Vincent, P.; McHugh, M.; et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004, 64, 7099–7109. [Google Scholar] [CrossRef] [Green Version]
- Bruix, J.; Raoul, J.L.; Sherman, M.; Mazzaferro, V.; Bolondi, L.; Craxi, A.; Galle, P.R.; Santoro, A.; Beaugrand, M.; Sangiovanni, A.; et al. Efficacy and safety of sorafenib in patients with advanced hepatocellular carcinoma: Subanalyses of a phase III trial. J. Hepatol. 2012, 57, 821–829. [Google Scholar] [CrossRef] [Green Version]
- Abou-Alfa, G.K.; Johnson, P.; Knox, J.J.; Capanu, M.; Davidenko, I.; Lacava, J.; Leung, T.; Gansukh, B.; Saltz, L.B. Doxorubicin plus sorafenib vs. doxorubicin alone in patients with advanced hepatocellular carcinoma: A randomized trial. J. Am. Med. Assoc. 2010, 304, 2154–2160. [Google Scholar] [CrossRef] [Green Version]
- Goyal, L.; Zheng, H.; Abrams, T.A.; Miksad, R.; Bullock, A.J.; Allen, J.N.; Yurgelun, M.B.; Clark, J.W.; Kambadakone, A.; Muzikansky, A.; et al. A phase II and biomarker study of sorafenib combined with modified FOLFOX in patients with advanced hepatocellular carcinoma. Clin. Cancer Res. 2019, 25, 80–89. [Google Scholar] [CrossRef] [Green Version]
- Chao, Y.; Chung, Y.H.; Han, G.; Yoon, J.H.; Yang, J.; Wang, J.; Shao, G.L.; Kim, B.I.; Lee, T.Y. The combination of transcatheter arterial chemoembolization and sorafenib is well tolerated and effective in Asian patients with hepatocellular carcinoma: Final results of the START trial. Int. J. Cancer 2015, 136, 1458–1467. [Google Scholar] [CrossRef]
- Kotsifa, E.; Vergadis, C.; Vailas, M.; Machairas, N.; Kykalos, S.; Damaskos, C.; Garmpis, N.; Lianos, G.D.; Schizas, D. Transarterial chemoembolization for hepatocellular carcinoma: Why, when, how? J. Pers. Med. 2022, 12, 436. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Dai, Y. Sorafenib combined with transarterial chemoembolization prolongs survival of patients with advanced hepatocellular carcinoma. J. Buon. 2020, 25, 945–951. [Google Scholar] [PubMed]
- Kudo, M.; Imanaka, K.; Chida, N.; Nakachi, K.; Tak, W.Y.; Takayama, T.; Yoon, J.H.; Hori, T.; Kumada, H.; Hayashi, N.; et al. Phase III study of sorafenib after transarterial chemoembolisation in Japanese and Korean patients with unresectable hepatocellular carcinoma. Eur. J. Cancer 2011, 47, 2117–2127. [Google Scholar] [CrossRef] [PubMed]
- Koyama, N.; Saito, K.; Nishioka, Y.; Yusa, W.; Yamamoto, N.; Yamada, Y.; Nokihara, H.; Koizumi, F.; Nishio, K.; Tamura, T. Pharmacodynamic change in plasma angiogenic proteins: A dose-escalation phase 1 study of the multi-kinase inhibitor lenvatinib. BMC Cancer 2014, 14, 530. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, K.; Kudo, M.; Kawazoe, S.; Osaki, Y.; Ikeda, M.; Okusaka, T.; Tamai, T.; Suzuki, T.; Hisai, T.; Hayato, S.; et al. Phase 2 study of lenvatinib in patients with advanced hepatocellular carcinoma. J. Gastroenterol. 2017, 52, 512–519. [Google Scholar] [CrossRef]
- Kudo, M.; Finn, R.S.; Qin, S.; Han, K.H.; Ikeda, K.; Piscaglia, F.; Baron, A.; Park, J.W.; Han, G.; Jassem, J.; et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: A randomised phase 3 non-inferiority trial. Lancet 2018, 391, 1163–1173. [Google Scholar] [CrossRef] [Green Version]
- Briggs, A.; Daniele, B.; Dick, K.; Evans, T.R.J.; Galle, P.R.; Hubner, R.A.; Lopez, C.; Siebert, U.; Tremblay, G. Covariate-adjusted analysis of the Phase 3 REFLECT study of lenvatinib versus sorafenib in the treatment of unresectable hepatocellular carcinoma. Br. J. Cancer 2020, 122, 1754–1759. [Google Scholar] [CrossRef] [Green Version]
- Faivre, S.; Raymond, E.; Boucher, E.; Douillard, J.; Lim, H.Y.; Skim, J.; Zappa, M.; Lanzalone, S.; Lin, X.; Deprimo, S.; et al. Safety and efficacy of sunitinib in patients with advanced hepatocellular carcinoma: An open-label, multicentre, phase II study. Lancet Oncol. 2009, 10, 794–800. [Google Scholar] [CrossRef]
- Zhu, A.X.; Sahani, D.V.; Duda, D.G.; di Tomaso, E.; Ancukiewicz, M.; Catalano, O.A.; Sindhwani, V.; Blaszkowsky, L.S.; Yoon, S.S.; Lahdenranta, J.; et al. Efficacy, safety, and potential biomarkers of sunitinib monotherapy in advanced hepatocellular carcinoma: A phase II study. J. Clin. Oncol. 2009, 27, 3027–3035. [Google Scholar] [CrossRef] [Green Version]
- Koeberle, D.; Montemurro, M.; Samaras, P.; Majno, P.; Simcock, M.; Limacher, A.; Lerch, S.; Kovàcs, K.; Inauen, R.; Hess, V.; et al. Continuous sunitinib treatment in patients with advanced hepatocellular carcinoma: A Swiss Group for Clinical Cancer Res (SAKK) and Swiss Association for the Study of the Liver (SASL) multicenter phase II trial (SAKK 77/06). Oncologist 2010, 15, 285–292. [Google Scholar] [CrossRef] [Green Version]
- Cheng, A.L.; Kang, Y.K.; Lin, D.Y.; Park, J.W.; Kudo, M.; Qin, S.; Chung, H.C.; Song, X.; Xu, J.; Poggi, G.; et al. Sunitinib versus sorafenib in advanced hepatocellular cancer: Results of a randomized phase III trial. J. Clin. Oncol. 2013, 31, 4067–4075. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhou, C.; Long, Y.; Yin, X. Sunitinib combined with transarterial chemoembolization versus transarterial chemoembolization alone for advanced-stage hepatocellular carcinoma: A propensity score matching study. Tumour Biol. 2015, 36, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Pokuri, V.K.; Tomaszewski, G.M.; Ait-Oudhia, S.; Groman, A.; Khushalani, N.I.; Lugade, A.A.; Thanavala, Y.; Ashton, E.A.; Grande, C.; Fetterly, G.J.; et al. Efficacy, safety, and potential biomarkers of sunitinib and transarterial chemoembolization (TACE) combination in advanced hepatocellular carcinoma (HCC): Phase II trial. Am. J. Clin. Oncol. 2018, 41, 332–338. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Huang, Y.; Shi, H.; Song, Q.; Xu, Y. Sunitinib versus sorafenib plus transarterial chemoembolization for inoperable hepatocellular carcinoma patients. J. Buon. 2018, 23, 193–199. [Google Scholar]
- Patt, Y.Z.; Murad, W.; Fekrazad, M.H.; Baron, A.D.; Bansal, P.; Boumber, Y.; Steinberg, K.; Lee, S.J.; Bedrick, E.; Du, R.; et al. INST OX-05-024: First line gemcitabine, oxaliplatin, and erlotinib for primary hepatocellular carcinoma and bile duct cancers: A multicenter Phase II trial. Cancer Med. 2017, 6, 2042–2051. [Google Scholar] [CrossRef]
- Qin, S.; Bi, F.; Gu, S.; Bai, Y.; Chen, Z.; Wang, Z.; Ying, J.; Lu, Y.; Meng, Z.; Pan, H.; et al. Donafenib versus sorafenib in first-line treatment of unresectable or metastatic hepatocellular carcinoma: A randomized, open-label, parallel-controlled phase II-III trial. J. Clin. Oncol. 2021, 39, 3002–3011. [Google Scholar] [CrossRef]
- Demetri, G.D.; Reichardt, P.; Kang, Y.K.; Blay, J.Y.; Rutkowski, P.; Gelderblom, H.; Hohenberger, P.; Leahy, M.; von Mehren, M.; Joensuu, H.; et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): An international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 2013, 381, 295–302. [Google Scholar] [CrossRef] [Green Version]
- Bruix, J.; Tak, W.Y.; Gasbarrini, A.; Santoro, A.; Colombo, M.; Lim, H.Y.; Mazzaferrog, V.; Wiesth, R.; Reiga, M.; Wagneri, A.; et al. Regorafenib as second-line therapy for intermediate or advanced hepatocellular carcinoma: Multicentre, open-label, phase II safety study. Eur. J. Cancer 2013, 49, 3412–3419. [Google Scholar] [CrossRef]
- Kelley, R.K.; Verslype, C.; Cohn, A.L.; Yang, T.S.; Su, W.C.; Burris, H.; Braiteh, F.; Vogelzang, N.; Spira, A.; Foster, P.; et al. Cabozantinib in hepatocellular carcinoma: Results of a phase 2 placebo-controlled randomized discontinuation study. Ann. Oncol. 2017, 28, 528–534. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Meyer, T.; Cheng, A.L.; El-Khoueiry, A.B.; Rimassa, L.; Ryoo, B.Y.; Cicin, I.; Merle, P.; Park, J.W.; Blanc, J.F.; et al. Cabozantinib (C) versus placebo (P) in patients (pts) with advanced hepatocellular carcinoma (HCC) who have received prior sorafenib: Results from the randomized phase III CELESTIAL trial. J. Clin. Oncol. 2018, 36, 207. [Google Scholar] [CrossRef]
- Kelley, R.K.; Ryoo, B.Y.; Merle, P.; Park, J.W.; Bolondi, L.; Chan, S.L.; Lim, H.Y.; Baron, A.D.; Parnis, F.; Knox, J.; et al. Second-line cabozantinib after sorafenib treatment for advanced hepatocellular carcinoma: A subgroup analysis of the phase 3 CELESTIAL trial. ESMO Open 2020, 5, e000714. [Google Scholar] [CrossRef] [PubMed]
- Adjei, A.A.; Schwartz, B.; Garmey, E. Early clinical development of ARQ 197, a selective, non-ATP-competitive inhibitor targeting MET tyrosine kinase for the treatment of advanced cancers. Oncologist 2011, 16, 788–799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santoro, A.; Rimassa, L.; Borbath, I.; Daniele, B.; Salvagni, S.; van Laethem, J.L.; van Vlierberghe, H.; Trojan, J.; Kolligs, F.T.; Weiss, A.; et al. Tivantinib for second-line treatment of advanced hepatocellular carcinoma: A randomised, placebo-controlled phase 2 study. Lancet Oncol. 2013, 14, 55–63. [Google Scholar] [CrossRef]
- Rimassa, L.; Santoro, A.; Daniele, B.; Germano, D.; Gasbarrini, A.; Salvagni, S.; Masi, G.; Abbadessa, G.; Lamar, M.; Goldberg, T.; et al. Tivantinib, a new option for second-line treatment of advanced hepatocellular carcinoma? The experience of Italian centers. Tumori 2015, 101, 139–143. [Google Scholar] [CrossRef]
- Rimassa, L.; Assenat, E.; Peck-Radosavljevic, M.; Pracht, M.; Zagonel, V.; Mathurin, P.; Rota Caremoli, E.; Porta, C.; Daniele, B.; Bolondi, L.; et al. Tivantinib for second-line treatment of MET-high, advanced hepatocellular carcinoma (METIV-HCC): A final analysis of a phase 3, randomised, placebo-controlled study. Lancet Oncol. 2018, 19, 682–693. [Google Scholar] [CrossRef]
- Kudo, M.; Morimoto, M.; Moriguchi, M.; Izumi, N.; Takayama, T.; Yoshiji, H.; Hino, K.; Oikawa, T.; Chiba, T.; Motomura, K.; et al. A randomized, double-blind, placebo-controlled, phase 3 study of tivantinib in Japanese patients with MET-high hepatocellular carcinoma. Cancer Sci. 2020, 111, 3759–3769. [Google Scholar] [CrossRef]
- McNamara, M.G.; Le, L.W.; Horgan, A.M.; Aspinall, A.; Burak, K.W.; Dhani, N.; Chen, E.; Sinaei, M.; Lo, G.; Kim, T.K.; et al. A phase II trial of second-line axitinib following prior antiangiogenic therapy in advanced hepatocellular carcinoma. Cancer 2015, 121, 1620–1627. [Google Scholar] [CrossRef]
- Kang, Y.K.; Yau, T.; Park, J.W.; Lim, H.Y.; Lee, T.Y.; Obi, S.; Chan, S.L.; Qin, S.; Kim, R.D.; Casey, M.; et al. Randomized phase II study of axitinib versus placebo plus best supportive care in second-line treatment of advanced hepatocellular carcinoma. Ann. Oncol. 2015, 26, 2457–2463. [Google Scholar] [CrossRef]
- Chan, S.L.; Yeo, W.; Mo, F.; Chan, A.W.H.; Koh, J.; Li, L.; Hui, E.P.; Chong, C.C.N.; Lai, P.B.S.; Mok, T.S.K.; et al. A phase 2 study of the efficacy and biomarker on the combination of transarterial chemoembolization and axitinib in the treatment of inoperable hepatocellular carcinoma. Cancer 2017, 123, 3977–3985. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Zhou, A.; Zhang, W.; Jiang, Z.; Chen, B.; Zhao, J.; Li, Z.; Wang, L.; Bi, X.; Zhao, H.; et al. Anlotinib in the treatment of advanced hepatocellular carcinoma: An open-label phase II study (ALTER-0802 study). Hepatol. Int. 2021, 15, 621–629. [Google Scholar] [CrossRef]
- Han, C.; Ye, S.; Hu, C.; Shen, L.; Qin, Q.; Bai, Y.; Yang, S.; Bai, C.; Zang, A.; Jiao, S.; et al. Clinical activity and safety of penpulimab (anti-PD-1) with anlotinib as first-line therapy for advanced hepatocellular carcinoma (HCC). J. Clin. Oncol. 2021, 11, 684867. [Google Scholar]
- Decaens, T.; Barone, C.; Assenat, E.; Wermke, M.; Fasolo, A.; Merle, P.; Blanc, J.F.; Grando, V.; Iacobellis, A.; Villa, E.; et al. Phase 1b/2 trial of tepotinib in sorafenib pretreated advanced hepatocellular carcinoma with MET overexpression. Br. J. Cancer 2021, 125, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Ryoo, B.Y.; Cheng, A.L.; Ren, Z.; Kim, T.Y.; Pan, H.; Rau, K.M.; Choi, H.J.; Park, J.W.; Kim, J.H.; Yen, C.J.; et al. Randomised phase 1b/2 trial of tepotinib vs sorafenib in Asian patients with advanced hepatocellular carcinoma with MET overexpression. Br. J. Cancer 2021, 125, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Spratlin, J.L.; Cohen, R.B.; Eadens, M.; Gore, L.; Camidge, D.R.; Diab, S.; Leong, S.; O’Bryant, C.; Chow, L.Q.; Serkova, N.J.; et al. Phase I pharmacologic and biologic study of ramucirumab (IMC-1121B), a fully human immunoglobulin G1 monoclonal antibody targeting the vascular endothelial growth factor receptor-2. J. Clin. Oncol. 2010, 28, 780–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, A.X.; Finn, R.S.; Mulcahy, M.; Gurtler, J.; Sun, W.; Schwartz, J.D.; Dalal, R.P.; Joshi, A.; Hozak, R.R.; Xu, Y.; et al. A phase II and biomarker study of ramucirumab, a human monoclonal antibody targeting the VEGF receptor-2, as first-line monotherapy in patients with advanced hepatocellular cancer. Clin. Cancer Res. 2013, 19, 6614–6623. [Google Scholar] [CrossRef] [Green Version]
- Zhu, A.X.; Park, J.O.; Ryoo, B.Y.; Yen, C.J.; Poon, R.; Pastorelli, D.; Blanc, J.F.; Chung, H.C.; Baron, A.D.; Pfiffer, T.E.; et al. Ramucirumab versus placebo as second-line treatment in patients with advanced hepatocellular carcinoma following first-line therapy with sorafenib (REACH): A randomised, double-blind, multicentre, phase 3 trial. Lancet Oncol. 2015, 16, 859–870. [Google Scholar] [CrossRef]
- Zhu, A.X.; Kang, Y.K.; Yen, C.J.; Finn, R.S.; Galle, P.R.; Llovet, J.M.; Assenat, E.; Brandi, G.; Pracht, M.; Lim, H.Y.; et al. Reach-2 study investigators. Ramucirumab after sorafenib in patients with advanced hepatocellular carcinoma and increased α-fetoprotein concentrations (REACH-2): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2019, 20, 282–296. [Google Scholar] [CrossRef]
- Harding, J.J.; Zhu, A.X.; Bauer, T.M.; Choueiri, T.K.; Drilon, A.; Voss, M.H.; Fuchs, C.S.; Abou-Alfa, G.K.; Wijayawardana, S.R.; Wang, X.A.; et al. A phase Ib/II study of ramucirumab in combination with emibetuzumab in patients with advanced cancer. Clin. Cancer Res. 2019, 25, 5202–5211. [Google Scholar] [CrossRef]
- Siegel, A.B.; Cohen, E.I.; Ocean, A.; Lehrer, D.; Goldenberg, A.; Knox, J.J.; Chen, H.; Clark-Garvey, S.; Weinberg, A.; Mandeli, J.; et al. Phase II trial evaluating the clinical and biologic effects of bevacizumab in unresectable hepatocellular carcinoma. J. Clin. Oncol. 2008, 26, 2992–2998. [Google Scholar] [CrossRef] [Green Version]
- Boige, V.; Malka, D.; Bourredjem, A.; Dromain, C.; Baey, C.; Jacques, N.; Pignon, J.P.; Vimond, N.; Bouvet-Forteau, N.; de Baere, T.; et al. Efficacy, safety, and biomarkers of single-agent bevacizumab therapy in patients with advanced hepatocellular carcinoma. Oncologist 2012, 17, 1063–1072. [Google Scholar] [CrossRef] [Green Version]
- Zhu, A.X.; Blaszkowsky, L.S.; Ryan, D.P.; Clark, J.W.; Muzikansky, A.; Horgan, K.; Sheehan, S.; Hale, K.E.; Enzinger, P.C.; Bhargava, P.; et al. Phase II study of gemcitabine and oxaliplatin in combination with bevacizumab in patients with advanced hepatocellular carcinoma. J. Clin. Oncol. 2006, 24, 1898–1903. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Sohal, D.; Haller, D.G.; Mykulowycz, K.; Rosen, M.; Soulen, M.C.; Caparro, M.; Teitelbaum, U.R.; Giantonio, B.; O’Dwyer, P.J.; et al. Phase 2 trial of bevacizumab, capecitabine, and oxaliplatin in treatment of advanced hepatocellular carcinoma. Cancer 2011, 117, 3187–3192. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.H.; Yang, T.S.; Hsu, C.; Toh, H.C.; Epstein, R.J.; Hsiao, L.T.; Chen, P.J.; Lin, Z.Z.; Chao, T.Y.; Cheng, A.L. Efficacy and tolerability of bevacizumab plus capecitabine as first-line therapy in patients with advanced hepatocellular carcinoma. Br. J. Cancer 2010, 102, 981–986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, M.B.; Morris, J.S.; Chadha, R.; Iwasaki, M.; Kaur, H.; Lin, E.; Kaseb, A.; Glover, K.; Davila, M.; Abbruzzese, J. Phase II trial of the combination of bevacizumab and erlotinib in patients who have advanced hepatocellular carcinoma. J. Clin. Oncol. 2009, 27, 843–850. [Google Scholar] [CrossRef] [PubMed]
- Kaseb, A.O.; Garrett-Mayer, E.; Morris, J.S.; Xiao, L.; Lin, E.; Onicescu, G.; Hassan, M.M.; Hassabo, H.M.; Iwasaki, M.; Deaton, F.L.; et al. Efficacy of bevacizumab plus erlotinib for advanced hepatocellular carcinoma and predictors of outcome: Final results of a phase II trial. Oncology 2012, 82, 67–74. [Google Scholar] [CrossRef]
- Yau, T.; Wong, H.; Chan, P.; Yao, T.J.; Pang, R.; Cheung, T.T.; Fan, S.T.; Poon, R.T. Phase II study of bevacizumab and erlotinib in the treatment of advanced hepatocellular carcinoma patients with sorafenib-refractory disease. Investig. New Drugs 2012, 30, 2384–2390. [Google Scholar] [CrossRef] [Green Version]
- Philip, P.A.; Mahoney, M.R.; Holen, K.D.; Northfelt, D.W.; Pitot, H.C.; Picus, J.; Flynn, P.J.; Erlichman, C. Phase 2 study of bevacizumab plus erlotinib in patients with advanced hepatocellular cancer. Cancer 2012, 118, 2424–2430. [Google Scholar] [CrossRef]
- Thomas, M.B.; Garrett-Mayer, E.; Anis, M.; Anderton, K.; Bentz, T.; Edwards, A.; Brisendine, A.; Weiss, G.; Siegel, A.B.; Bendell, J.; et al. A randomized phase II open-label multi-institution study of the combination of bevacizumab and erlotinib compared to sorafenib in the first-line treatment of patients with advanced hepatocellular carcinoma. Oncology 2018, 94, 329–339. [Google Scholar] [CrossRef]
- Buijs, M.; Reyes, D.K.; Pawlik, T.M.; Blackford, A.L.; Salem, R.; Messersmith, W.A.; Weekes, C.D.; Mulcahy, M.; Kamel, I.R.; Geschwind, J.F. Phase 2 trial of concurrent bevacizumab and transhepatic arterial chemoembolization in patients with unresectable hepatocellular carcinoma. Cancer 2013, 119, 1042–1049. [Google Scholar] [CrossRef]
- Britten, C.D.; Gomes, A.S.; Wainberg, Z.A.; Elashoff, D.; Amado, R.; Xin, Y.; Busuttil, R.W.; Slamon, D.J.; Finn, R.S. Transarterial chemoembolization plus or minus intravenous bevacizumab in the treatment of hepatocellular cancer: A pilot study. BMC Cancer 2012, 12, 16. [Google Scholar] [CrossRef] [Green Version]
- Pinter, M.; Ulbrich, G.; Sieghart, W.; Kölblinger, C.; Reiberger, T.; Li, S.; Ferlitsch, A.; Müller, C.; Lammer, J.; Peck-Radosavljevic, M. Hepatocellular carcinoma: A phase II randomized controlled double-blind trial of transarterial chemoembolization in combination with biweekly intravenous administration of bevacizumab or a placebo. Radiology 2015, 277, 903–912. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.S.; Ryoo, B.Y.; Hsu, C.H.; Numata, K.; Stein, S.; Verret, W.; Hack, S.P.; Spahn, J.; Liu, B.; Abdullah, H.; et al. Atezolizumab with or without bevacizumab in unresectable hepatocellular carcinoma (GO30140): An open-label, multicentre, phase 1b study. Lancet Oncol. 2020, 21, 808–820. [Google Scholar] [CrossRef]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Li, Q.; Gu, S.; Chen, X.; Lin, L.; Wang, Z.; Xu, A.; Chen, X.; Zhou, C.; Ren, Z.; et al. Apatinib as second-line or later therapy in patients with advanced hepatocellular carcinoma (AHELP): A multicentre, double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Gastroenterol. Hepatol. 2021, 6, 559–568. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, Y.; Jia, R.; Yue, C.; Chang, L.; Liu, R.; Zhang, G.; Zhao, C.; Zhang, Y.; Chen, C.; et al. Anti-PD-1 antibody SHR-1210 combined with apatinib for advanced hepatocellular carcinoma, gastric, or esophagogastric junction cancer: An open-label, dose escalation and expansion study. Clin. Cancer Res. 2019, 25, 515–523. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Shen, J.; Gu, S.; Zhang, Y.; Wu, L.; Wu, J.; Shao, G.; Zhang, Y.; Xu, L.; Yin, T.; et al. Camrelizumab in combination with apatinib in patients with advanced hepatocellular carcinoma (RESCUE): A nonrandomized, open-label, phase II trial. Clin. Cancer Res. 2021, 27, 1003–1011. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Jin, X.L.; Yang, C.; Du, P.; Jiang, F.Q.; Ma, J.P.; Yang, J.; Xie, P.; Zhang, Z. Comparison of efficacy between TACE combined with apatinib and TACE alone in the treatment of intermediate and advanced hepatocellular carcinoma: A single-center randomized controlled trial. Cancer Biol. Ther. 2017, 18, 433–438. [Google Scholar] [CrossRef]
- Gonzalez-Sanchez, E.; Vaquero, J.; Férnandez-Barrena, M.G.; Lasarte, J.J.; Avila, M.A.; Sarobe, P.; Reig, M.; Calvo, M.; Fabregat, I. The TGF-β pathway: A pharmacological target in hepatocellular carcinoma? Cancers 2021, 13, 3248. [Google Scholar] [CrossRef]
- Herbertz, S.; Sawyer, J.S.; Stauber, A.J.; Gueorguieva, I.; Driscoll, K.E.; Estrem, S.T.; Cleverly, A.L.; Desaiah, D.; Guba, S.C.; Benhadji, K.A.; et al. Clinical development of galunisertib (LY2157299 monohydrate), a small molecule inhibitor of transforming growth factor-beta signaling pathway. Drug Des. Devel. Ther. 2015, 9, 4479–4499. [Google Scholar]
- Faivre, S.; Santoro, A.; Kelley, R.K.; Gane, E.; Costentin, C.E.; Gueorguieva, I.; Smith, C.; Cleverly, A.; Lahn, M.M.; Raymond, E.; et al. Novel transforming growth factor beta receptor I kinase inhibitor galunisertib (LY2157299) in advanced hepatocellular carcinoma. Liver Int. 2019, 39, 1468–1477. [Google Scholar] [CrossRef]
- Kelley, R.K.; Gane, E.; Assenat, E.; Siebler, J.; Galle, P.R.; Merle, P.; Hourmand, I.O.; Cleverly, A.; Zhao, Y.; Gueorguieva, I.; et al. A phase 2 study of galunisertib (TGF-β1 receptor type I inhibitor) and sorafenib in patients with advanced hepatocellular carcinoma. Clin. Transl. Gastroenterol. 2019, 10, e00056. [Google Scholar] [CrossRef] [PubMed]
- Greenwald, R.J.; Freeman, G.J.; Sharpe, A.H. The B7 family revisited. Annu. Rev. Immunol. 2005, 23, 515–548. [Google Scholar] [CrossRef] [PubMed]
- Linsley, P.S.; Greene, J.L.; Tan, P.; Bradshaw, J.; Ledbetter, J.A.; Anasetti, C.; Damle, N.K. Coexpression and functional cooperation of CTLA-4 and CD28 on activated T lymphocytes. J. Exp. Med. 1992, 176, 1595–1604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krummel, M.F.; Allison, J.P. D28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J. Exp. Med. 1995, 182, 459–465. [Google Scholar] [CrossRef] [Green Version]
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 2000, 192, 1027–1034. [Google Scholar] [CrossRef] [Green Version]
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Robert, C.; Thomas, L.; Bondarenko, I.; O’Day, S.; Weber, J.; Garbe, C.; Lebbe, C.; Baurain, J.F.; Testori, A.; Grob, J.J.; et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N. Engl. J. Med. 2011, 364, 2517–2526. [Google Scholar] [CrossRef]
- Motzer, R.J.; Escudier, B.; McDermott, D.F.; George, S.; Hammers, H.J.; Srinivas, S.; Tykodi, S.S.; Sosman, J.A.; Procopio, G.; Plimack, E.R.; et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N. Engl. J. Med. 2015, 373, 1803–1813. [Google Scholar] [CrossRef] [Green Version]
- Powles, T.; Eder, J.P.; Fine, G.D.; Braiteh, F.S.; Loriot, Y.; Cruz, C.; Bellmunt, J.; Burris, H.A.; Petrylak, D.P.; Teng, S.; et al. MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature 2014, 515, 558–562. [Google Scholar] [CrossRef]
- Cohen, E.E.W.; Machiels, J.P.H.; Harrington, K.J.; Burtness, B.; Shin, S.W.; Gause, C.K.; Swift, A.M.; Brown, H.; Perrone, A.M.; Cheng, J.D.; et al. KEYNOTE-040: A phase III randomized trial of pembrolizumab (MK-3475) versus standard treatment in patients with recurrent or metastatic head and neck cancer. J. Clin. Oncol. 2015, 33, 6084. [Google Scholar] [CrossRef]
- Ribas, A.; Puzanov, I.; Dummer, R.; Schadendorf, D.; Hamid, O.; Robert, C.; Hodi, F.S.; Schachter, J.; Pavlick, A.C.; Lewis, K.D.; et al. Pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory melanoma (KEYNOTE-002): A randomised, controlled, phase 2 trial. Lancet Oncol. 2015, 16, 908–918. [Google Scholar] [CrossRef] [Green Version]
- Weber, J.S.; D’Angelo, S.P.; Minor, D.; Hodi, F.S.; Gutzmer, R.; Neyns, B.; Hoeller, C.; Khushalani, N.I.; Miller, W.H.; Lao, C.D.; et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): A randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2015, 16, 375–384. [Google Scholar] [CrossRef]
- Trifylli, E.M.; Koustas, E.; Papadopoulos, N.; Sarantis, P.; Aloizos, G.; Damaskos, C.; Garmpis, N.; Garmpi, A.; Karamouzis, M.V. An insight into the novel immunotherapy and targeted therapeutic strategies for hepatocellular carcinoma and cholangiocarcinoma. Life 2022, 12, 665. [Google Scholar] [CrossRef]
- Koustas, E.; Trifylli, E.M.; Sarantis, P.; Papadopoulos, N.; Karapedi, E.; Aloizos, G.; Damaskos, C.; Garmpis, N.; Garmpi, A.; Papavassiliou, K.A.; et al. Immunotherapy as a therapeutic strategy for gastrointestinal cancer-Current treatment options and future perspectives. Int. J. Mol. Sci. 2022, 23, 6664. [Google Scholar] [CrossRef]
- Elsegood, C.L.; Tirnitz-Parker, J.E.; Olynyk, J.K.; Yeoh, G.C. Immune checkpoint inhibition: Prospects for prevention and therapy of hepatocellular carcinoma. Clin. Transl. Immunol. 2017, 6, e161. [Google Scholar] [CrossRef]
- El-Khoueiry, A.B.; Sangro, B.; Yau, T.; Crocenzi, T.S.; Kudo, M.; Hsu, C.; Kim, T.Y.; Choo, S.P.; Trojan, J.; Welling, T.H.; et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): An open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet Oncol. 2017, 389, 2492–2502. [Google Scholar] [CrossRef]
- Kudo, M.; Matilla, A.; Santoro, A.; Melero, I.; Gracián, A.C.; Acosta-Rivera, M.; Choo, S.P.; El-Khoueiry, A.B.; Kuromatsu, R.; El-Rayes, B.; et al. CheckMate 040 cohort 5: A phase I/II study of nivolumab in patients with advanced hepatocellular carcinoma and Child-Pugh B cirrhosis. J. Hepatol. 2021, 75, 600–609. [Google Scholar] [CrossRef]
- Yau, T.; Kang, Y.K.; Kim, T.Y.; El-Khoueiry, A.B.; Santoro, A.; Sangro, B.; Melero, I.; Kudo, M.; Hou, M.M.; Matilla, A.; et al. Efficacy and safety of nivolumab plus ipilimumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib: The CheckMate 040 randomized clinical trial. J. Am. Med. Assoc. Oncol. 2020, 6, e204564. [Google Scholar] [CrossRef]
- Yau, T.; Park, J.W.; Finn, R.S.; Cheng, A.L.; Mathurin, P.; Edeline, J.; Kudo, M.; Harding, J.J.; Merle, P.; Rosmorduc, O.; et al. Nivolumab versus sorafenib in advanced hepatocellular carcinoma (CheckMate 459): A randomised, multicentre, open-label, phase 3 trial. Lancet Oncol. 2022, 23, 77–90. [Google Scholar] [CrossRef]
- Zhu, A.X.; Finn, R.S.; Edeline, J.; Cattan, S.; Ogasawara, S.; Palmer, D.; Verslype, C.; Zagonel, V.; Fartoux, L.; Vogel, A.; et al. KEYNOTE-224 investigators. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): A non-randomised, open-label phase 2 trial. Lancet Oncol. 2018, 19, 940–952. [Google Scholar] [CrossRef]
- Finn, R.S.; Ryoo, B.Y.; Merle, P.; Kudo, M.; Bouattour, M.; Lim, H.Y.; Breder, V.; Edeline, J.; Chao, Y.; Ogasawara, S.; et al. Pembrolizumab as second-line therapy in patients with advanced hepatocellular carcinoma in KEYNOTE-240: A randomized, double-blind, phase III trial. J. Clin. Oncol. 2020, 38, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Ryoo, B.Y.; Merle, P.; Kulkarni, A.S.; Cheng, A.L.; Bouattour, M.; Lim, H.Y.; Breder, V.; Edeline, J.; Chao, Y.; Ogasawara, S.; et al. Health-related quality-of-life impact of pembrolizumab versus best supportive care in previously systemically treated patients with advanced hepatocellular carcinoma: KEYNOTE-240. Cancer 2021, 127, 865–874. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.W.; Cho, E.J.; Lee, J.H.; Yu, S.J.; Kim, Y.J.; Yoon, J.H.; Kim, T.Y.; Han, S.W.; Oh, D.Y.; Im, S.A.; et al. Phase II study of avelumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib. Clin. Cancer Res. 2021, 27, 713–718. [Google Scholar] [CrossRef] [PubMed]
- Kelley, R.K.; Sangro, B.; Harris, W.; Ikeda, M.; Okusaka, T.; Kang, Y.K.; Qin, S.; Tai, D.W.; Lim, H.Y.; Yau, T.; et al. Safety, efficacy, and pharmacodynamics of tremelimumab plus durvalumab for patients with unresectable hepatocellular carcinoma: Randomized expansion of a Phase I/II study. J. Clin. Oncol. 2021, 39, 2991–3001. [Google Scholar] [CrossRef]
- Ren, Z.; Xu, J.; Bai, Y.; Xu, A.; Cang, S.; Du, C.; Li, Q.; Lu, Y.; Chen, Y.; Guo, Y.; et al. Sintilimab plus a bevacizumab biosimilar (IBI305) versus sorafenib in unresectable hepatocellular carcinoma (ORIENT-32): A randomised, open-label, phase 2-3 study. Lancet Oncol. 2021, 22, 977–990. [Google Scholar] [CrossRef]
- Dal Bo, M.; De Mattia, E.; Baboci, L.; Mezzalira, S.; Cecchin, E.; Assaraf, Y.G.; Toffoli, G. New insights into the pharmacological, immunological, and CAR-T-cell approaches in the treatment of hepatocellular carcinoma. Drug Resist. Updates 2020, 51, 100702. [Google Scholar] [CrossRef]
- Dai, H.; Tong, C.; Shi, D.; Chen, M.; Guo, Y.; Chen, D.; Han, X.; Wang, H.; Wang, Y.; Shen, P. Efficacy and biomarker analysis of CD133-directed CAR T cells in advanced hepatocellular carcinoma: A single-arm, open-label, phase II trial. Oncoimmunology 2020, 9, 1846926. [Google Scholar] [CrossRef]
- El-Khoueiry, A. Atezolizumab and Bevacizumab Combination Therapy for Hepatocellular Carcinoma. Gastroenterol. Hepatol. 2020, 3, 145–148. [Google Scholar]
- Woei-A.-Jin, F.J.S.H.; Weijl, N.I.; Burgmans, M.C.; Fariña Sarasqueta, A.; van Wezel, J.T.; Wasser, M.N.J.M.; Coenraad, M.J.; Burggraaf, J.; Osanto, S. Neoadjuvant treatment with angiogenesis-inhibitor dovitinib prior to local therapy in hepatocellular carcinoma: A phase II study. Oncologist 2021, 26, 854–864. [Google Scholar]
- Jenkins, R.W.; Fisher, D.E. Treatment of advanced melanoma in 2020 and beyond. J. Investig. Dermatol. 2021, 141, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Sarantis, P.; Tzanetatou, E.D.; Ioakeimidou, E.; Vallilas, C.; Androutsakos, T.; Damaskos, C.; Garmpis, N.; Garmpi, A.; Papavassiliou, A.G.; Karamouzis, M.V. Cholangiocarcinoma: The role of genetic and epigenetic factors; current and prospective treatment with checkpoint inhibitors and immunotherapy. Am. J. Transl. Res. 2021, 13, 13246–13260. [Google Scholar] [PubMed]
- Chen, Y.; Chang-Yong, E.; Gong, Z.W.; Liu, S.; Wang, Z.X.; Yang, Y.S.; Zhang, X.W. Chimeric antigen receptor-engineered T-cell therapy for liver cancer. Hepatobiliary Pancreat. Dis. Int. 2018, 17, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Li, N.; Zhang, Y.F.; Fu, H.; Feng, M.; Schneider, D.; Su, L.; Wu, X.; Zhou, J.; Mackay, S.; et al. Persistent polyfunctional chimeric antigen receptor T cells that target glypican 3 eliminate orthotopic hepatocellular carcinomas in mice. Gastroenterology 2020, 158, 2250–2265. [Google Scholar] [CrossRef]
- Shi, D.; Shi, Y.; Kaseb, A.O.; Qi, X.; Zhang, Y.; Chi, J.; Lu, Q.; Gao, H.; Jiang, H.; Wang, H.; et al. Chimeric antigen receptor-glypican-3 T-cell therapy for advanced hepatocellular carcinoma: Results of phase I trials. Clin. Cancer Res. 2020, 26, 3979–3989. [Google Scholar] [CrossRef]
- Assenat, E.; Pageaux, G.P.; Thézenas, S.; Peron, J.M.; Bécouarn, Y.; Seitz, J.F.; Merle, P.; Blanc, J.F.; Bouché, O.; Ramdani, M.; et al. Sorafenib alone vs. sorafenib plus GEMOX as 1st-line treatment for advanced HCC: The phase II orafenib PRODIGE 10 trial. Br. J. Cancer 2019, 120, 896–902. [Google Scholar] [CrossRef]
- Han, T.S.; Ban, H.S.; Hur, K.; Cho, H.S. The epigenetic regulation of HCC metastasis. Int. J. Mol. Sci. 2018, 19, 3978. [Google Scholar] [CrossRef] [Green Version]
- Wei, H.; Xu, Z.; Chen, L.; Wei, Q.; Huang, Z.; Liu, G.; Li, W.; Wang, J.; Tang, Q.; Pu, J. Long non-coding RNA PAARH promotes hepatocellular carcinoma progression and angiogenesis via upregulating HOTTIP and activating HIF-1α/VEGF signaling. Cell Death Dis. 2022, 13, 102. [Google Scholar] [CrossRef]
- Damaskos, C.; Garmpis, N.; Dimitroulis, D.; Garmpi, A.; Diamantis, E.; Sarantis, P.; Georgakopoulou, V.E.; Patsouras, A.; Despotidis, M.; Prevezanos, D.; et al. The role of SNHG15 in the pathogenesis of hepatocellular carcinoma. J. Pers. Med. 2022, 12, 753. [Google Scholar] [CrossRef]
- Hong, D.S.; Kang, Y.K.; Borad, M.; Sachdev, J.; Ejadi, S.; Lim, H.Y.; Brenner, A.J.; Park, K.; Lee, J.L.; Kim, T.Y.; et al. Phase 1 study of MRX34, a liposomal miR-34a mimic, in patients with advanced solid tumours. Br. J. Cancer 2020, 122, 1630–1637. [Google Scholar] [CrossRef]
- Teufel, M.; Seidel, H.; Köchert, K.; Meinhardt, G.; Finn, R.S.; Llovet, J.M.; Bruix, J. Biomarkers associated with response to regorafenib in patients with hepatocellular carcinoma. Gastroenterology 2019, 156, 1731–1741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, R.; Psarelli, E.E.; Berhane, S.; Khan, H.; Johnson, P. Impact of viral status on survival in patients receiving sorafenib for advanced hepatocellular cancer: A meta-analysis of randomized phase III trials. J. Clin. Oncol. 2017, 35, 622–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, A.L.; Guan, Z.; Chen, Z.; Tsao, C.J.; Qin, S.; Kim, J.S.; Yang, T.S.; Tak, W.Y.; Pan, H.; Yu, S.; et al. Efficacy and safety of sorafenib in patients with advanced hepatocellular carcinoma according to baseline status: Subset analyses of the phase III sorafenib Asia-Pacific trial. Eur. J. Cancer 2012, 48, 1452–1465. [Google Scholar] [CrossRef] [PubMed]
- Pfister, D.; Núñez, N.G.; Pinyol, R.; Govaere, O.; Pinter, M.; Szydlowska, M.; Gupta, R.; Qiu, M.; Deczkowska, A.; Weiner, A.; et al. NASH limits anti-tumour surveillance in immunotherapy-treated HCC. Nature 2021, 592, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Albano, E.; Sutti, S. The paradox role of cytotoxic T-lymphocytes in NAFLD-associated hepatocellular carcinoma. Hepatobiliary Surg. Nutr. 2021, 10, 705–707. [Google Scholar] [CrossRef] [PubMed]
- Kudo, M. Lack of response to immunotherapy in non-alcoholic steatohepatitis related hepatocellular carcinoma. Hepatobiliary Surg. Nutr. 2021, 10, 522–525. [Google Scholar] [CrossRef] [PubMed]
- Lencioni, R.; Kudo, M.; Ye, S.L.; Bronowicki, J.P.; Chen, X.P.; Dagher, L.; Furuse, J.; Geschwind, J.F.; de Guevara, L.L.; Papandreou, C.; et al. GIDEON (Global Investigation of therapeutic DEcisions in hepatocellular carcinoma and of its treatment with sorafeNib): Second interim analysis. Int. J. Clin. Pract. 2014, 68, 609–617. [Google Scholar] [CrossRef] [Green Version]
- Ye, S.L.; Chen, X.; Yang, J.; Bie, P.; Zhang, S.; Liu, F.; Liu, L.; Zhou, J.; Dou, K.; Hao, C.; et al. Safety and efficacy of sorafenib therapy in patients with hepatocellular carcinoma: Final outcome from the Chinese patient subset of the GIDEON study. Oncotarget 2016, 7, 6639–6648. [Google Scholar] [CrossRef]
- McNamara, M.G.; Slagter, A.E.; Nuttall, C.; Frizziero, M.; Pihlak, R.; Lamarca, A.; Tariq, N.; Valle, J.W.; Hubner, R.A.; Knox, J.J.; et al. Sorafenib as first-line therapy in patients with advanced Child-Pugh B hepatocellular carcinoma-a meta-analysis. Eur. J. Cancer 2018, 105, 1–9. [Google Scholar] [CrossRef]
- Raoul, J.L.; Bruix, J.; Greten, T.F.; Sherman, M.; Mazzaferro, V.; Hilgard, P.; Scherubl, H.; Scheulen, M.E.; Germanidis, G.; Dominguez, S.; et al. Relationship between baseline hepatic status and outcome, and effect of sorafenib on liver function: SHARP trial subanalyses. J. Hepatol. 2012, 56, 1080–1088. [Google Scholar] [CrossRef]
- Harding, J.J.; Nandakumar, S.; Armenia, J.; Khalil, D.N.; Albano, M.; Ly, M.; Shia, J.; Hechtman, J.F.; Kundra, R.; El Dika, I.; et al. Prospective genotyping of hepatocellular carcinoma: Clinical implications of next-generation sequencing for matching patients to targeted and immune therapies. Clin. Cancer Res. 2019, 25, 2116–2126. [Google Scholar] [CrossRef] [Green Version]
- Huang, A.; Yang, X.R.; Chung, W.Y.; Dennison, A.R.; Zhou, J. Targeted therapy for hepatocellular carcinoma. Signal Transduct. Target. Ther. 2020, 11, 146. [Google Scholar] [CrossRef] [PubMed]
- Niu, M.; Yi, M.; Li, N.; Wu, K.; Wu, K. Advances of Targeted Therapy for Hepatocellular Carcinoma. Front. Oncol. 2021, 11, 719896. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, W.; Jiang, L.; Chen, Y. Recent advances in systemic therapy for hepatocellular carcinoma. Biomark. Res. 2022, 10, 3. [Google Scholar] [CrossRef] [PubMed]
- Tella, S.H.; Kommalapati, A.; Mahipal, A. Systemic therapy for advanced hepatocellular carcinoma: Targeted therapies. Chin. Clin. Oncol. 2021, 10, 10. [Google Scholar] [CrossRef] [PubMed]
- Stotz, M.; Gerger, A.; Haybaeck, J.; Kiesslich, T.; Bullock, M.D.; Pichler, M. Molecular Targeted Therapies in Hepatocellular Carcinoma: Past, Present and Future. Anticancer Res. 2015, 3, 5737–5744. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study | Agent | N | OS or PE | HR | p Value | Result | |||
|---|---|---|---|---|---|---|---|---|---|
| Name | Author | Year | |||||||
| 1 | SHARP | Llovet et al. [82] | 2008 | Sorafenib vs. placebo | 602 | 10.7 vs. 7.9 | 0.69 (0.55–0.87) | <0.001 | Confirmed efficacy |
| 2 | ASIA-PACIFIC | Cheng et al. [83] | 2009 | Sorafenib vs. placebo | 271 | 6.5 vs. 4.2 | 0.68 (0.50–0.93) | 0.014 | Confirmed efficacy |
| 3 | SUN | Cheng et al. [101] | 2013 | Sunitinib vs. Sorafenib | 1074 | 7.9 vs. 10.2 | 1.30 (1.13–1.50) | 0.0014 | Negative |
| 4 | SEARCH | Zhu et al. [51] | 2015 | Sorafenib plus Erlotinb vs. Sorafenib plus placebo | 720 | 9.5 vs. 8.5 | 0.93 (0.78–1.11) | 0.408 | Negative |
| 5 | LIGHT | Cainap et al. [67] | 2015 | Linifanib vs. Sorafenib | 1035 | 9.1 vs. 9.8 | 1.04 (0.896–1.221) | - | Negative |
| 6 | REFLECT | Kudo et al. [95] | 2018 | Lenvatinib vs. Sorafenib | 954 | 13.6 vs. 12.3 | 0.92 (0.79–1.06) | - | Confirmed efficacy |
| 7 | IMBRAVE150 | Finn et al. [145] | 2020 | Bevacizumab plus Atezolizumab vs. Sorafenib | 501 | 12 m OS: 67.2% vs. 54.6% | 0.58 (0.42–0.79) | <0.001 | Confirmed efficacy |
| 8 | - | Qin et al. [108] | 2021 | Donafenib vs. Sorafenib | 659 | 12.1 vs. 10.3 | 0.831 (0.699–0.988) | 0.0245 | Confirmed non-inferiority as well as superiority of Donafenib |
| 9 | ORIENT-32 | Ren et al. [178] | 2021 | Sintilimab plus Bevacizumab | 571 | Not reached vs. 10.4 | 0.57 (0.43–0.75) | <0.0001 | Confirmed efficacy |
| 10 | CheckMate 459 | Yau et al. [172] | 2022 | Nivolumab vs. Sorafenib | 743 | 16.4 vs. 14.7 | 0.85 (0.72–1.02) | 0.075 | No superiority of Nivolumab |
| Study | Agent | N | OS or PE | HR | p Value | Result | |||
|---|---|---|---|---|---|---|---|---|---|
| Name | Author | Year | |||||||
| 1 | METIV-HCC | Santoro et al. [117] | 2013 | Tivantinib vs. placebo | 340 | 8.4 vs. 9.1 | 0.97 (0.75–1.25) | 0.81 | Negative |
| 2 | REACH | Zhu et al. [128] | 2015 | Ramucirumab vs. placebo | 565 | 9.2 vs. 7.6 | 0.87 | 0.14 | No superiority of Ramucirumab |
| 3 | RESORCE | Bruix et al. [101] | 2017 | Regorafenib vs. placebo | 573 | 10.6 vs. 7.8 | 0.63 (0.50–0.79) | <0.001 | Confirmed efficacy |
| 4 | CALESTIAL | Abou-Alfa et al. [112] | 2018 | Cabozantinib vs. placebo | 707 | 20.2 vs. 8.0 | 0.76 (0.63–0.92) | 0.0049 | Confirmed efficacy |
| 5 | REACH-2 | Zhu et al. [129] | 2019 | Ramucirumab vs. placebo | 295 | 8.5 vs. 7.3 | 0.71 (0.531–0.949) | 0.0199 | Confirmed efficacy |
| 6 | JET-HCC | Kudo et al. [118] | 2020 | Tivantinib vs. placebo | 195 | 10.3 vs. 8.5 | 0.82 (0.58–1.15) | 0.082 | Negative |
| 7 | KEYNOTE-240 | Finn et al. [145] | 2020 | Pembrolizumab vs. placebo | 413 | 13.9 vs. 10.6 | 0.781 (0.6–0.998) | 0.238 | Efficacy not confirmed in terms of OS |
| 8 | AHELP | Qin et al. [146] | 2021 | Apatinib vs. placebo | 393 | 8.7 vs. 6.8 | 0.785 (0.617–0.998) | 0.048 | Confirmed efficacy |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Damaskos, C.; Garmpis, N.; Dimitroulis, D.; Garmpi, A.; Psilopatis, I.; Sarantis, P.; Koustas, E.; Kanavidis, P.; Prevezanos, D.; Kouraklis, G.; et al. Targeted Therapies for Hepatocellular Carcinoma Treatment: A New Era Ahead—A Systematic Review. Int. J. Mol. Sci. 2022, 23, 14117. https://doi.org/10.3390/ijms232214117
Damaskos C, Garmpis N, Dimitroulis D, Garmpi A, Psilopatis I, Sarantis P, Koustas E, Kanavidis P, Prevezanos D, Kouraklis G, et al. Targeted Therapies for Hepatocellular Carcinoma Treatment: A New Era Ahead—A Systematic Review. International Journal of Molecular Sciences. 2022; 23(22):14117. https://doi.org/10.3390/ijms232214117
Chicago/Turabian StyleDamaskos, Christos, Nikolaos Garmpis, Dimitrios Dimitroulis, Anna Garmpi, Iason Psilopatis, Panagiotis Sarantis, Evangelos Koustas, Prodromos Kanavidis, Dionysios Prevezanos, Gregory Kouraklis, and et al. 2022. "Targeted Therapies for Hepatocellular Carcinoma Treatment: A New Era Ahead—A Systematic Review" International Journal of Molecular Sciences 23, no. 22: 14117. https://doi.org/10.3390/ijms232214117