
Polyphosphate Activates von Willebrand Factor Interaction with Glycoprotein Ib in the Absence of Factor VIII In Vitro

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
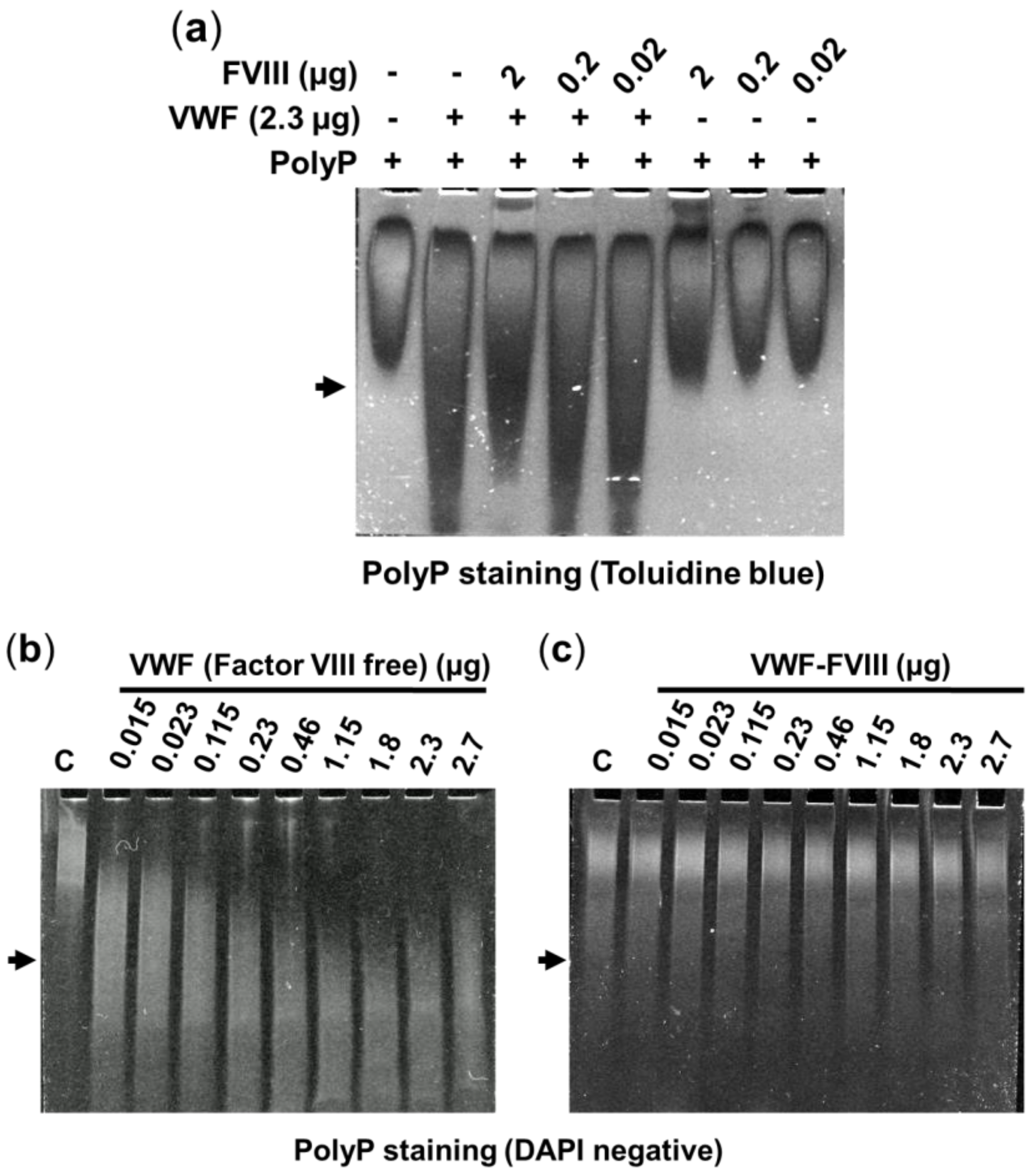
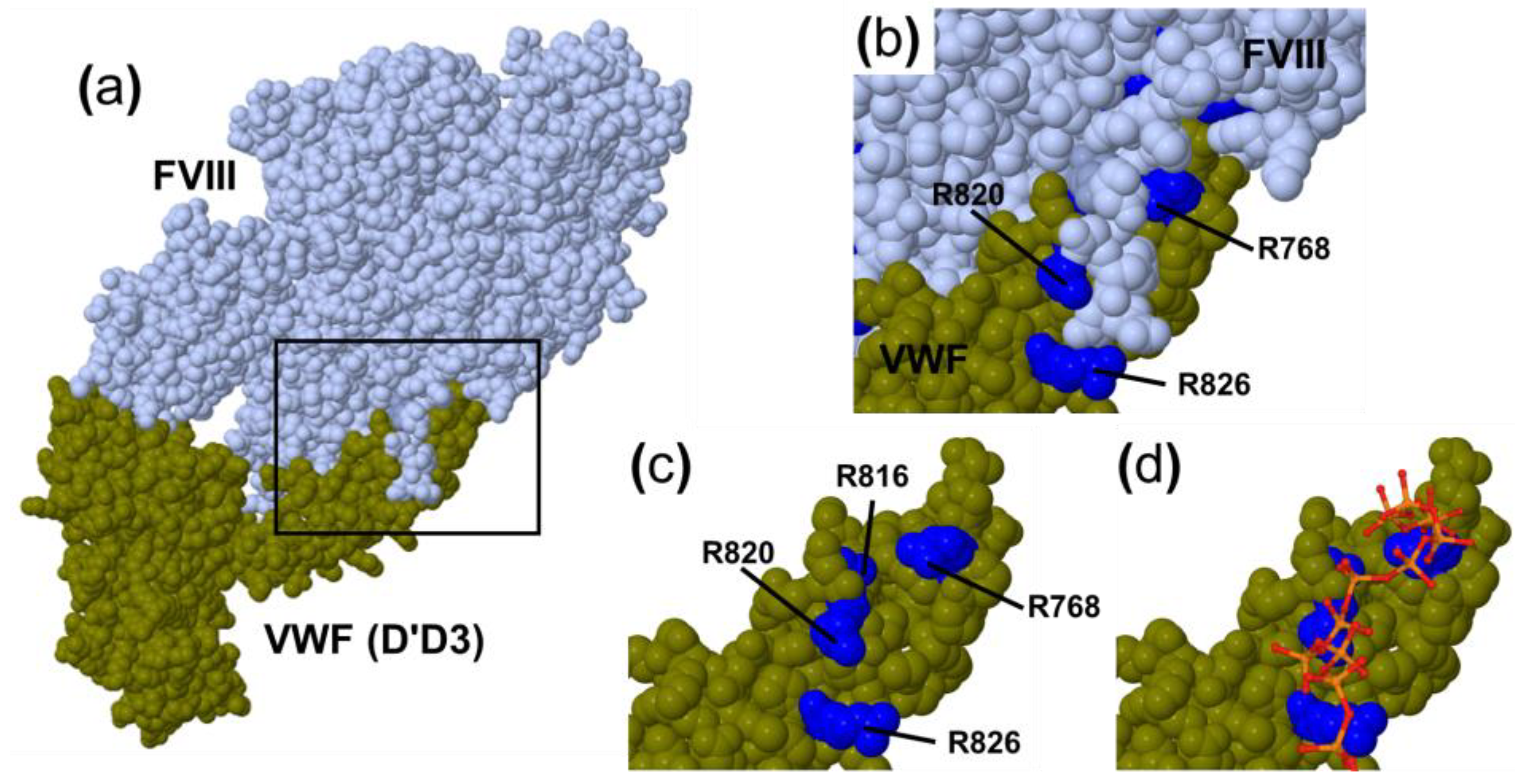
2.1. Effect of Factor VIII on the Interaction between VWF and PolyP
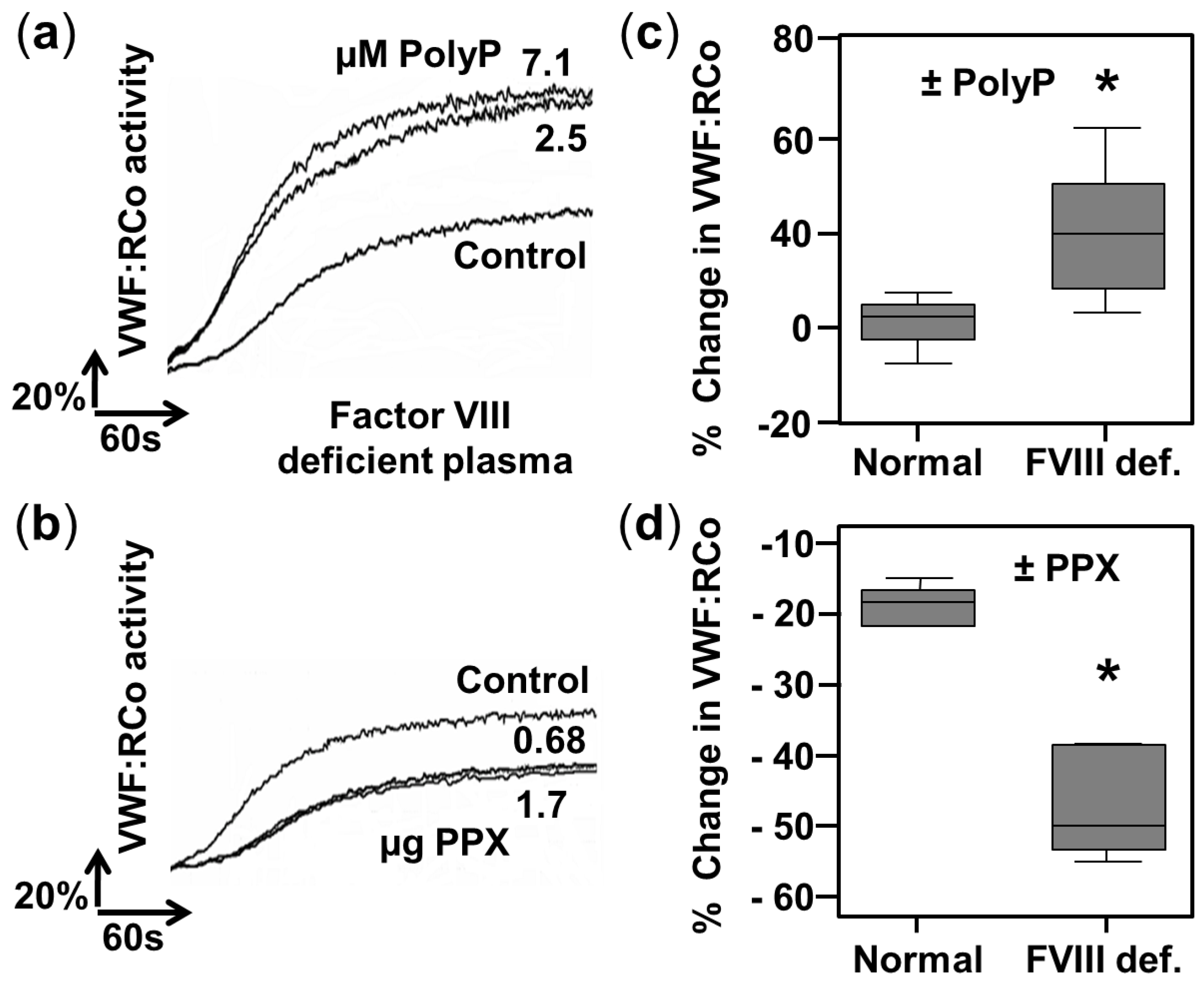
2.2. PolyP Increases VWF Ristocetin Co-factor Activity in Factor VIII-Deficient Plasmas
2.3. PolyP Increases VWF Ristocetin Co-factor Activity in Severe Hemophilia A Plasmas
2.4. Exopolyphosphatase Cannot Inhibit VWF Ristocetin Co-factor Activity in the Presence of Factor VIII
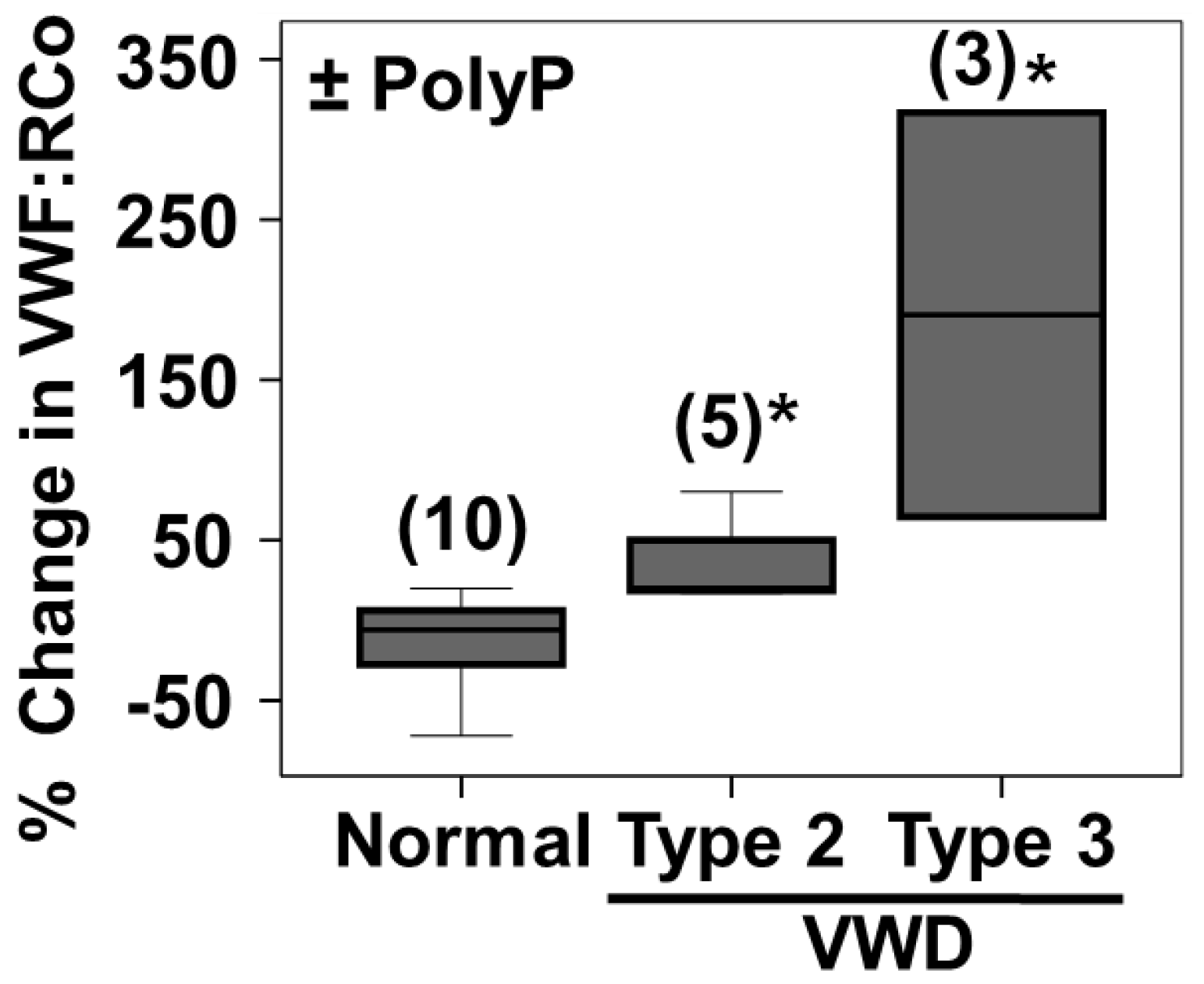
2.5. PolyP Increases VWF Ristocetin Co-factor Activity on Type 2 and Type 3 VWD
3. Discussion
4. Materials and Methods
4.1. Plasma Samples
4.2. Exopolyphosphatase Isolation and PolyP Measurements
4.3. VWF Ristocetin Co-factor (VWF:RCo) Activity
4.4. Surface Plasmon Resonance
4.5. Separation of PolyP by Urea–Polyacrylamide Gel Electrophoresis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ruiz, F.A.; Lea, C.R.; Oldfield, E.; Docampo, R. Human Platelet Dense Granules Contain Polyphosphate and Are Similar to Acidocalcisomes of Bacteria and Unicellular Eukaryotes. J. Biol. Chem. 2004, 279, 44250–44257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno-Sanchez, D.; Hernandez-Ruiz, L.; Ruiz, F.A.; Docampo, R. Polyphosphate Is a Novel Pro-inflammatory Regulator of Mast Cells and Is Located in Acidocalcisomes. J. Biol. Chem. 2012, 287, 28435–28444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrissey, J.H.; Smith, S.A. Polyphosphate as modulator of hemostasis, thrombosis, and inflammation. J. Thromb. Haemost. 2015, 13 (Suppl. 1), S92–S97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, S.A.; Mutch, N.J.; Baskar, D.; Rohloff, P.; Docampo, R.; Morrissey, J.H. Polyphosphate modulates blood coagulation and fibrinolysis. Proc. Natl. Acad. Sci. USA 2006, 103, 903–908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, S.H.; Smith, S.A.; Morrissey, J.H. Polyphosphate is a cofactor for the activation of factor XI by thrombin. Blood 2011, 118, 6963–6970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, S.H.; Smith, S.A.; Morrissey, J.H. Polyphosphate accelerates factor V activation by factor XIa. Thromb. Haemost. 2015, 113, 599–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mutch, N.J.; Myles, T.; Leung, L.L.K.; Morrissey, J.H. Polyphosphate binds with high affinity to exosite II of thrombin. J. Thromb. Haemost. 2010, 8, 548–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puy, C.; Tucker, E.I.; Ivanov, I.S.; Gailani, D.; Smith, S.A.; Morrissey, J.H.; Gruber, A.; McCarty, O.J.T. Platelet-Derived Short-Chain Polyphosphates Enhance the Inactivation of Tissue Factor Pathway Inhibitor by Activated Coagulation Factor XI. PLoS ONE 2016, 11, e0165172. [Google Scholar] [CrossRef] [Green Version]
- Mutch, N.J.; Engel, R.; de Willige, S.U.; Philippou, H.; Ariëns, R.A.S. Polyphosphate modifies the fibrin network and down-regulates fibrinolysis by attenuating binding of tPA and plasminogen to fibrin. Blood 2010, 115, 3980–3988. [Google Scholar] [CrossRef] [Green Version]
- Verhoef, J.J.F.; Barendrecht, A.D.; Nickel, K.F.; Dijkxhoorn, K.; Kenne, E.; Labberton, L.; Mccarty, O.J.T.; Schiffelers, R.; Heijnen, H.F.; Hendrickx, A.P.; et al. Polyphosphate nanoparticles on the platelet surface trigger contact system activation. Blood 2017, 129, 1707–1717. [Google Scholar] [CrossRef]
- Wijeyewickrema, L.C.; Lameignere, E.; Hor, L.; Duncan, R.C.; Shiba, T.; Travers, R.J.; Kapopara, P.R.; Lei, V.; Smith, S.A.; Kim, H.; et al. Polyphosphate is a novel cofactor for regulation of complement by a serpin, C1 inhibitor. Blood 2016, 128, 1766–1776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, S.A.; Morrissey, J.H. Polyphosphate enhances fibrin clot structure. Blood 2008, 112, 2810–2816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montilla, M.; Hernández-Ruiz, L.; García-Cozar, F.J.; Alvarez-Laderas, I.; Rodríguez-Martorell, J.; Ruiz, F.A. Polyphosphate binds to human von Willebrand factor in vivo and modulates its interaction with glycoprotein Ib. J. Thromb. Haemost. 2012, 10, 2315–2323. [Google Scholar] [CrossRef] [PubMed]
- Bryckaert, M.; Rosa, J.-P.; Denis, C.V.; Lenting, P.J. Of von Willebrand factor and platelets. Cell. Mol. Life Sci. 2014, 72, 307–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Favaloro, E.J.; Pasalic, L.; Curnow, J. Laboratory tests used to help diagnose von Willebrand disease: An update. Pathology 2016, 48, 303–318. [Google Scholar] [CrossRef]
- Skornova, I.; Simurda, T.; Stasko, J.; Zolkova, J.; Sokol, J.; Holly, P.; Dobrotova, M.; Plamenova, I.; Hudecek, J.; Brunclikova, M.; et al. Multimer Analysis of Von Willebrand Factor in Von Willebrand Disease with a Hydrasys Semi-Automatic Analyzer—Single-Center Experience. Diagnostics 2021, 11, 2153. [Google Scholar] [CrossRef]
- Favaloro, E.J.; Oliver, S.; Mohammed, S.; Vong, R. Comparative assessment of von Willebrand factor multimers vs activity for von Willebrand disease using modern contemporary methodologies. Haemophilia 2020, 26, 503–512. [Google Scholar] [CrossRef]
- Sadler, J.E. von Willebrand factor assembly and secretion. J. Thromb. Haemost. 2009, 7 (Suppl. 1), 24–27. [Google Scholar] [CrossRef]
- Wagner, D.D. Cell Biology of von Willebrand Factor. Annu. Rev. Cell Biol. 1990, 6, 217–242. [Google Scholar] [CrossRef]
- Federici, A.B. The factor VIII/von Willebrand factor complex: Basic and clinical issues. Haematologica 2003, 88, EREP02. [Google Scholar]
- Peake, I.; Goodeve, A. Type 1 von Willebrand disease. J. Thromb. Haemost. 2007, 5 (Suppl. 1), 7–11. [Google Scholar] [CrossRef] [PubMed]
- Simurda, T.; Dobrotova, M.; Skornova, I.; Sokol, J.; Kubisz, P.; Stasko, J. Successful Use of a Highly Purified Plasma von Willebrand Factor Concentrate Containing Little FVIII for the Long-Term Prophylaxis of Severe (Type 3) von Willebrand’s Disease. Semin. Thromb. Hemost. 2017, 43, 639–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.-F.; Eng, E.T.; Zhu, J.; Lu, C.; Walz, T.; Springer, T.A. Sequence and structure relationships within von Willebrand factor. Blood 2012, 120, 449–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuller, J.R.; Knockenhauer, K.E.; Leksa, N.C.; Peters, R.T.; Batchelor, J.D. Molecular determinants of the factor VIII/von Willebrand factor complex revealed by BIVV001 cryo-electron microscopy. Blood 2021, 137, 2970–2980. [Google Scholar] [CrossRef] [PubMed]
- Sehnal, D.; Bittrich, S.; Deshpande, M.; Svobodová, R.; Berka, K.; Bazgier, V.; Velankar, S.; Burley, S.K.; Koča, J.; Rose, A.S. Mol* Viewer: Modern web app for 3D visualization and analysis of large biomolecular structures. Nucleic Acids Res. 2021, 49, W431–W437. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.; Choi, S.H.; Davis-Harrison, R.; Huyck, J.; Boettcher, J.; Rienstra, C.M.; Morrissey, J. Polyphosphate exerts differential effects on blood clotting, depending on polymer size. Blood 2010, 116, 4353–4359. [Google Scholar] [CrossRef]
- Pipe, S.W.; Montgomery, R.R.; Pratt, K.P.; Lenting, P.J.; Lillicrap, D. Life in the shadow of a dominant partner: The FVIII-VWF association and its clinical implications for hemophilia A. Blood 2016, 128, 2007–2016. [Google Scholar] [CrossRef] [Green Version]
- Shi, Q.; Kuether, E.L.; Schroeder, J.A.; Perry, C.L.; Fahs, S.A.; Gill, J.C.; Montgomery, R.R. Factor VIII inhibitors: Von Willebrand factor makes a difference in vitro and in vivo. J. Thromb. Haemost. 2012, 10, 2328–2337. [Google Scholar] [CrossRef] [Green Version]
- Iorio, A.; Halimeh, S.; Holzhauer, S.; Goldenberg, N.; Marchesini, E.; Marcucci, M.; Young, G.; Bidlingmaier, C.; Brandao, L.R.; Ettingshausen, C.E.; et al. Rate of inhibitor development in previously untreated hemophilia A patients treated with plasma-derived or recombinant factor VIII concentrates: A systematic review. J. Thromb. Haemost. 2010, 8, 1256–1265. [Google Scholar] [CrossRef]
- Smith, S.A.; Morrissey, J.H. Polyphosphate as a general procoagulant agent. J. Thromb. Haemost. 2008, 6, 1750–1756. [Google Scholar] [CrossRef] [Green Version]
- Weiss, H.J. Abnormalities of factor VIII and platelet aggregation—Use of ristocetin in diagnosing the von Willebrand syndrome. Blood 1975, 45, 403–412. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, Y.; Nogami, K.; Yada, K.; Kawamura, T.; Ogiwara, K.; Furukawa, S.; Shimonishi, N.; Takeyama, M.; Shima, M. Evaluation of clinical severity in patients with type 2N von Willebrand disease using microchip-based flow-chamber system. Int. J. Hematol. 2020, 111, 369–377. [Google Scholar] [CrossRef]
- Hrdinova, J.; Fernández, D.I.; Ercig, B.; Tullemans, B.M.E.; Suylen, D.P.L.; Agten, S.M.; Jurk, K.; Hackeng, T.M.; Vanhoorelbeke, K.; Voorberg, J.; et al. Structure-Based Cyclic Glycoprotein Ibα-Derived Peptides Interfering with von Willebrand Factor-Binding, Affecting Platelet Aggregation under Shear. Int. J. Mol. Sci. 2022, 23, 2046. [Google Scholar] [CrossRef]
- Okhota, S.; Melnikov, I.; Avtaeva, Y.; Kozlov, S.; Gabbasov, Z. Shear Stress-Induced Activation of von Willebrand Factor and Cardiovascular Pathology. Int. J. Mol. Sci. 2020, 21, 7804. [Google Scholar] [CrossRef] [PubMed]
- Vollack-Hesse, N.; Oleshko, O.; Werwitzke, S.; Solecka-Witulska, B.; Kannicht, C.; Tiede, A. Recombinant VWF fragments improve bioavailability of subcutaneous factor VIII in hemophilia A mice. Blood 2021, 137, 1072–1081. [Google Scholar] [CrossRef] [PubMed]
- Morfini, M.; Auerswald, G.; Kobelt, R.A.; Rivolta, G.F.; Rodríguez-Martorell, J.; Scaraggi, F.A.; Altisent, C.; Blatný, J.; Borel-Derlon, A.; Rossi, V. Prophylactic treatment of haemophilia patients with inhibitors: Clinical experience with recombinant factor VIIa in European Haemophilia Centres. Haemophilia 2007, 13, 502–507. [Google Scholar] [CrossRef] [PubMed]
- Álvarez-Laderas, I.; Núñez, R.; Jiménez-Bárcenas, R.; Martorell, F.J.R.; Garcia-Lozano, J.R.; de Cos, C.; Garrido, R.P. The spectrum of mutations in Southern Spanish patients with von Willebrand disease. Haemophilia 2015, 21, e240–e242. [Google Scholar] [CrossRef]
- Wurst, H.; Kornberg, A. A soluble exopolyphosphatase of Saccharomyces cerevisiae. Purification and characterization. J. Biol. Chem. 1994, 269, 10996–11001. [Google Scholar] [CrossRef]
- Hernandez-Ruiz, L.; González-García, I.; Castro, C.; A Brieva, J.; A Ruiz, F. Inorganic polyphosphate and specific induction of apoptosis in human plasma cells. Haematologica 2006, 91, 1180–1186. [Google Scholar]
- Smith, S.A.; Morrissey, J.H. Sensitive fluorescence detection of polyphosphate in polyacrylamide gels using 4′,6-diamidino-2-phenylindol. Electrophoresis 2007, 28, 3461–3465. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Montilla, M.; Atienza-Navarro, I.; García-Cozar, F.J.; Castro, C.; Rodríguez-Martorell, F.J.; Ruiz, F.A. Polyphosphate Activates von Willebrand Factor Interaction with Glycoprotein Ib in the Absence of Factor VIII In Vitro. Int. J. Mol. Sci. 2022, 23, 14118. https://doi.org/10.3390/ijms232214118
Montilla M, Atienza-Navarro I, García-Cozar FJ, Castro C, Rodríguez-Martorell FJ, Ruiz FA. Polyphosphate Activates von Willebrand Factor Interaction with Glycoprotein Ib in the Absence of Factor VIII In Vitro. International Journal of Molecular Sciences. 2022; 23(22):14118. https://doi.org/10.3390/ijms232214118
Chicago/Turabian StyleMontilla, Marcela, Isabel Atienza-Navarro, Francisco Jose García-Cozar, Carmen Castro, Francisco Javier Rodríguez-Martorell, and Felix A. Ruiz. 2022. "Polyphosphate Activates von Willebrand Factor Interaction with Glycoprotein Ib in the Absence of Factor VIII In Vitro" International Journal of Molecular Sciences 23, no. 22: 14118. https://doi.org/10.3390/ijms232214118