


SWATH Mass Spectrometry-Based CSF Proteome Profile of GBA-Linked Parkinson’s Disease Patients
 , , , ,
, , , , 
Abstract
:1. Introduction
2. Results
2.1. GBA-Mutation-Linked Proteomic Alterations in PD Patients
2.2. Pathway Analysis and Functional Characterization
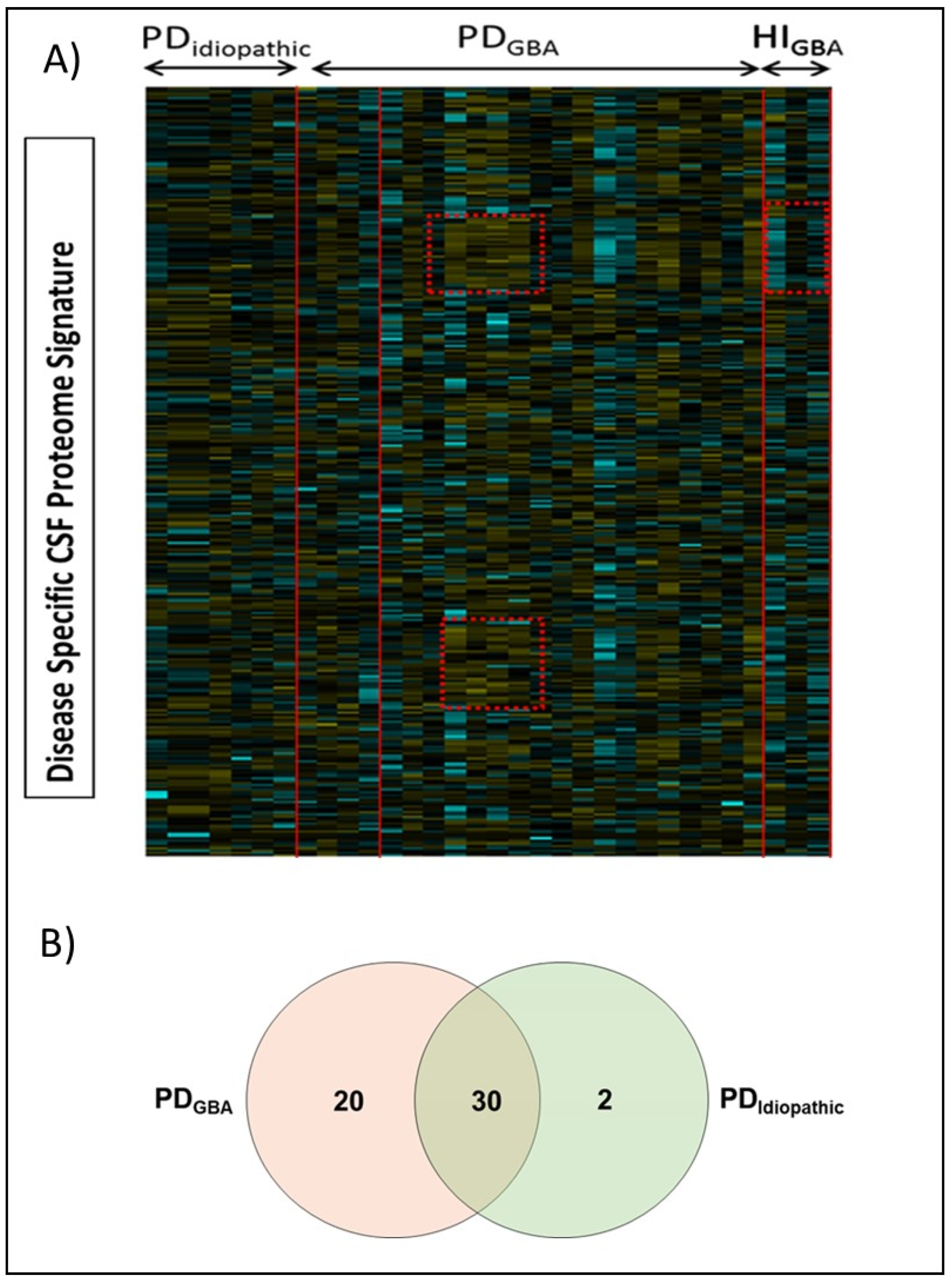
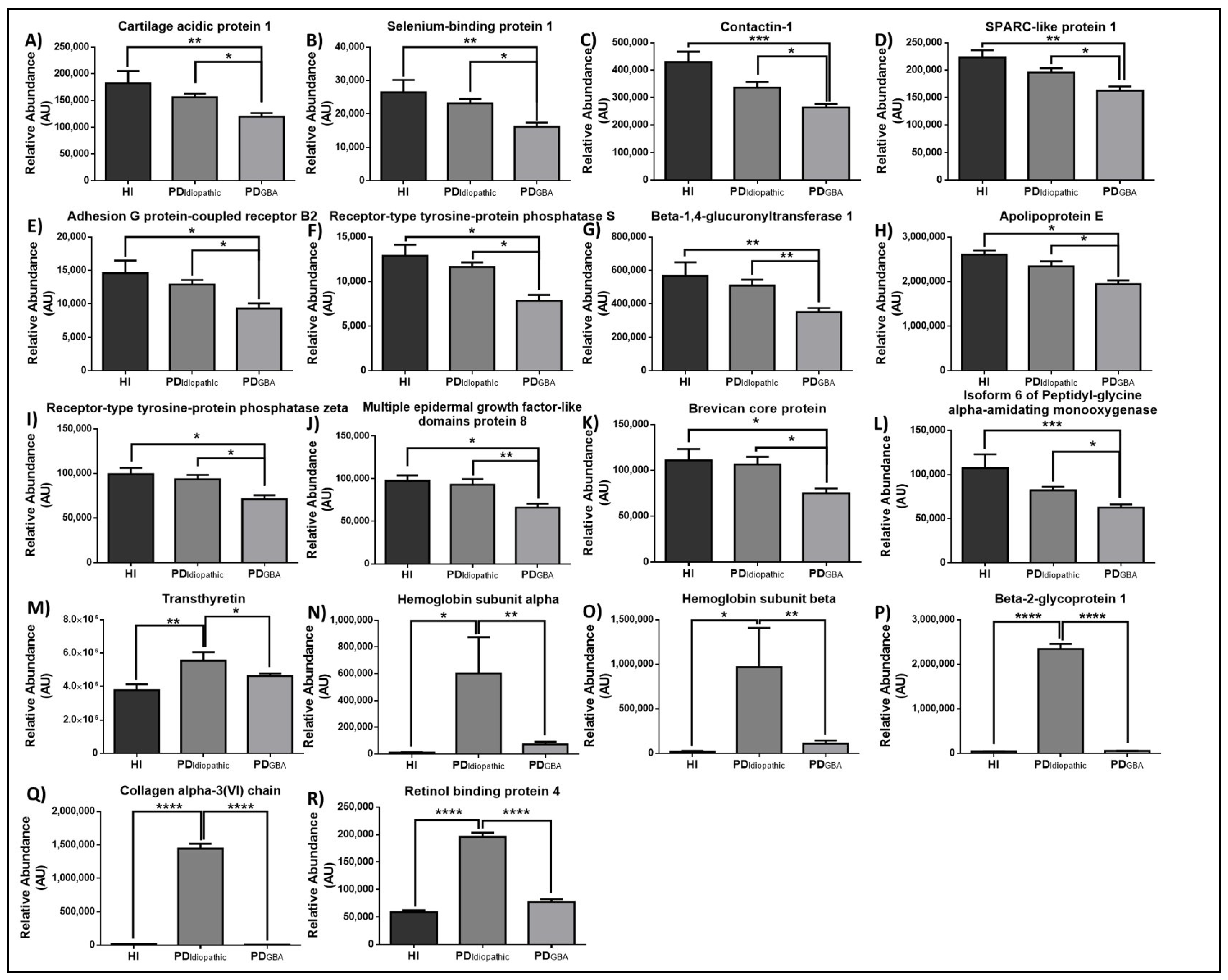
2.3. Proteomic Alterations in Response to GBA-Mutation in PD Patients
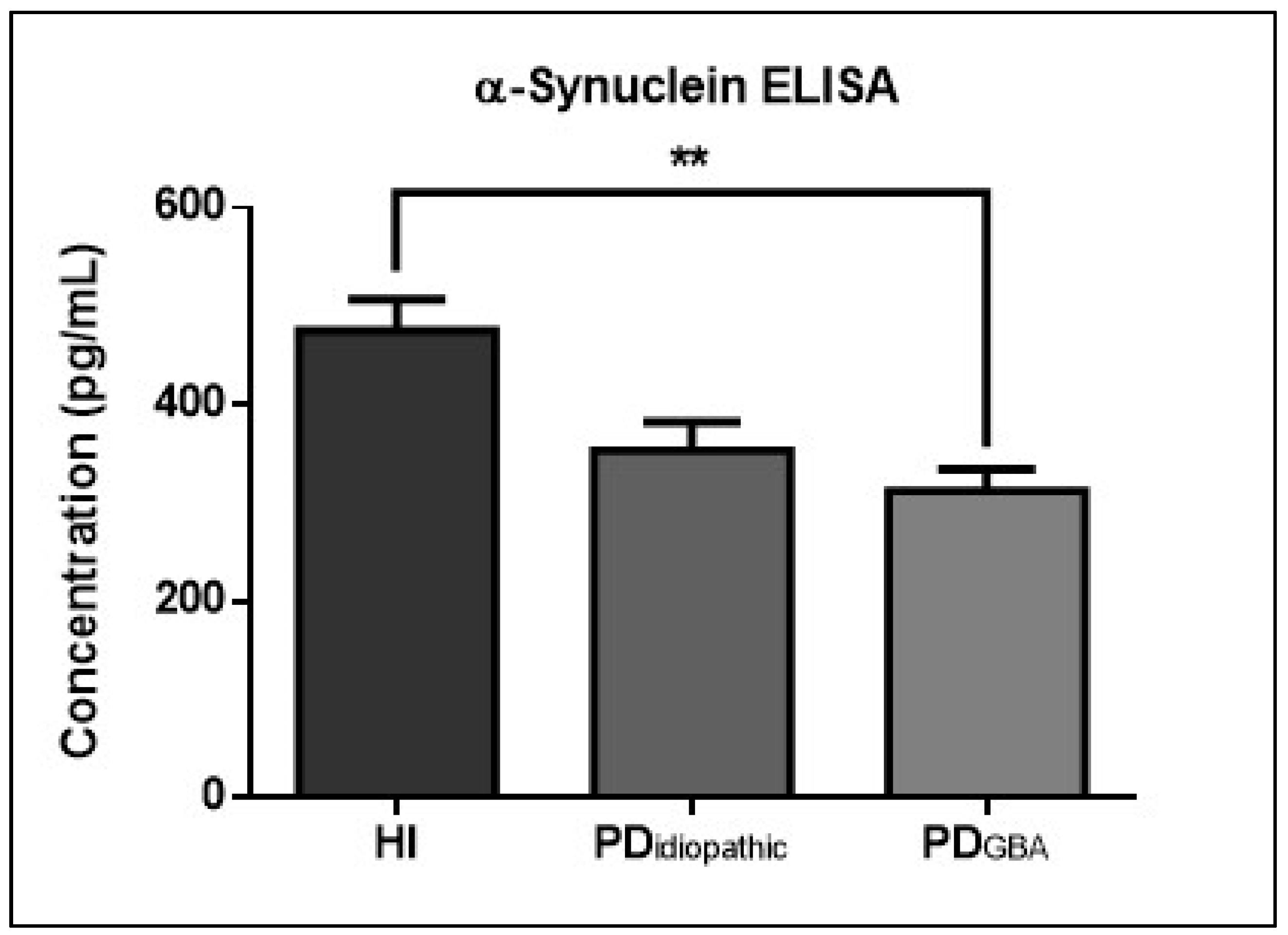
2.4. CSF α-Syn Profiles in PD Patients with GBA Mutations
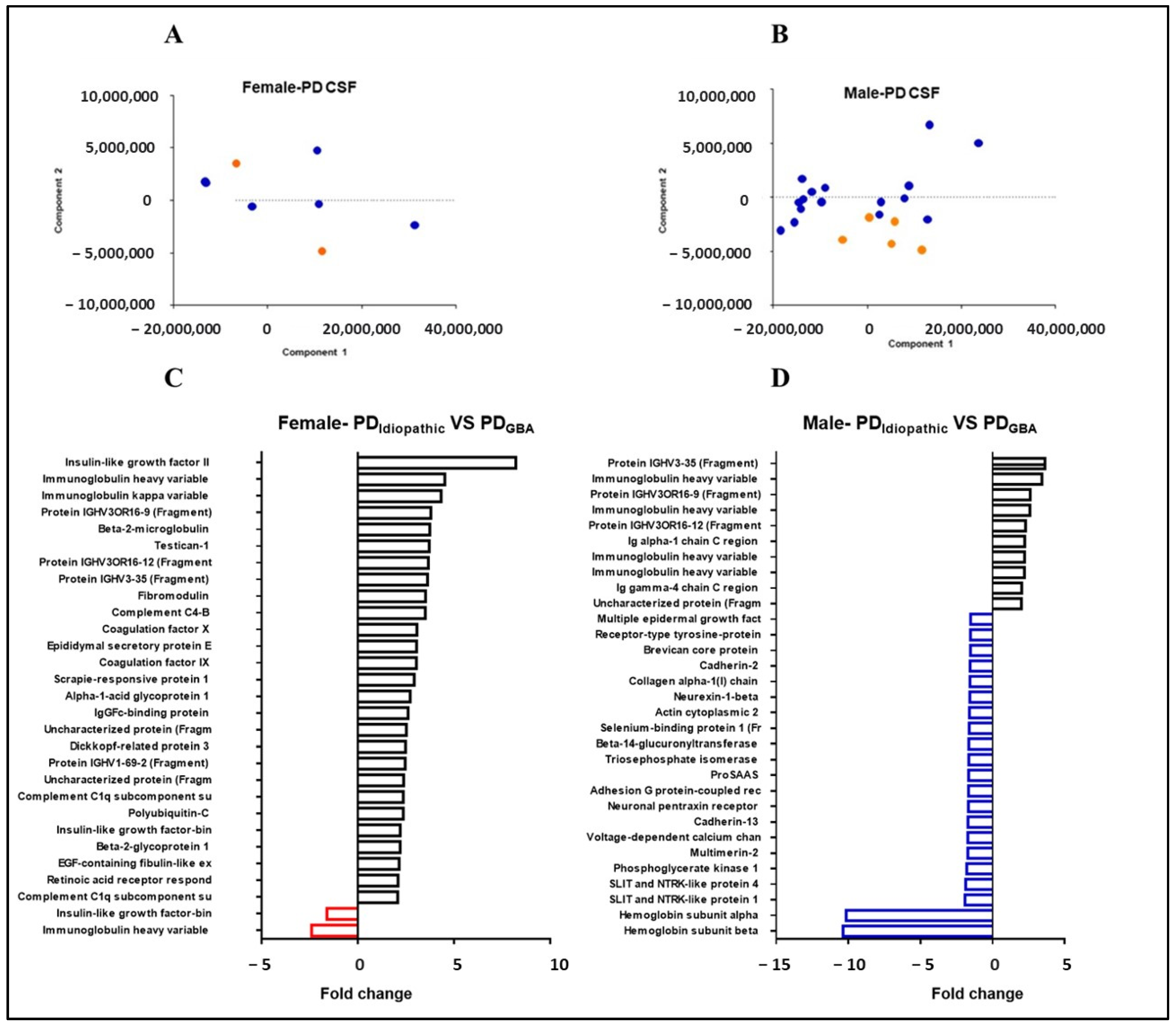
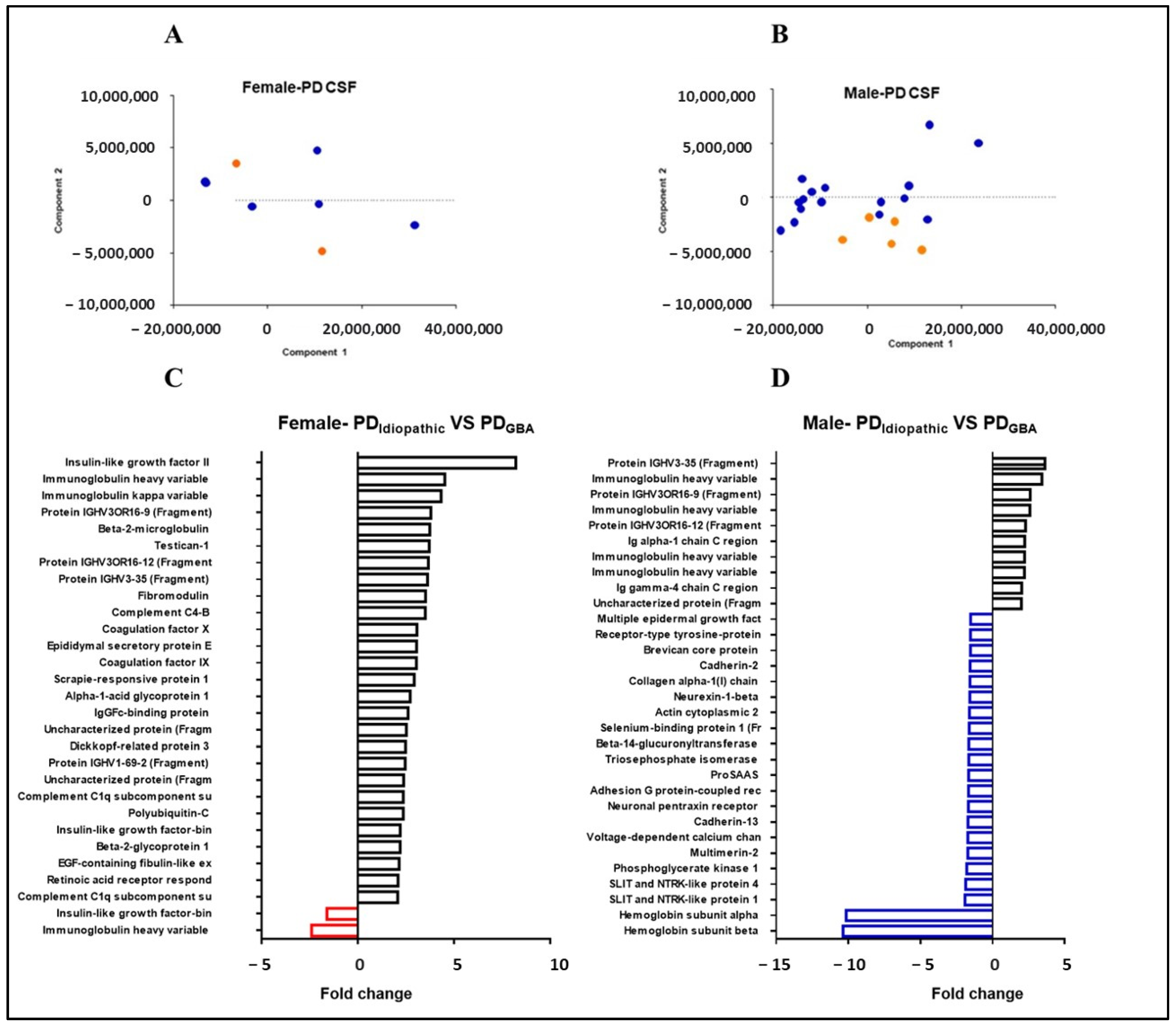
2.5. Gender-Specific Proteome Alterations in PD Patients with GBA Mutations
3. Discussion
4. Materials and Methods
4.1. Participants and Ethical Approval
4.2. Sample Collection, Preparation and In-Gel Tryptic Digestion
4.3. Quantitative SWATH Analysis
4.4. Protein Annotation and Functional Characterization
4.5. ELISA
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tysnes, O.B.; Storstein, A. Epidemiology of Parkinson’s disease. J. Neural Transm. Suppl. 2017, 124, 901–905. [Google Scholar] [CrossRef]
- Parkinson, J. An essay on the shaking palsy. J. Neuropsychiatry Clin. Neurosci. 2002, 14, 223–236. [Google Scholar] [CrossRef] [PubMed]
- Baba, M.; Nakajo, S.; Tu, P.H.; Tomita, T.; Nakaya, K.; Lee, V.M.; Trojanowski, J.Q.; Iwatsubo, T. Aggregation of alpha-synuclein in Lewy bodies of sporadic Parkinson’s disease and dementia with Lewy bodies. Am. J. Pathol. 1998, 152, 879. [Google Scholar] [PubMed]
- Jankovic, J. Parkinson’s disease: Clinical features and diagnosis. J. Neurol. Neurosurg. Psychiatry 2008, 79, 368–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frank, C.; Pari, G.; Rossiter, J.P. Approach to diagnosis of Parkinson disease. Can. Fam. Physician 2006, 52, 862–868. [Google Scholar]
- McCormack, A.L.; Thiruchelvam, M.; Manning-Bog, A.B.; Thiffaulta, C.; Langstona, J.W.; Cory-Slechta, D.A.; Di Monte, D.A. Environmental risk factors and Parkinson’s disease: Selective degeneration of nigral dopaminergic neurons caused by the herbicide paraquat. Neurobiol. Dis. 2002, 10, 119–127. [Google Scholar] [CrossRef] [Green Version]
- Satake, W.; Nakabayashi, Y.; Mizuta, I.; Hirota, Y.; Ito, C.; Kubo, M.; Kawaguchi, T.; Tsunoda, T.; Watanabe, M.; Takeda, A.; et al. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat. Genet. 2009, 41, 1303–1307. [Google Scholar] [CrossRef]
- Collier, T.J.; Kanaan, N.M.; Kordower, J.H. Aging and Parkinson’s disease: Different sides of the same coin? Mov. Disord. 2017, 32, 983–990. [Google Scholar] [CrossRef] [Green Version]
- Goker-Alpan, O.; Schiffmann, R.; LaMarca, M.E.; Nussbaum, R.L.; McInerney-Leo, A.; Sidransky, E. Parkinsonism among Gaucher disease carriers. J. Med. Genet. 2004, 41, 937–940. [Google Scholar] [CrossRef] [Green Version]
- Klein, C.; Westenberger, A. Genetics of Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2. [Google Scholar] [CrossRef] [Green Version]
- Grabowski, G.A.; Dinur, T.; Osiecki, K.M.; Kruse, J.R.; Legler, G.; Gatt, S. Gaucher disease types 1, 2, and 3: Differential mutations of the acid beta-glucosidase active site identified with conduritol B epoxide derivatives and sphingosine. Am. J. Hum. Genet. 1985, 37, 499. [Google Scholar] [PubMed]
- Hruska, K.S.; LaMarca, M.E.; Scott, C.R.; Sidransky, E. Gaucher disease: Mutation and polymorphism spectrum in the glucocerebrosidase gene (GBA). Hum. Mutat. 2008, 29, 567–583. [Google Scholar] [CrossRef] [PubMed]
- O’Regan, G.; deSouza, R.M.; Balestrino, R.; Schapira, A.H. Glucocerebrosidase mutations in Parkinson disease. J. Parkinsons. Dis. 2017, 7, 411–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neumann, J.; Bras, J.; Deas, E.; O’Sullivan, S.S.; Parkkinen, L.; Lachmann, R.H.; Li, A.; Holton, J.; Guerreiro, R.; Paudel, R.; et al. Glucocerebrosidase mutations in clinical and pathologically proven Parkinson’s disease. Brain 2009, 132, 1783–1794. [Google Scholar] [CrossRef] [Green Version]
- Gan-Or, Z.; Amshalom, I.; Kilarski, L.L.; Bar-Shira, A.; Gana-Weisz, M.; Mirelman, A.; Marder, K.; Bressman, S.; Giladi, N.; Orr-Urtreger, A. Differential effects of severe vs mild GBA mutations on Parkinson disease. Neurology 2015, 84, 880–887. [Google Scholar] [CrossRef] [Green Version]
- Du, T.-T.; Wang, L.; Duan, C.-L.; Lu, L.-L.; Zhang, J.-L.; Gao, G.; Qiu, X.-B.; Wang, X.-M.; Yang, H. GBA deficiency promotes SNCA/α-synuclein accumulation through autophagic inhibition by inactivated PPP2A. Autophagy 2015, 11, 1803–1820. [Google Scholar] [CrossRef] [Green Version]
- Chahine, L.M.; Qiang, J.; Ashbridge, E.; Minger, J.; Yearout, D.; Horn, S.; Colcher, A.; Hurtig, H.I.; Lee, V.M.-Y.; Van Deerlin, V.M.; et al. Clinical and biochemical differences in patients having Parkinson disease with vs without GBA mutations. JAMA Neurol. 2013, 70, 852–858. [Google Scholar] [CrossRef] [Green Version]
- Mata, I.F.; Leverenz, J.B.; Weintraub, D.; Trojanowski, J.Q.; Chen-Plotkin, A.; Van Deerlin, V.M.; Ritz, B.; Rausch, R.; Do, S.A.F.; Wood-Siverio, C.; et al. GBA Variants are associated with a distinct pattern of cognitive deficits in Parkinson’s disease. Mov. Disord. 2016, 31, 95–102. [Google Scholar] [CrossRef] [Green Version]
- Davis, M.Y.; Johnson, C.O.; Leverenz, J.B.; Weintraub, D.; Trojanowski, J.Q.; Chen-Plotkin, A.; Van Deerlin, V.M.; Quinn, J.F.; Chung, K.A.; Peterson-Hiller, A.L.; et al. Association of GBA mutations and the E326K polymorphism with motor and cognitive progression in Parkinson disease. JAMA Neurol. 2016, 73, 1217–1224. [Google Scholar] [CrossRef] [Green Version]
- Brockmann, K.; Srulijes, K.; Pflederer, S.; Hauser, A.; Schulte, C.; Maetzler, W.; Gasser, T.; Berg, D. GBA-associated Parkinson’s disease: Reduced survival and more rapid progression in a prospective longitudinal study. Move. Disord. 2015, 30, 407–411. [Google Scholar] [CrossRef]
- Guedes, L.C.; Chan, R.B.; Gomes, M.A.; Conceição, V.A.; Machado, R.B.; Soares, T.; Xu, Y.; Gaspar, P.; Carriço, J.A.; Alcalay, R.N.; et al. Serum lipid alterations in GBA-associated Parkinson’s disease. Park. Relat. Disord. 2017, 44, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Adler, C.H.; Beach, T.G.; Shill, H.A.; Caviness, J.N.; Driver-Dunckley, E.; Sabbagh, M.N.; Patel, A.; Sue, L.I.; Serrano, G.; Jacobson, S.A.; et al. GBA mutations in Parkinson disease: Earlier death but similar neuropathological features. Eur. J. Neurol. 2017, 24, 1363–1368. [Google Scholar] [CrossRef] [PubMed]
- Gillet, L.C.; Navarro, P.; Tate, S.; Röst, H.; Selevsek, N.; Reiter, L.; Bonner, R.; Aebersold, R. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: A new concept for consistent and accurate proteome analysis. Mol. Cell Proteom. 2012, 11, O111.016717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winder-Rhodes, S.E.; Evans, J.R.; Ban, M.; Mason, S.L.; Williams-Gray, C.; Foltynie, T.; Duran, R.; Mencacci, N.E.; Sawcer, S.J.; Barker, R.A. Glucocerebrosidase mutations influence the natural history of Parkinson’s disease in a community-based incident cohort. Brain 2013, 136, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Anjo, S.I.; Santa, C.; Manadas, B. Short GeLC-SWATH: A fast and reliable quantitative approach for proteomic screenings. Proteomics 2015, 15, 757–762. [Google Scholar] [CrossRef]
- Miller, I.N.; Cronin-Golomb, A. Gender differences in Parkinson’s disease: Clinical characteristics and cognition. Mov. Disord. 2010, 25, 2695–2703. [Google Scholar] [CrossRef] [Green Version]
- De Deyn, P.P.; Hiramatsu, M.; Borggreve, F.; Goeman, J.; Dʼhooge, R.; Saerens, J.; Mori, A. Superoxide dismutase activity in cerebrospinal fluid of patients with dementia and some other neurological disorders. Alzheimer Dis. Assoc. Disord. 1998, 12, 26–32. [Google Scholar] [CrossRef]
- Kim, H.J.; Jeon, B.; Song, J.; Lee, W.W.; Park, H.; Shin, C. Leukocyte glucocerebrosidase and β-hexosaminidase activity in sporadic and genetic Parkinson disease. Park. Relat. Disord. 2016, 23, 99–101. [Google Scholar] [CrossRef]
- Famitafreshi, H.; Karimian, M. Prostaglandins as the agents that modulate the course of brain disorders. Degener. Neurol. Neuromuscul. Dis. 2020, 10, 1–13. [Google Scholar] [CrossRef]
- Singh, K.; Jayaram, M.; Kaare, M.; Leidmaa, E.; Jagomäe, T.; Heinla, I.; Hickey, M.; Kaasik, A.; Schäfer, M.K.; Innos, J.; et al. Neural cell adhesion molecule Negr1 deficiency in mouse results in structural brain endophenotypes and behavioural deviations related to psychiatric disorders. Sci. Rep. 2019, 9, 5457. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Yin, L.; Chen, Z. Neuroprotective role of fibronectin in neuron-glial extrasynaptic transmission. Neural Regen. Res. 2013, 8, 376. [Google Scholar] [CrossRef] [PubMed]
- Welander, H.; Bontha, S.V.; Näsström, T.; Karlsson, M.; Nikolajeff, F.; Danzer, K.; Kostka, M.; Kalimo, H.; Lannfelt, L.; Ingelsson, M.; et al. Gelsolin co-occurs with Lewy bodies in vivo and accelerates α-synuclein aggregation in vitro. Biochem. Biophys. Res. Commun. 2011, 412, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Xiao, M.F.; Xu, J.C.; Tereshchenko, Y.; Novak, D.; Schachner, M.; Kleene, R. Neural cell adhesion molecule modulates dopaminergic signaling and behavior by regulating dopamine D2 receptor internalization. J. Neurosci. 2009, 29, 14752–14763. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Tang, P.; Li, X.; Chong, L.; Zhang, X.; Li, R.; Xingzhi, G.; Peng, T.; Xiaoqing, L.; Li, C.; et al. Association between two α-2-macroglobulin gene polymorphisms and Parkinson’s disease: A meta-analysis. Int. J. Neurosci. 2016, 126, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, M.; van Steenoven, I.; Huisman, E.; Oosterveld, L.; Berendse, H.; Van Der Flier, W.M.; Del Campo, M.; Lemstra, A.W.; Van De Berg, W.D.J.; Teunissen, C.E. Contactin-1 is reduced in cerebrospinal fluid of parkinson’s disease patients and is present within lewy bodies. Biomolecules 2020, 10, 1177. [Google Scholar] [CrossRef] [PubMed]
- Ellwanger, J.H.; Franke, S.I.; Bordin, D.L.; Prá, D.; Henriques, J.A. Biological functions of selenium and its potential influence on Parkinson’s disease. An. Acad. Bras. Cienc. 2016, 88, 1655–1674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.S.; Bae, W.Y.; Nam, S.; Jeong, J.-W. New targets for Parkinson’s disease: Adhesion G protein-coupled receptor B1 is downregulated by AMP-activated protein kinase activation. OMICS J. Integr. Biol. 2018, 22, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Fyfe, I. APOE* ε4 promotes synucleinopathy. Nat. Rev. Neurol. 2020, 16, 185. [Google Scholar] [CrossRef]
- Shohimardonov, S.H.; Daminova, K. Examining level of haemoglobin in patients with Parkinson’s disease and the role of haemoglobin level in the disease course. Mov. Disord. 2019, 34, 1075. [Google Scholar]
- Gao, L.; Tang, H.; Nie, K.; Wang, L.; Zhao, J.; Gan, R.; Huang, J.; Zhu, R.; Feng, S.; Duan, Z.; et al. Cerebrospinal fluid alpha-synuclein as a biomarker for Parkinson’s disease diagnosis: A systematic review and meta-analysis. Int. J. Neurosci. 2015, 125, 645–654. [Google Scholar] [CrossRef]
- Stav, A.L.; Aarsland, D.; Johansen, K.K.; Hessen, E.; Auning, E.; Fladby, T. Amyloid-β and α-synuclein cerebrospinal fluid biomarkers and cognition in early Parkinson’s disease. Park. Relat. Disord. 2015, 21, 758–764. [Google Scholar] [CrossRef] [PubMed]
- Thaler, A.; Bregman, N.; Gurevich, T.; Shiner, T.; Dror, Y.; Zmira, O.; Gan-Or, Z.; Bar-Shira, A.; Gana-Weisz, M.; Orr-Urtreger, A.; et al. Parkinson’s disease phenotype is influenced by the severity of the mutations in the GBA gene. Park. Relat. Disord. 2018, 55, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Lerche, S.; Wurster, I.; Roeben, B.; Zimmermann, M.; Riebenbauer, B.; Deuschle, C.; Hauser, A.; Schulte, C.; Berg, D.; Maetzler, W.; et al. Parkinson’s disease: Glucocerebrosidase 1 mutation severity is associated with CSF alpha-synuclein profiles. Mov. Disord. 2020, 35, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Brockmann, K.; Quadalti, C.; Lerche, S.; Rossi, M.; Wurster, I.; Baiardi, S.; Roeben, B.; Mammana, A.; Zimmermann, M.; Hauser, A.-K.; et al. Association between CSF alpha-synuclein seeding activity and genetic status in Parkinson’s disease and dementia with Lewy bodies. Acta Neuropathol. Commun. 2021, 9, 175. [Google Scholar] [CrossRef] [PubMed]
- Brockmann, K.; Schulte, C.; Deuschle, C.; Hauser, A.-K.; Heger, T.; Gasser, T.; Maetzler, W.; Berg, D. Neurodegenerative CSF markers in genetic and sporadic PD: Classification and prediction in a longitudinal study. Park. Relat. Disord. 2015, 21, 1427–1434. [Google Scholar] [CrossRef] [PubMed]
- Brockmann, K.; Srulijes, K.; Hauser, A.K.; Schulte, C.; Csoti, I.; Gasser, T.; Berg, D. GBA-associated PD presents with nonmotor characteristics. Neurology 2011, 77, 276–280. [Google Scholar] [CrossRef]
- Schmidt, C.; Urlaub, H. iTRAQ-labeling of in-gel digested proteins for relative quantification. In Proteomics; Humana Press: Totowa, NJ, USA, 2009; pp. 207–226. [Google Scholar] [CrossRef] [Green Version]
- Thomas, P.D.; Campbell, M.J.; Kejariwal, A.; Mi, H.; Karlak, B.; Daverman, R.; Diemer, K.; Muruganujan, A.; Narechania, A. PANTHER: A library of protein families and subfamilies indexed by function. Genome Res. 2003, 13, 2129–2141. [Google Scholar] [CrossRef] [Green Version]
- Metsalu, T.; Vilo, J. ClustVis: A web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Res. 2015, 43, 566–570. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Patient ID | Age | Gender | Age at Onset |
|---|---|---|---|---|
| PDIdiopathic (Mean age 63.4 ± 7.78 years) | ||||
| 1 | PDIdiopathic-1 | 66 | Male | 63 |
| 2 | PDIdiopathic-2 | 74 | Male | 62 |
| 3 | PDIdiopathic-3 | 62 | Female | 53 |
| 4 | PDIdiopathic-4 | 53 | Male | 42 |
| 5 | PDIdiopathic-5 | 60 | Male | 51 |
| 6 | PDIdiopathic-6 | 74 | Female | 67 |
| 7 | PDIdiopathic-7 | 55 | Male | 50 |
| PDGBA (Mean age 60.5 ± 9.97 years) | ||||
| 8 | PDGBA-1 | 76 | Female | 47 |
| 9 | PDGBA-2 | 58 | Male | 50 |
| 10 | PDGBA-3 | 65 | Female | 55 |
| 11 | PDGBA-4 | 61 | Male | 45 |
| 12 | PDGBA-5 | 44 | Male | 28 |
| 13 | PDGBA-6 | 60 | Male | 49 |
| 14 | PDGBA-7 | 59 | Male | 52 |
| 15 | PDGBA-8 | 51 | Male | 41 |
| 16 | PDGBA-9 | 50 | Male | 44 |
| 17 | PDGBA-10 | 75 | Male | 70 |
| 18 | PDGBA-11 | 41 | Male | 39 |
| 19 | PDGBA-12 | 67 | Female | 56 |
| 20 | PDGBA-13 | 73 | Female | 65 |
| 21 | PDGBA-14 | 63 | Male | 58 |
| 22 | PDGBA-15 | 58 | Female | 51 |
| 23 | PDGBA-16 | 70 | Male | 60 |
| 24 | PDGBA-17 | 67 | Male | 63 |
| 25 | PDGBA-18 | 42 | Male | 40 |
| 26 | PDGBA-19 | 64 | Female | 51 |
| 27 | PDGBA-20 | 59 | Male | 51 |
| 28 | PDGBA-21 | 68 | Male | 56 |
| 29 | PDGBA-22 | 62 | Male | 52 |
| HIGBA (Mean age 52.6 ± 13.76 years) | ||||
| 30 | HIGBA-1 | 72 | Female | - |
| 31 | HIGBA-2 | 45 | Male | - |
| 32 | HIGBA-3 | 41 | Female | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zafar, S.; Noor, A.; Younas, N.; Shafiq, M.; Schmitz, M.; Wurster, I.; Brockmann, K.; Gasser, T.; Zerr, I. SWATH Mass Spectrometry-Based CSF Proteome Profile of GBA-Linked Parkinson’s Disease Patients. Int. J. Mol. Sci. 2022, 23, 14166. https://doi.org/10.3390/ijms232214166
Zafar S, Noor A, Younas N, Shafiq M, Schmitz M, Wurster I, Brockmann K, Gasser T, Zerr I. SWATH Mass Spectrometry-Based CSF Proteome Profile of GBA-Linked Parkinson’s Disease Patients. International Journal of Molecular Sciences. 2022; 23(22):14166. https://doi.org/10.3390/ijms232214166
Chicago/Turabian StyleZafar, Saima, Aneeqa Noor, Neelam Younas, Mohsin Shafiq, Matthias Schmitz, Isabel Wurster, Kathrin Brockmann, Thomas Gasser, and Inga Zerr. 2022. "SWATH Mass Spectrometry-Based CSF Proteome Profile of GBA-Linked Parkinson’s Disease Patients" International Journal of Molecular Sciences 23, no. 22: 14166. https://doi.org/10.3390/ijms232214166