
Alzheimer’s Disease Seen through the Eye: Ocular Alterations and Neurodegeneration

 , ,
, ,  ,
, 
Abstract
:1. Introduction
2. Cognitive Alterations and Visual Repercussions Related to AD
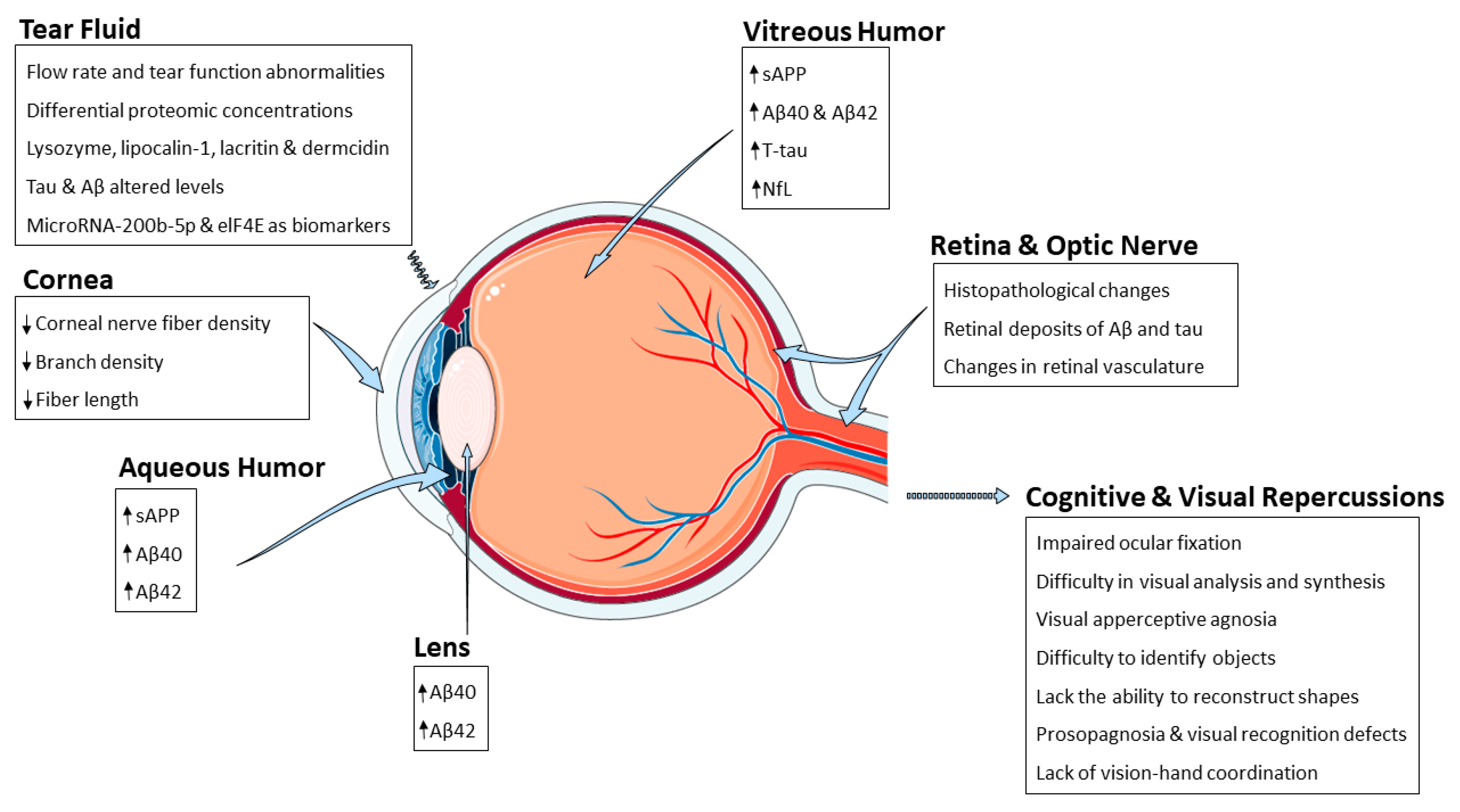
3. Ocular Alterations Related to AD
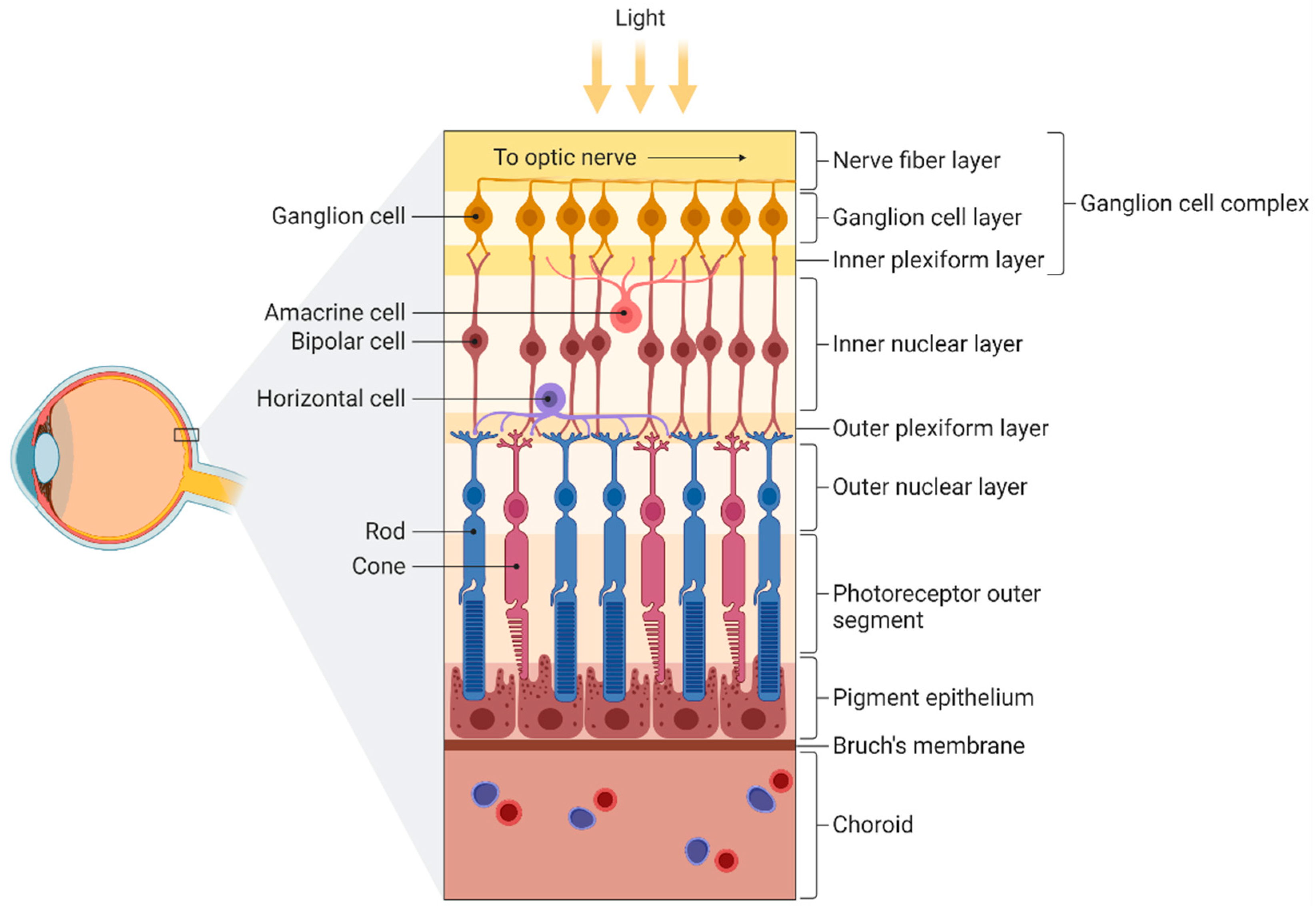
3.1. Retina and Optic Nerve
3.1.1. Histopathological Changes Associated with Progression of Cognitive Impairment
3.1.2. Aβ and Tau Accumulation in AD Retinas
3.1.3. Changes in Retinal Vasculature Associated with Progression of Cognitive Impairment
3.2. Tear Fluid
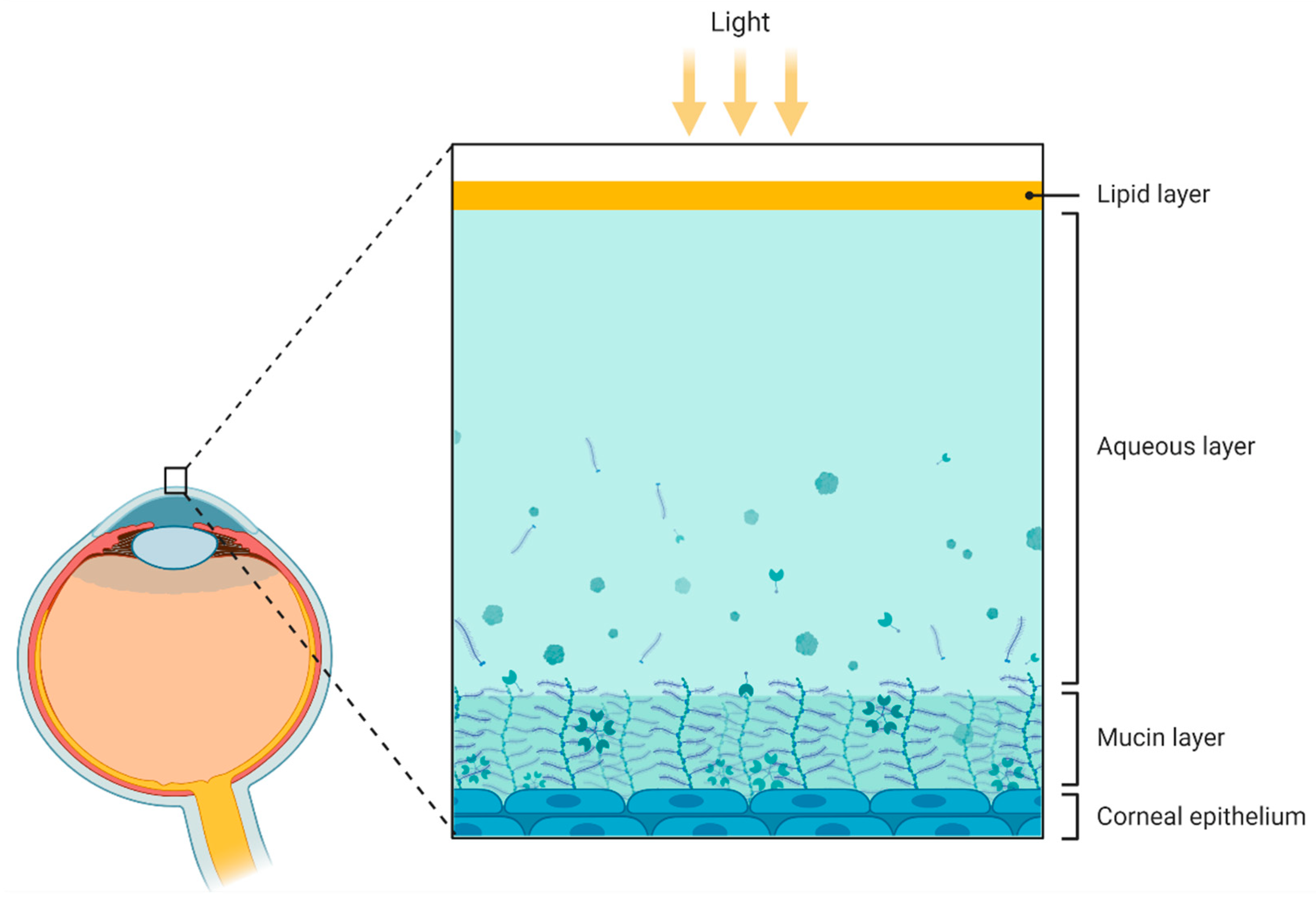
3.3. Cornea
3.4. Lens
3.5. Aqueous and Vitreous Humor
4. Limitations
5. Concluding Remarks and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s Disease. Eur. J. Neurol. 2018, 25, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Toldi, J.; Vécsei, L. Exploring the Etiological Links behind Neurodegenerative Diseases: Inflammatory Cytokines and Bioactive Kynurenines. Int. J. Mol. Sci. 2020, 21, 2431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Battaglia, S.; Garofalo, S.; di Pellegrino, G. Context-Dependent Extinction of Threat Memories: Influences of Healthy Aging. Sci. Rep. 2018, 8, 12592. [Google Scholar] [CrossRef]
- Kamboh, M.I. Genomics and Functional Genomics of Alzheimer’s Disease. Neurother. J. Am. Soc. Exp. NeuroTherapeutics 2021. [Google Scholar] [CrossRef] [PubMed]
- Giri, M.; Shah, A.; Upreti, B.; Rai, J.C. Unraveling the Genes Implicated in Alzheimer’s Disease. Biomed. Rep. 2017, 7, 105–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blennow, K.; de Leon, M.J.; Zetterberg, H. Alzheimer’s Disease. Lancet 2006, 368, 387–403. [Google Scholar] [CrossRef]
- Custodia, A.; Ouro, A.; Romaus-Sanjurjo, D.; Pías-Peleteiro, J.M.; de Vries, H.E.; Castillo, J.; Sobrino, T. Endothelial Progenitor Cells and Vascular Alterations in Alzheimer’s Disease. Front. Aging Neurosci. 2022, 13, 13. [Google Scholar] [CrossRef]
- Deture, M.A.; Dickson, D.W. The Neuropathological Diagnosis of Alzheimer’s Disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef] [Green Version]
- Unschuld, P.G. Prevention of Alzheimer’s Disease: Medical and Lifestyle Interventions. Rev. Med. Suisse 2021, 17, 1614–1616. [Google Scholar]
- Veitch, D.P.; Weiner, M.W.; Aisen, P.S.; Beckett, L.A.; DeCarli, C.; Green, R.C.; Harvey, D.; Jack, C.R., Jr.; Jagust, W.; Landau, S.M.; et al. Using the Alzheimer’s Disease Neuroimaging Initiative to Improve Early Detection, Diagnosis, and Treatment of Alzheimer’s Disease. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2021. [Google Scholar] [CrossRef]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R., Jr.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The Diagnosis of Dementia Due to Alzheimer’s Disease: Recommendations from the National Institute on Aging-Alzheimer’s Association Workgroups on Diagnostic Guidelines for Alzheimer’s Disease. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2011, 7, 263. [Google Scholar] [CrossRef] [Green Version]
- Sanches, C.; Stengel, C.; Godard, J.; Mertz, J.; Teichmann, M.; Migliaccio, R.; Valero-Cabré, A. Past, Present, and Future of Non-Invasive Brain Stimulation Approaches to Treat Cognitive Impairment in Neurodegenerative Diseases: Time for a Comprehensive Critical Review. Front. Aging Neurosci. 2021, 12, 378. [Google Scholar] [CrossRef]
- Chang, C.H.; Lane, H.Y.; Lin, C.H. Brain Stimulation in Alzheimer’s Disease. Front. Psychiatry 2018, 9, 201. [Google Scholar] [CrossRef] [Green Version]
- Borgomaneri, S.; Battaglia, S.; Avenanti, A.; di Pellegrino, G. Don’t Hurt Me No More: State-Dependent Transcranial Magnetic Stimulation for the Treatment of Specific Phobia. J. Affect. Disord. 2021, 286, 78–79. [Google Scholar] [CrossRef]
- Borgomaneri, S.; Battaglia, S.; Sciamanna, G.; Tortora, F.; Laricchiuta, D. Memories Are Not Written in Stone: Re-Writing Fear Memories by Means of Non-Invasive Brain Stimulation and Optogenetic Manipulations. Neurosci. Biobehav. Rev. 2021, 127, 334–352. [Google Scholar] [CrossRef]
- Buss, S.S.; Fried, P.J.; Pascual-Leone, A. Therapeutic Noninvasive Brain Stimulation in Alzheimer’s Disease and Related Dementias. Curr. Opin. Neurol. 2019, 32, 292–304. [Google Scholar] [CrossRef]
- Sperling, R.; Aisen, P.; Beckett, L.; Bennett, D.; Craft, S.; Fagan, A.; Iwatsubo, T.; Jack, C.; Kaye, J.; Montine, T.; et al. Toward Defining the Preclinical Stages of Alzheimer’s Disease: Recommendations from the National Institute on Aging-Alzheimer’s Association Workgroups on Diagnostic Guidelines for Alzheimer’s Disease. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2011, 7, 280. [Google Scholar] [CrossRef] [Green Version]
- Albert, M.; DeKosky, S.; Dickson, D.; Dubois, B.; Feldman, H.; Fox, N.; Gamst, A.; Holtzman, D.; Jagust, W.; Petersen, R.; et al. The Diagnosis of Mild Cognitive Impairment Due to Alzheimer’s Disease: Recommendations from the National Institute on Aging-Alzheimer’s Association Workgroups on Diagnostic Guidelines for Alzheimer’s Disease. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2011, 7, 270. [Google Scholar] [CrossRef] [Green Version]
- London, A.; Benhar, I.; Schwartz, M. The Retina as a Window to the Brain-from Eye Research to CNS Disorders. Nat. Rev. Neurol. 2013, 9, 44–53. [Google Scholar] [CrossRef]
- Yolcu, U.; Sahin, O.F.; Gundogan, F.C. Imaging in Ophthalmology. In Ophthalmology—Current Clinical and Research Updates; InTech: London, UK, 2014. [Google Scholar]
- Webb, R.H.; Hughes, G.W.; Delori, F.C. Confocal Scanning Laser Ophthalmoscope. Appl. Opt. 1987, 26, 1492. [Google Scholar] [CrossRef]
- Huang, D.; Swanson, E.A.; Lin, C.P.; Schuman, J.S.; Stinson, W.G.; Chang, W.; Hee, M.R.; Flotte, T.; Gregory, K.; Puliafito, C.A.; et al. Optical coherence tomography. Science 1991, 254, 1178–1181. [Google Scholar] [CrossRef] [Green Version]
- Tippett, W.J.; Black, S.E. Regional Cerebral Blood Flow Correlates of Visuospatial Tasks in Alzheimer’s Disease. J. Int. Neuropsychol. Soc. JINS 2008, 14, 1034–1045. [Google Scholar] [CrossRef] [Green Version]
- Rizzo, M.; Anderson, S.W.; Dawson, J.; Nawrot, M. Vision and Cognition in Alzheimer’s Disease. Neuropsychologia 2000, 38, 1157–1169. [Google Scholar] [CrossRef]
- Ko, F.; Muthy, Z.; Gallacher, J.; Sudlow, C.; Rees, G.; Yang, Q.; Keane, P.; Petzold, A.; Khaw, P.T.; Reisman, C.; et al. Association of Retinal Nerve Fiber Layer Thinning With Current and Future Cognitive Decline: A Study Using Optical Coherence Tomography. JAMA Neurol. 2018, 75, 1198. [Google Scholar] [CrossRef] [PubMed]
- Ponirakis, G.; Al Hamad, H.; Sankaranarayanan, A.; Khan, A.; Chandran, M.; Ramadan, M.; Tosino, R.; Gawhale, P.; Alobaidi, M.; AlSulaiti, E.; et al. Association of Corneal Nerve Fiber Measures with Cognitive Function in Dementia. Ann. Clin. Transl. Neurol. 2019, 6, 689–697. [Google Scholar] [CrossRef] [PubMed]
- Khawaja, A.P.; Chan, M.P.Y.; Yip, J.L.Y.; Broadway, D.C.; Garway-Heath, D.F.; Luben, R.; Hayat, S.; Matthews, F.E.; Brayne, C.; Khaw, K.-T.; et al. Retinal Nerve Fiber Layer Measures and Cognitive Function in the EPIC-Norfolk Cohort Study. Investig. Ophthalmol. Vis. Sci. 2016, 57, 1921. [Google Scholar] [CrossRef] [Green Version]
- Leung, K.K.; Bartlett, J.W.; Barnes, J.; Manning, E.N.; Ourselin, S.; Fox, N.C.; Alzheimer’s Disease Neuroimaging Initiative. Cerebral Atrophy in Mild Cognitive Impairment and Alzheimer Disease: Rates and Acceleration. Neurology 2013, 80, 648. [Google Scholar] [CrossRef] [Green Version]
- Lenoir, H.; Siéroff, É. Visual Perceptual Disorders in Alzheimer’s Disease. Geriatr. Psychol. Neuropsychiatr. Vieil. 2019, 17, 307–316. [Google Scholar] [CrossRef]
- Frith, C.D.; Frith, U. Implicit and Explicit Processes in Social Cognition. Neuron 2008, 60, 503–510. [Google Scholar] [CrossRef] [Green Version]
- Folstein, M.F.; Folstein, S.E.; McHugh, P.R. “Mini-Mental State”. A Practical Method for Grading the Cognitive State of Patients for the Clinician. J. Psychiatr. Res. 1975, 12, 189–198. [Google Scholar] [CrossRef]
- Blessed, G.; Tomlinson, B.E.; Roth, M. The Association between Quantitative Measures of Dementia and of Senile Change in the Cerebral Grey Matter of Elderly Subjects. Br. J. Psychiatry J. Ment. Sci. 1968, 114, 797–811. [Google Scholar] [CrossRef]
- Rossetti, H.; Lacritz, L.; Cullum, C.; Weiner, M. Normative Data for the Montreal Cognitive Assessment (MoCA) in a Population-Based Sample. Neurology 2011, 77, 1272–1275. [Google Scholar] [CrossRef]
- Vos, S.J.B.; van Rossum, I.A.; Verhey, F.; Knol, D.L.; Soininen, H.; Wahlund, L.O.; Hampel, H.; Tsolaki, M.; Minthon, L.; Frisoni, G.B.; et al. Prediction of Alzheimer Disease in Subjects with Amnestic and Nonamnestic MCI. Neurology 2013, 80, 1124–1132. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Das, S.; Hyman, B.T. APOE and Alzheimer’s Disease: Advances in Genetics, Pathophysiology, and Therapeutic Approaches. Lancet Neurol. 2021, 20, 68–80. [Google Scholar] [CrossRef]
- Spoletini, I.; Marra, C.; Di Iulio, F.; Gianni, W.; Sancesario, G.; Giubilei, F.; Trequattrini, A.; Bria, P.; Caltagirone, C.; Spalletta, G. Facial Emotion Recognition Deficit in Amnestic Mild Cognitive Impairment and Alzheimer Disease. Am. J. Geriatr. Psychiatry 2008, 16, 389–398. [Google Scholar] [CrossRef]
- Kaeser, P.F.; Ghika, J.; Borruat, F.X. Visual Signs and Symptoms in Patients with the Visual Variant of Alzheimer Disease. BMC Ophthalmol. 2015, 15, 65. [Google Scholar] [CrossRef] [Green Version]
- Mashige, K.P.; Oduntan, O.A. Retinal nerve fibre layer thickness values and their associations with ocular and systemic parameters in Black South Africans. Afr Health Sci. 2016, 16, 1188–1194. [Google Scholar] [CrossRef] [Green Version]
- Yan, W.; Peng, Y.R.; van Zyl, T.; Regev, A.; Shekhar, K.; Juric, D.; Sanes, J.R. Cell Atlas of The Human Fovea and Peripheral Retina. Sci. Rep. 2020, 10, 9802. [Google Scholar] [CrossRef]
- Hinton, D.R.; Sadun, A.A.; Blanks, J.C.; Miller, C.A. Optic-Nerve Degeneration in Alzheimer’s Disease. N. Engl. J. Med. 1986, 315, 485–487. [Google Scholar] [CrossRef]
- Blanks, J.C.; Hinton, D.R.; Sadun, A.A.; Miller, C.A. Retinal Ganglion Cell Degeneration in Alzheimer’s Disease. Brain Res. 1989, 501, 364–372. [Google Scholar] [CrossRef]
- Sadun, A.A.; Borchert, M.; DeVita, E.; Hinton, D.R.; Bassi, C.J. Assessment of Visual Impairment in Patients with Alzheimer’s Disease. Am. J. Ophthalmol. 1987, 104, 113–120. [Google Scholar] [CrossRef]
- Katz, B.; Rimmer, S.; Iragui, V.; Katzman, R. Abnormal Pattern Electroretinogram in Alzheimer’s Disease: Evidence for Retinal Ganglion Cell Degeneration? Ann. Neurol. 1989, 26, 221–225. [Google Scholar] [CrossRef]
- Trick, G.L.; Trick, L.R.; Morris, P.; Wolf, M. Visual field loss in senile dementia of the Alzheimer’s type. Neurology 1995, 45, 68–74. [Google Scholar] [CrossRef]
- Asanad, S.; Ross-Cisneros, F.N.; Nassisi, M.; Barron, E.; Karanjia, R.; Sadun, A.A. The Retina in Alzheimer’s Disease: Histomorphometric Analysis of an Ophthalmologic Biomarker. Investig. Ophthalmol. Vis. Sci. 2019, 60, 1491–1500. [Google Scholar] [CrossRef] [Green Version]
- Ong, Y.T.; Hilal, S.; Cheung, C.Y.; Venketasubramanian, N.; Niessen, W.J.; Vrooman, H.; Anuar, A.R.; Chew, M.; Chen, C.; Wong, T.Y.; et al. Retinal Neurodegeneration on Optical Coherence Tomography and Cerebral Atrophy. Neurosci. Lett. 2015, 584, 12–16. [Google Scholar] [CrossRef]
- Méndez-Gómez, J.L.; Rougier, M.B.; Tellouck, L.; Korobelnik, J.F.; Schweitzer, C.; Delyfer, M.N.; Amieva, H.; Dartigues, J.F.; Delcourt, C.; Helmer, C. Peripapillary Retinal Nerve Fiber Layer Thickness and the Evolution of Cognitive Performance in an Elderly Population. Front. Neurol. 2017, 8, 93. [Google Scholar] [CrossRef] [Green Version]
- Méndez-Gómez, J.L.; Pelletier, A.; Rougier, M.B.; Korobelnik, J.F.; Schweitzer, C.; Delyfer, M.N.; Catheline, G.; Monfermé, S.; Dartigues, J.F.; Delcourt, C.; et al. Association of Retinal Nerve Fiber Layer Thickness with Brain Alterations in the Visual and Limbic Networks in Elderly Adults without Dementia. JAMA Netw. Open 2018, 1, e184406. [Google Scholar] [CrossRef] [Green Version]
- Mutlu, U.; Colijn, J.M.; Ikram, M.A.; Bonnemaijer, P.W.M.; Licher, S.; Wolters, F.J.; Tiemeier, H.; Koudstaal, P.J.; Klaver, C.C.W.; Ikram, M.K. Association of Retinal Neurodegeneration on Optical Coherence Tomography with Dementia: A Population-Based Study. JAMA Neurol. 2018, 75, 1256–1263. [Google Scholar] [CrossRef]
- Golzan, S.M.; Goozee, K.; Georgevsky, D.; Avolio, A.; Chatterjee, P.; Shen, K.; Gupta, V.; Chung, R.; Savage, G.; Orr, C.F.; et al. Retinal Vascular and Structural Changes Are Associated with Amyloid Burden in the Elderly: Ophthalmic Biomarkers of Preclinical Alzheimer’s Disease. Alzheimer’s Res. Ther. 2017, 9, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van de Kreeke, J.A.; Nguyen, H.T.; den Haan, J.; Konijnenberg, E.; Tomassen, J.; den Braber, A.; ten Kate, M.; Collij, L.; Yaqub, M.; van Berckel, B.; et al. Retinal Layer Thickness in Preclinical Alzheimer’s Disease. Acta Ophthalmol. 2019, 97, 798–804. [Google Scholar] [CrossRef] [PubMed]
- Santos, C.Y.; Johnson, L.N.; Sinoff, S.E.; Festa, E.K.; Heindel, W.C.; Snyder, P.J. Change in Retinal Structural Anatomy during the Preclinical Stage of Alzheimer’s Disease. Alzheimer’s Dement. Diagn. Assess. Dis. Monit. 2018, 10, 196–209. [Google Scholar] [CrossRef] [PubMed]
- Cheng, D.L.; Thompson, L.; Snyder, P.J. A Potential Association Between Retinal Changes, Subjective Memory Impairment, and Anxiety in Older Adults at Risk for Alzheimer’s Disease: A 27-Month Pilot Study. Front. Aging Neurosci. 2019, 11, 288. [Google Scholar] [CrossRef] [PubMed]
- Marquié, M.; Valero, S.; Castilla-Marti, M.; Martínez, J.; Rodríguez-Gómez, O.; Sanabria, Á.; Tartari, J.P.; Monté-Rubio, G.C.; Sotolongo-Grau, O.; Alegret, M.; et al. Association between Retinal Thickness and β-Amyloid Brain Accumulation in Individuals with Subjective Cognitive Decline: Fundació ACE Healthy Brain Initiative. Alzheimer’s Res. Ther. 2020, 12, 37. [Google Scholar] [CrossRef] [Green Version]
- Paquet, C.; Boissonnot, M.; Roger, F.; Dighiero, P.; Gil, R.; Hugon, J. Abnormal Retinal Thickness in Patients with Mild Cognitive Impairment and Alzheimer’s Disease. Neurosci. Lett. 2007, 420, 97–99. [Google Scholar] [CrossRef]
- Kesler, A.; Vakhapova, V.; Korczyn, A.D.; Naftaliev, E.; Neudorfer, M. Retinal Thickness in Patients with Mild Cognitive Impairment and Alzheimer’s Disease. Clin. Neurol. Neurosurg. 2011, 113, 523–526. [Google Scholar] [CrossRef]
- Ascaso, F.J.; Cruz, N.; Modrego, P.J.; Lopez-Anton, R.; Santabárbara, J.; Pascual, L.F.; Lobo, A.; Cristóbal, J.A. Retinal Alterations in Mild Cognitive Impairment and Alzheimer’s Disease: An Optical Coherence Tomography Study. J. Neurol. 2014, 261, 1522–1530. [Google Scholar] [CrossRef]
- Gao, L.Y.; Liu, Y.; Li, X.H.; Bai, Q.H.; Liu, P. Abnormal Retinal Nerve Fiber Layer Thickness and Macula Lutea in Patients with Mild Cognitive Impairment and Alzheimer’s Disease. Arch. Gerontol. Geriatr. 2015, 60, 162–167. [Google Scholar] [CrossRef]
- Tao, R.; Lu, Z.; Ding, D.; Fu, S.; Hong, Z.; Liang, X.; Zheng, L.; Xiao, Y.; Zhao, Q. Perifovea Retinal Thickness as an Ophthalmic Biomarker for Mild Cognitive Impairment and Early Alzheimer’s Disease. Alzheimer’s Dement. Diagn. Assess. Dis. Monit. 2019, 11, 405–414. [Google Scholar] [CrossRef]
- Cheung, C.Y.L.; Ong, Y.T.; Hilal, S.; Ikram, M.K.; Low, S.; Ong, Y.L.; Venketasubramanian, N.; Yap, P.; Seow, D.; Chen, C.L.H.; et al. Retinal Ganglion Cell Analysis Using High-Definition Optical Coherence Tomography in Patients with Mild Cognitive Impairment and Alzheimer’s Disease. J. Alzheimer’s Dis. 2015, 45, 45–56. [Google Scholar] [CrossRef] [Green Version]
- Almeida, A.L.M.; Pires, L.A.; Figueiredo, E.A.; Costa-Cunha, L.V.F.; Zacharias, L.C.; Preti, R.C.; Monteiro, M.L.R.; Cunha, L.P. Correlation between Cognitive Impairment and Retinal Neural Loss Assessed by Swept-Source Optical Coherence Tomography in Patients with Mild Cognitive Impairment. Alzheimer’s Dement. Diagn. Assess. Dis. Monit. 2019, 11, 659–669. [Google Scholar] [CrossRef]
- Sánchez, D.; Castilla-Marti, M.; Rodríguez-Gómez, O.; Valero, S.; Piferrer, A.; Martínez, G.; Martínez, J.; Serra, J.; Moreno-Grau, S.; Hernández-Olasagarre, B.; et al. Usefulness of Peripapillary Nerve Fiber Layer Thickness Assessed by Optical Coherence Tomography as a Biomarker for Alzheimer’s Disease. Sci. Rep. 2018, 8, 16345. [Google Scholar] [CrossRef] [PubMed]
- López-de-Eguileta, A.; Lage, C.; López-García, S.; Pozueta, A.; García-Martínez, M.; Kazimierczak, M.; Bravo, M.; de Arcocha-Torres, M.; Banzo, I.; Jimenez-Bonilla, J.; et al. Ganglion Cell Layer Thinning in Prodromal Alzheimer’s Disease Defined by Amyloid PET. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2019, 5, 570–578. [Google Scholar] [CrossRef] [PubMed]
- Lad, E.M.; Mukherjee, D.; Stinnett, S.S.; Cousins, S.W.; Potter, G.G.; Burke, J.R.; Farsiu, S.; Whitson, H.E. Evaluation of Inner Retinal Layers as Biomarkers in Mild Cognitive Impairment to Moderate Alzheimer’s Disease. PLoS ONE 2018, 13, e0192646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, S.P.; Grewal, D.S.; Thompson, A.C.; Polascik, B.W.; Dunn, C.; Burke, J.R.; Fekrat, S. Retinal Microvascular and Neurodegenerative Changes in Alzheimer’s Disease and Mild Cognitive Impairment Compared with Control Participants. Ophthalmol. Retin. 2019, 3, 489–499. [Google Scholar] [CrossRef]
- Shi, Z.; Wu, Y.; Wang, M.; Cao, J.; Feng, W.; Cheng, Y.; Li, C.; Shen, Y. Greater Attenuation of Retinal Nerve Fiber Layer Thickness in Alzheimer’s Disease Patients. J. Alzheimer’s Dis. 2014, 40, 277–283. [Google Scholar] [CrossRef]
- Choi, S.H.; Park, S.J.; Kim, N.R. Macular Ganglion Cell -Inner Plexiform Layer Thickness Is Associated with Clinical Progression in Mild Cognitive Impairment and Alzheimers Disease. PLoS ONE 2016, 11, e0162202. [Google Scholar] [CrossRef]
- Berisha, F.; Feke, G.T.; Trempe, C.L.; McMeel, J.W.; Schepens, C.L. Retinal Abnormalities in Early Alzheimer’s Disease. Investig. Ophthalmol. Vis. Sci. 2007, 48, 2285–2289. [Google Scholar] [CrossRef] [Green Version]
- Bambo, M.P.; Garcia-Martin, E.; Pinilla, J.; Herrero, R.; Satue, M.; Otin, S.; Fuertes, I.; Marques, M.L.; Pablo, L.E. Detection of Retinal Nerve Fiber Layer Degeneration in Patients with Alzheimer’s Disease Using Optical Coherence Tomography: Searching New Biomarkers. Acta Ophthalmol. 2014, 92, e581–e582. [Google Scholar] [CrossRef]
- Tzekov, R.; Mullan, M. Vision Function Abnormalities in Alzheimer Disease. Surv. Ophthalmol. 2014, 59, 414–433. [Google Scholar] [CrossRef]
- Trebbastoni, A.; D’Antonio, F.; Bruscolini, A.; Marcelli, M.; Cecere, M.; Campanelli, A.; Imbriano, L.; de Lena, C.; Gharbiya, M. Retinal Nerve Fibre Layer Thickness Changes in Alzheimer’s Disease: Results from a 12-Month Prospective Case Series. Neurosci. Lett. 2016, 629, 165–170. [Google Scholar] [CrossRef]
- Haan, J.D.; van de Kreeke, J.A.; Konijnenberg, E.; ten Kate, M.; den Braber, A.; Barkhof, F.; van Berckel, B.N.; Teunissen, C.E.; Scheltens, P.; Visser, P.J.; et al. Retinal Thickness as a Potential Biomarker in Patients with Amyloid-Proven Early- and Late-Onset Alzheimer’s Disease. Alzheimer’s Dement. Diagn. Assess. Dis. Monit. 2019, 11, 463–471. [Google Scholar] [CrossRef]
- Haan, J.D.; Janssen, S.F.; Van De Kreeke, J.A.; Scheltens, P.; Verbraak, F.D.; Bouwman, F.H. Retinal Thickness Correlates with Parietal Cortical Atrophy in Early-Onset Alzheimer’s Disease and Controls. Alzheimer’s Dement. (Amst. Neth.) 2017, 10, 49–55. [Google Scholar] [CrossRef]
- Iseri, P.K.; Altintas, O.; Tokay, T.; Yüksel, N. Relationship between cognitive impairment and retinal morphological and visual functional abnormalities in Alzheimer disease. J Neuroophthalmol. 2006, 26, 18–24. [Google Scholar] [CrossRef]
- Garcia-Martin, E.; Bambo, M.P.; Marques, M.L.; Satue, M.; Otin, S.; Larrosa, J.M.; Polo, V.; Pablo, L.E. Ganglion Cell Layer Measurements Correlate with Disease Severity in Patients with Alzheimer’s Disease. Acta Ophthalmol. 2016, 94, e454–e459. [Google Scholar] [CrossRef]
- La Morgia, C.; Ross-Cisneros, F.N.; Koronyo, Y.; Hannibal, J.; Gallassi, R.; Cantalupo, G.; Sambati, L.; Pan, B.X.; Tozer, K.R.; Barboni, P.; et al. Melanopsin Retinal Ganglion Cell Loss in Alzheimer Disease. Ann. Neurol. 2016, 79, 90–109. [Google Scholar] [CrossRef]
- Polo, V.; Garcia-Martin, E.; Bambo, M.P.; Pinilla, J.; Larrosa, J.M.; Satue, M.; Otin, S.; Pablo, L.E. Reliability and Validity of Cirrus and Spectralis Optical Coherence Tomography for Detecting Retinal Atrophy in Alzheimer’s Disease. Eye (Basingstoke) 2014, 28, 680–690. [Google Scholar] [CrossRef] [Green Version]
- Cunha, L.P.; Lopes, L.C.; Costa-Cunha, L.V.; Costa, C.F.; Pires, L.A.; Almeida, A.L.; Monteiro, M.L. Macular Thickness Measurements with Frequency Domain-OCT for Quantification of Retinal Neural Loss and Its Correlation with Cognitive Impairment in Alzheimer’s Disease. PLoS ONE 2016, 11, e0153830. [Google Scholar] [CrossRef] [Green Version]
- Parisi, V.; Restuccia, R.; Fattapposta, F.; Mina, C.; Bucci, M.G.; Pierelli, F.; Sapienza, R.L. Morphological and functional retinal impairment in Alzheimer’s disease patients. Clin. Neurophysiol. 2001, 112, 1860–1867. [Google Scholar] [CrossRef]
- Marziani, E.; Pomati, S.; Ramolfo, P.; Cigada, M.; Giani, A.; Mariani, C.; Staurenghi, G. Evaluation of Retinal Nerve Fiber Layer and Ganglion Cell Layer Thickness in Alzheimer’s Disease Using Spectral- Domain Optical Coherence Tomography. Investig. Ophthalmol. Vis. Sci. 2013, 54, 5953–5958. [Google Scholar] [CrossRef]
- Querques, G.; Borrelli, E.; Sacconi, R.; de Vitis, L.; Leocani, L.; Santangelo, R.; Magnani, G.; Comi, G.; Bandello, F. Functional and Morphological Changes of the Retinal Vessels in Alzheimer’s Disease and Mild Cognitive Impairment. Sci. Rep. 2019, 9, 63. [Google Scholar] [CrossRef] [Green Version]
- Bayhan, H.A.; Aslan Bayhan, S.; Celikbilek, A.; Tanik, N.; Gürdal, C. Evaluation of the Chorioretinal Thickness Changes in Alzheimer’s Disease Using Spectral-Domain Optical Coherence Tomography. Clin. Exp. Ophthalmol. 2015, 43, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.Y.; Troncoso, J.C.; Knox, D.; Stark, W.; Eberhart, C.G. Beta-Amyloid, Phospho-Tau and Alpha-Synuclein Deposits Similar to Those in the Brain Are Not Identified in the Eyes of Alzheimer’s and Parkinson’s Disease Patients. Brain Pathol. (Zur. Switz.) 2014, 24, 25–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leger, F.; Fernagut, P.O.; Canron, M.H.; Léoni, S.; Vital, C.; Tison, F.; Bezard, E.; Vital, A. Protein Aggregation in the Aging Retina. J. Neuropathol. Exp. Neurol. 2011, 70, 63–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schön, C.; Hoffmann, N.A.; Ochs, S.M.; Burgold, S.; Filser, S.; Steinbach, S.; Seeliger, M.W.; Arzberger, T.; Goedert, M.; Kretzschmar, H.A.; et al. Long-Term in Vivo Imaging of Fibrillar Tau in the Retina of P301S Transgenic Mice. PLoS ONE 2012, 7, e53547. [Google Scholar] [CrossRef]
- Chidlow, G.; Wood, J.P.M.; Manavis, J.; Finnie, J.; Casson, R.J. Investigations into Retinal Pathology in the Early Stages of a Mouse Model of Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 56, 655–675. [Google Scholar] [CrossRef] [Green Version]
- Dutescu, R.M.; Li, Q.X.; Crowston, J.; Masters, C.L.; Baird, P.N.; Culvenor, J.G. Amyloid Precursor Protein Processing and Retinal Pathology in Mouse Models of Alzheimer’s Disease. Graefe’s Arch. Clin. Exp. Ophthalmol. 2009, 247, 1213–1221. [Google Scholar] [CrossRef]
- Koronyo-Hamaoui, M.; Koronyo, Y.; Ljubimov, A.V.; Miller, C.A.; Ko, M.K.; Black, K.L.; Schwartz, M.; Farkas, D.L. Identification of Amyloid Plaques in Retinas from Alzheimer’s Patients and Noninvasive in Vivo Optical Imaging of Retinal Plaques in a Mouse Model. NeuroImage 2011, 54 (Suppl. 1), S204–S217. [Google Scholar] [CrossRef] [Green Version]
- Koronyo, Y.; Biggs, D.; Barron, E.; Boyer, D.S.; Pearlman, J.A.; Au, W.J.; Kile, S.J.; Blanco, A.; Fuchs, D.T.; Ashfaq, A.; et al. Retinal Amyloid Pathology and Proof-of-Concept Imaging Trial in Alzheimer’s Disease. JCI Insight 2017, 2, 2. [Google Scholar] [CrossRef]
- Lee, S.; Jiang, K.; McIlmoyle, B.; To, E.; Xu, Q.A.; Hirsch-Reinshagen, V.; Mackenzie, I.R.; Hsiung, G.R.; Eadie, B.D.; Sarunic, M.V.; et al. Amyloid Beta Immunoreactivity in the Retinal Ganglion Cell Layer of the Alzheimer’s Eye. Front. Neurosci. 2020, 14, 758. [Google Scholar] [CrossRef]
- den Haan, J.; Morrema, T.H.J.; Verbraak, F.D.; de Boer, J.F.; Scheltens, P.; Rozemuller, A.J.; Bergen, A.A.B.; Bouwman, F.H.; Hoozemans, J.J. Amyloid-Beta and Phosphorylated Tau in Post-Mortem Alzheimer’s Disease Retinas. Acta Neuropathol. Commun. 2018, 6, 147. [Google Scholar] [CrossRef]
- More, S.S.; Vince, R. Hyperspectral Imaging Signatures Detect Amyloidopathy in Alzheimer’s Mouse Retina Well before Onset of Cognitive Decline. ACS Chem. Neurosci. 2015, 6, 306–315. [Google Scholar] [CrossRef] [Green Version]
- More, S.S.; Beach, J.M.; Vince, R. Early Detection of Amyloidopathy in Alzheimer’s Mice by Hyperspectral Endoscopy. Investig. Ophthalmol. Vis. Sci. 2016, 57, 3231–3238. [Google Scholar] [CrossRef] [Green Version]
- Ning, A.; Cui, J.; To, E.; Ashe, K.H.; Matsubara, J. Amyloid-Beta Deposits Lead to Retinal Degeneration in a Mouse Model of Alzheimer Disease. Investig. Ophthalmol. Vis. Sci. 2008, 49, 5136–5143. [Google Scholar] [CrossRef] [Green Version]
- Perez, S.E.; Lumayag, S.; Kovacs, B.; Mufson, E.J.; Xu, S. Beta-Amyloid Deposition and Functional Impairment in the Retina of the APPswe/PS1DeltaE9 Transgenic Mouse Model of Alzheimer’s Disease. Investig. Ophthalmol. Vis. Sci. 2009, 50, 793–800. [Google Scholar] [CrossRef]
- Grimaldi, A.; Brighi, C.; Peruzzi, G.; Ragozzino, D.; Bonanni, V.; Limatola, C.; Ruocco, G.; di Angelantonio, S. Inflammation, Neurodegeneration and Protein Aggregation in the Retina as Ocular Biomarkers for Alzheimer’s Disease in the 3xTg-AD Mouse Model. Cell Death Dis. 2018, 9, 685. [Google Scholar] [CrossRef] [Green Version]
- Bevan, R.J.; Hughes, T.R.; Williams, P.A.; Good, M.A.; Morgan, B.P.; Morgan, J.E. Retinal Ganglion Cell Degeneration Correlates with Hippocampal Spine Loss in Experimental Alzheimer’s Disease. Acta Neuropathol. Commun. 2020, 8, 3. [Google Scholar] [CrossRef]
- Rodrigues-Neves, A.C.; Carecho, R.; Correia, S.C.; Carvalho, C.; Campos, E.J.; Baptista, F.I.; Moreira, P.I.; Ambrósio, A.F. Retina and Brain Display Early and Differential Molecular and Cellular Changes in the 3xTg-AD Mouse Model of Alzheimer’s Disease. Mol. Neurobiol. 2021, 58, 3043–3060. [Google Scholar] [CrossRef]
- Gasparini, L.; Anthony Crowther, R.; Martin, K.R.; Berg, N.; Coleman, M.; Goedert, M.; Spillantini, M.G. Tau Inclusions in Retinal Ganglion Cells of Human P301S Tau Transgenic Mice: Effects on Axonal Viability. Neurobiol. Aging 2011, 32, 419–433. [Google Scholar] [CrossRef]
- Mazzaro, N.; Barini, E.; Spillantini, M.G.; Goedert, M.; Medini, P.; Gasparini, L. Tau-Driven Neuronal and Neurotrophic Dysfunction in a Mouse Model of Early Tauopathy. J. Neurosci. 2016, 36, 2086–2100. [Google Scholar] [CrossRef] [Green Version]
- Bull, N.D.; Guidi, A.; Goedert, M.; Martin, K.R.; Spillantini, M.G. Reduced Axonal Transport and Increased Excitotoxic Retinal Ganglion Cell Degeneration in Mice Transgenic for Human Mutant P301s Tau. PLoS ONE 2012, 7, e34724. [Google Scholar] [CrossRef]
- Zhao, H.; Chang, R.; Che, H.; Wang, J.; Yang, L.; Fang, W.; Xia, Y.; Li, N.; Ma, Q.; Wang, X. Hyperphosphorylation of Tau Protein by Calpain Regulation in Retina of Alzheimer’s Disease Transgenic Mouse. Neurosci. Lett. 2013, 551, 12–16. [Google Scholar] [CrossRef] [PubMed]
- Chiasseu, M.; Alarcon-Martinez, L.; Belforte, N.; Quintero, H.; Dotigny, F.; Destroismaisons, L.; Vande Velde, C.; Panayi, F.; Louis, C.; Di Polo, A. Tau Accumulation in the Retina Promotes Early Neuronal Dysfunction and Precedes Brain Pathology in a Mouse Model of Alzheimer’s Disease. Mol. Neurodegener. 2017, 12, 58. [Google Scholar] [CrossRef] [PubMed]
- Latina, V.; Giacovazzo, G.; Cordella, F.; Balzamino, B.O.; Micera, A.; Varano, M.; Marchetti, C.; Malerba, F.; Florio, R.; Ercole, B.B.; et al. Systemic Delivery of a Specific Antibody Targeting the Pathological N-Terminal Truncated Tau Peptide Reduces Retinal Degeneration in a Mouse Model of Alzheimer’s Disease. Acta Neuropathol. Commun. 2021, 9, 38. [Google Scholar] [CrossRef] [PubMed]
- Romaus-Sanjurjo, D.; Custodia, A.; Aramburu-Núñez, M.; Posado-Fernández, A.; Vázquez-Vázquez, L.; Camino-Castiñeiras, J.; Leira, Y.; Manuel Pías-Peleteiro, J.; Aldrey, J.M.; Ouro, A.; et al. Symmetric and Asymmetric Synapses Driving Neurodegenerative Disorders. Symmetry 2021, 13, 2333. [Google Scholar] [CrossRef]
- Zlokovic, B.V. Neurovascular Pathways to Neurodegeneration in Alzheimer’s Disease and Other Disorders. Nat. Rev. Neurosci. 2011, 12, 723–738. [Google Scholar] [CrossRef]
- Zlokovic, B.V. Neurovascular Mechanisms of Alzheimer’s Neurodegeneration. Trends Neurosci. 2005, 28, 202–208. [Google Scholar] [CrossRef]
- Teja, K.V.R.; Berendschot, T.T.; Steinbusch, H.; Webers, A.C.; Murthy, R.P.; Mathuranath, P. Cerebral and Retinal Neurovascular Changes: A Biomarker for Alzheimer’s Disease. J. Gerontol. Geriatr. Res. 2017, 6, 6. [Google Scholar] [CrossRef]
- de Jong, F.J.; Schrijvers, E.M.C.; Ikram, M.K.; Koudstaal, P.J.; de Jong, P.T.V.M.; Hofman, P.A.; Vingerling, J.R.; Breteler, M.M.B. Retinal vascular caliber and risk of dementia: The Rotterdam study. Neurology 2011, 76, 816–821. [Google Scholar] [CrossRef] [Green Version]
- O’Bryhim, B.E.; Apte, R.S.; Kung, N.; Coble, D.; Van Stavern, G.P. Association of preclinical Alzheimer disease with optical coherence tomographic angiography findings. JAMA Ophthalmol. 2018, 136, 1242–1248. [Google Scholar] [CrossRef]
- van de Kreeke, J.A.; Nguyen, H.T.; Konijnenberg, E.; Tomassen, J.; Den Braber, A.; Kate, M.T.; Yaqub, M.; van Berckel, B.; Lammertsma, A.A.; Boomsma, D.I.; et al. Optical Coherence Tomography Angiography in Preclinical Alzheimer’s Disease. Br. J. Ophthalmol. 2020, 104, 157–161. [Google Scholar] [CrossRef]
- Frost, S.; Kanagasingam, Y.; Sohrabi, H.; Vignarajan, J.; Bourgeat, P.; Salvado, O.; Villemagne, V.; Rowe, C.C.; Macaulay, S.L.; Szoeke, C.; et al. Retinal Vasc. Biomarkers for Early Detection and Monitoring of Alzheimer’s Disease. Transl. Psychiatry 2013, 3, e233. [Google Scholar] [CrossRef]
- Feke, G.T.; Hyman, B.T.; Stern, R.A.; Pasquale, L.R. Retinal Blood Flow in Mild Cognitive Impairment and Alzheimer’s Disease. Alzheimer’s Dement. Diagn. Assess. Dis. Monit. 2015, 1, 144–151. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Wei, Y.; Shi, Y.; Wright, C.B.; Sun, X.; Gregori, G.; Zheng, F.; Vanner, E.A.; Lam, B.L.; Rundek, T.; et al. Altered Macular Microvasculature in Mild Cognitive Impairment and Alzheimer Disease. J. Neuro-Ophthalmol. Off. J. N. Am. Neuro-Ophthalmol. Soc. 2018, 38, 292–298. [Google Scholar] [CrossRef]
- Gameiro, G.R.; Jiang, H.; Liu, Y.; Deng, Y.; Sun, X.; Nascentes, B.; Baumel, B.; Rundek, T.; Wang, J. Retinal Tissue Hypoperfusion in Patients with Clinical Alzheimer’s Disease. Eye Vis. 2018, 5, 21. [Google Scholar] [CrossRef]
- Den Haan, J.; van de Kreeke, J.A.; van Berckel, B.N.; Barkhof, F.; Teunissen, C.E.; Scheltens, P.; Verbraak, F.D.; Bouwman, F.H. Is Retinal Vasculature a Biomarker in Amyloid Proven Alzheimer’s Disease? Alzheimer’s Dement. Diagn. Assess. Dis. Monit. 2019, 11, 383–391. [Google Scholar] [CrossRef]
- Chua, J.; Hu, Q.; Ke, M.; Tan, B.; Hong, J.; Yao, X.; Hilal, S.; Venketasubramanian, N.; Garhöfer, G.; Cheung, C.Y.; et al. Retinal Microvasculature Dysfunction Is Associated with Alzheimer’s Disease and Mild Cognitive Impairment. Alzheimer’s Res. Ther. 2020, 12, 161. [Google Scholar] [CrossRef]
- Cheung, C.Y.L.; Ong, Y.T.; Ikram, M.K.; Ong, S.Y.; Li, X.; Hilal, S.; Catindig, J.A.S.; Venketasubramanian, N.; Yap, P.; Seow, D.; et al. Microvascular Network Alterations in the Retina of Patients with Alzheimer’s Disease. Alzheimer’s Dement. 2014, 10, 135–142. [Google Scholar] [CrossRef]
- Jung, N.Y.; Han, J.C.; Ong, Y.T.; Cheung, C.Y.-L.; Chen, C.P.; Wong, T.Y.; Kim, H.J.; Kim, Y.J.; Lee, J.; Lee, J.S.; et al. Retinal Microvasculature Changes in Amyloid-Negative Subcortical Vascular Cognitive Impairment Compared to Amyloid-Positive Alzheimer’s Disease. J. Neurol. Sci. 2019, 396, 94–101. [Google Scholar] [CrossRef]
- Bulut, M.; Kurtuluş, F.; Gözkaya, O.; Erol, M.K.; Cengiz, A.; Akıdan, M.; Yaman, A. Evaluation of Optical Coherence Tomography Angiographic Findings in Alzheimer’s Type Dementia. Br. J. Ophthalmol. 2018, 102, 233–237. [Google Scholar] [CrossRef]
- Grewal, D.S.; Polascik, B.W.; Hoffmeyer, G.C.; Fekrat, S. Assessment of Differences in Retinal Microvasculature Using OCT Angiography in Alzheimer’s Disease: A Twin Discordance Report. Ophthalmic Surg. Lasers Imaging Retin. 2018, 49, 440–444. [Google Scholar] [CrossRef]
- Thambisetty, M.; Beason-Held, L.; An, Y.; Kraut, M.A.; Resnick, S.M. APOE Ε4 Genotype and Longitudinal Changes in Cerebral Blood Flow in Normal Aging. Arch. Neurol. 2010, 67, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Nättinen, J.; Aapola, U.; Jylhä, A.; Vaajanen, A.; Uusitalo, H. Comparison of Capillary and Schirmer Strip Tear Fluid Sampling Methods Using SWATH-MS Proteomics Approach. Transl. Vis. Sci. Technol. 2020, 9, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.R.; Chuang, H.C.; Tripathi, A.; Wang, Y.L.; Ko, M.L.; Chuang, C.C.; Chen, J.C. High-Sensitivity and Trace-Amount Specimen Electrochemical Sensors for Exploring the Levels of β-Amyloid in Human Blood and Tears. Anal. Chem. 2021, 93, 8099–8106. [Google Scholar] [CrossRef] [PubMed]
- Hagan, S.; Martin, E.; Enríquez-de-Salamanca, A. Tear Fluid Biomarkers in Ocular and Systemic Disease: Potential Use for Predictive, Preventive and Personalised Medicine. EPMA J. 2016, 7, 126628. [Google Scholar] [CrossRef] [Green Version]
- López-López, M.; Regueiro, U.; Bravo, S.B.; Chantada-Vázquez, M.D.P.; Varela-Fernández, R.; Ávila-Gómez, P.; Hervella, P.; Lema, I. Tear Proteomics in Keratoconus: A Quantitative SWATH-MS Analysis. Investig. Ophthalmol. Vis. Sci. 2021, 62, 30. [Google Scholar] [CrossRef]
- Örnek, N.; Dağ, E.; Örnek, K. Corneal Sensitivity and Tear Function in Neurodegenerative Diseases. Curr. Eye Res. 2015, 40, 423–428. [Google Scholar] [CrossRef]
- Kalló, G.; Emri, M.; Varga, Z.; Ujhelyi, B.; Tozsér, J.; Csutak, A.; Csosz, É. Changes in the Chemical Barrier Composition of Tears in Alzheimer’s Disease Reveal Potential Tear Diagnostic Biomarkers. PLoS ONE 2016, 11, e0158000. [Google Scholar] [CrossRef] [Green Version]
- Gijs, M.; Ramakers, I.; Visser, P.J.; Verhey, F.; Waarenburg, M.; Schalkwijk, C.; Nuijts, R.; Webers, C. Detection of Amyloid-Beta and Tau in Tear Fluid of Patients with Alzheimer’s Disease. 2020. [Google Scholar] [CrossRef]
- Gijs, M.; Nuijts, R.M.; Ramakers, I.; Verhey, F.; Webers, C.A.B. Differences in Tear Protein Biomarkers between Patients with Alzheimer’s Disease and Controls. Investig. Ophthalmol. Vis. Sci. 2019, 60, 1744. [Google Scholar]
- Gijs, M.; Ramakers, I.H.G.B.; Visser, P.J.; Verhey, F.R.J.; van de Waarenburg, M.P.H.; Schalkwijk, C.G.; Nuijts, R.M.M.A.; Webers, C.A.B. Association of Tear Fluid Amyloid and Tau Levels with Disease Severity and Neurodegeneration. Sci. Rep. 2021, 11, 22675. [Google Scholar] [CrossRef]
- Kenny, A.; Jiménez-Mateos, E.M.; Zea-Sevilla, M.A.; Rábano, A.; Gili-Manzanaro, P.; Prehn, J.H.M.; Henshall, D.C.; Ávila, J.; Engel, T.; Hernández, F. Proteins and MicroRNAs Are Differentially Expressed in Tear Fluid from Patients with Alzheimer’s Disease. Sci. Rep. 2019, 9, 15437. [Google Scholar] [CrossRef] [Green Version]
- Eghrari, A.; Riazuddin, S.; Gottsch, J. Overview of the Cornea: Structure, Function, and Development. Prog. Mol. Biol. Transl. Sci. 2015, 134, 7–23. [Google Scholar] [CrossRef]
- DelMonte, D.W.; Kim, T. Anatomy and Physiology of the Cornea. J. Cataract Refract. Surg. 2011, 37, 588–598. [Google Scholar] [CrossRef]
- Al-Aqaba, M.A.; Dhillon, V.K.; Mohammed, I.; Said, D.G.; Dua, H.S. Corneal Nerves in Health and Disease. Prog. Retin. Eye Res. 2019, 73, 100762. [Google Scholar] [CrossRef]
- Marfurt, C.F.; Cox, J.; Deek, S.; Dvorscak, L. Anatomy of the Human Corneal Innervation. Exp. Eye Res. 2010, 90, 478–492. [Google Scholar] [CrossRef]
- Bonini, S.; Rama, P.; Olzi, D.; Lambiase, A. Neurotrophic Keratitis. Eye (Lond. Engl.) 2003, 17, 989–995. [Google Scholar] [CrossRef] [Green Version]
- Ferrari, G.; Grisan, E.; Scarpa, F.; Fazio, R.; Comola, M.; Quattrini, A.; Comi, G.; Rama, P.; Riva, N. Corneal Confocal Microscopy Reveals Trigeminal Small Sensory Fiber Neuropathy in Amyotrophic Lateral Sclerosis. Front. Aging Neurosci. 2014, 6, 278. [Google Scholar] [CrossRef] [Green Version]
- Petropoulos, I.; Kamran, S.; Li, Y.; Khan, A.; Ponirakis, G.; Akhtar, N.; Deleu, D.; Shuaib, A.; Malik, R. Corneal Confocal Microscopy: An Imaging Endpoint for Axonal Degeneration in Multiple Sclerosis. Investig. Ophthalmol. Vis. Sci. 2017, 58, 3677–3681. [Google Scholar] [CrossRef] [Green Version]
- Messmer, E.M.; Schmid-Tannwald, C.; Zapp, D.; Kampik, A. In Vivo Confocal Microscopy of Corneal Small Fiber Damage in Diabetes Mellitus. Graefe’s Arch. Clin. Exp. Ophthalmol. 2010, 248, 1307–1312. [Google Scholar] [CrossRef]
- Kalteniece, A.; Ferdousi, M.; Adam, S.; Schofield, J.; Azmi, S.; Petropoulos, I.; Soran, H.; Malik, R.A. Corneal Confocal Microscopy Is a Rapid Reproducible Ophthalmic Technique for Quantifying Corneal Nerve Abnormalities. PLoS ONE 2017, 12, e0183040. [Google Scholar] [CrossRef]
- Swanevelder, S.K.; Misra, S.L.; Tyler, E.F.; McGhee, C.N. Precision, Agreement and Utility of a Contemporary Non-Contact Corneal Aesthesiometer. Clin. Exp. Optom. 2020, 103, 798–803. [Google Scholar] [CrossRef]
- McPheeters, M.T.; Blackburn, B.J.; Dupps, W.J., Jr.; Rollins, A.M.; Jenkins, M.W. Genetically Encoded Calcium Indicators for In Situ Functional Studies of Corneal Nerves. Investig. Ophthalmol. Vis. Sci. 2020, 61, 10. [Google Scholar] [CrossRef] [PubMed]
- VanGuilder, H.D.; Farley, J.A.; Yan, H.; van Kirk, C.A.; Mitschelen, M.; Sonntag, W.E.; Freeman, W.M. Hippocampal Dysregulation of Synaptic Plasticity-Associated Proteins with Age-Related Cognitive Decline. Neurobiol. Dis. 2011, 43, 201–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schliebs, R.; Arendt, T. The Cholinergic System in Aging and Neuronal Degeneration. Behav. Brain Res. 2011, 221, 555–563. [Google Scholar] [CrossRef] [PubMed]
- Dehghani, C.; Frost, S.; Jayasena, R.; Masters, C.L.; Kanagasingam, Y. Ocular Biomarkers of Alzheimer’s Disease: The Role of Anterior Eye and Potential Future Directions. Investig. Ophthalmol. Vis. Sci. 2018, 59, 3554–3563. [Google Scholar] [CrossRef] [Green Version]
- Stix, B.; Leber, M.; Bingemer, P.; Gross, C.; Rüschoff, J.; Fändrich, M.; Schorderet, D.F.; Vorwerk, C.K.; Zacharias, M.; Roessner, A.; et al. Hereditary Lattice Corneal Dystrophy Is Associated with Corneal Amyloid Deposits Enclosing C-Terminal Fragments of Keratoepithelin. Investig. Ophthalmol. Vis. Sci. 2005, 46, 1133–1139. [Google Scholar] [CrossRef] [Green Version]
- Frederikse, P.H.; Ren, X.O. Lens Defects and Age-Related Fiber Cell Degeneration in a Mouse Model of Increased AbetaPP Gene Dosage in Down Syndrome. Am. J. Pathol. 2002, 161, 1985–1990. [Google Scholar] [CrossRef]
- Shen, J.; Wu, J. Nicotinic Cholinergic Mechanisms in Alzheimer’s Disease. Int. Rev. Neurobiol. 2015, 124, 275–292. [Google Scholar] [CrossRef]
- Goldstein, L.E.; Muffat, J.A.; Cherny, R.A.; Moir, R.D.; Ericsson, M.H.; Huang, X.; Mavros, C.; Coccia, J.A.; Faget, K.Y.; Fitch, K.A.; et al. Cytosolic Beta-Amyloid Deposition and Supranuclear Cataracts in Lenses from People with Alzheimer’s Disease. Lancet (Lond. Engl.) 2003, 361, 1258–1265. [Google Scholar] [CrossRef]
- Bei, L.; Shui, Y.B.; Bai, F.; Nelson, S.K.; Van Stavern, G.P.; Beebe, D.C. A Test of Lens Opacity as an Indicator of Preclinical Alzheimer Disease. Exp. Eye Res. 2015, 140, 117. [Google Scholar] [CrossRef] [Green Version]
- Kerbage, C.; Sadowsky, C.; Tariot, P.; Agronin, M.; Alva, G.; Turner, F.; Nilan, D.; Cameron, A.; Cagle, G.D.; Hartung, P.D. Detection of Amyloid β Signature in the Lens and Its Correlation in the Brain to Aid in the Diagnosis of Alzheimer’s Disease. Am. J. Alzheimer’s Dis. Other Dement. 2015, 30, 738–745. [Google Scholar] [CrossRef]
- Jun, G.; Moncaster, J.A.; Koutras, C.; Seshadri, S.; Buros, J.; McKee, A.C.; Levesque, G.; Wolf, P.A.; George-Hyslop, P.S.; Goldstein, L.E.; et al. δ-Catenin Is Genetically and Biologically Associated with Cortical Cataract and Future Alzheimer-Related Structural and Functional Brain Changes. PLoS ONE 2012, 7, e43728. [Google Scholar] [CrossRef]
- Moncaster, J.A.; Pineda, R.; Moir, R.D.; Lu, S.; Burton, M.A.; Ghosh, J.G.; Ericsson, M.; Soscia, S.J.; Mocofanescu, A.; Folkerth, R.D.; et al. Alzheimer’s Disease Amyloid-Beta Links Lens and Brain Pathology in Down Syndrome. PLoS ONE 2010, 5, e10659. [Google Scholar] [CrossRef] [Green Version]
- Williams, E.A.; McGuone, D.; Frosch, M.P.; Hyman, B.T.; Laver, N.; Stemmer-Rachamimov, A. Absence of Alzheimer Disease Neuropathologic Changes in Eyes of Subjects With Alzheimer Disease. J. Neuropathol. Exp. Neurol. 2017, 76, 376–383. [Google Scholar] [CrossRef]
- Fereshetian, S.; Agranat, J.S.; Siegel, N.; Ness, S.; Stein, T.D.; Subramanian, M.L. Protein and Imaging Biomarkers in the Eye for Early Detection of Alzheimer’s Disease. J. Alzheimer’s Dis. Rep. 2021, 5, 375–387. [Google Scholar] [CrossRef]
- Chowdhury, U.R.; Madden, B.J.; Charlesworth, M.C.; Fautsch, M.P. Proteome Analysis of Human Aqueous Humor. Investig. Ophthalmol. Vis. Sci. 2010, 51, 4921–4931. [Google Scholar] [CrossRef]
- Wilmott, L.A.; Grambergs, R.C.; Allegood, J.C.; Lyons, T.J.; Mandal, N. Analysis of Sphingolipid Composition in Human Vitreous from Control and Diabetic Individuals. J. Diabetes Its Complicat. 2019, 33, 195. [Google Scholar] [CrossRef]
- Lim, J.K.H.; Li, Q.X.; He, Z.; Vingrys, A.J.; Wong, V.H.Y.; Currier, N.; Mullen, J.; Bui, B.V.; Nguyen, C.T.O. The Eye As a Biomarker for Alzheimer’s Disease. Front. Neurosci. 2016, 10, 536. [Google Scholar] [CrossRef] [Green Version]
- Prakasam, A.; Muthuswamy, A.; Ablonczy, Z.; Greig, N.H.; Fauq, A.; Rao, K.J.; Pappolla, M.A.; Sambamurti, K. Differential Accumulation of Secreted APP Metabolites in Ocular Fluids. J. Alzheimer’s Dis. 2010, 20, 1243. [Google Scholar] [CrossRef] [Green Version]
- Wright, L.M.; Stein, T.D.; Jun, G.; Chung, J.; McConnell, K.; Fiorello, M.; Siegel, N.; Ness, S.; Xia, W.; Turner, K.L.; et al. Association of Cognitive Function with Amyloid-β and Tau Proteins in the Vitreous Humor. J. Alzheimer’s Dis. JAD 2019, 68, 1429–1438. [Google Scholar] [CrossRef]
- Subramanian, M.L.; Vig, V.; Chung, J.; Fiorello, M.G.; Xia, W.; Zetterberg, H.; Blennow, K.; Zetterberg, M.; Shareef, F.; Siegel, N.H.; et al. Neurofilament Light Chain in the Vitreous Humor of the Eye. Alzheimer’s Res. Ther. 2020, 12, 111. [Google Scholar] [CrossRef]
- Gaetani, L.; Blennow, K.; Calabresi, P.; Di Filippo, M.; Parnetti, L.; Zetterberg, H. Neurofilament Light Chain as a Biomarker in Neurological Disorders. J. Neurol. Neurosurg. Psychiatry 2019, 90, 870–881. [Google Scholar] [CrossRef]
- Marchesi, N.; Fahmideh, F.; Boschi, F.; Pascale, A.; Barbieri, A. Ocular Neurodegenerative Diseases: Interconnection between Retina and Cortical Areas. Cells 2021, 10, 2394. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Publication | Study | Results |
|---|---|---|
| Kalló et al. [128] | Examination of changes in tear protein composition from patients with AD compared to control |
|
| Gijs et al. [129] | Examination of AD-specific biomarkers in tear fluid from SCD, MCI, and AD patients |
|
| Gijs et al. [130] | Testing the diagnostic potential of tears as a source of AD biomarkers |
|
| Gijs et al. [131] | Observational study to investigate AD-specific biomarkers in tear fluid |
|
| Kenny et al. [132] | Examination of tear fluid to discover disease-specific protein and microRNA-based biomarkers for AD |
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Romaus-Sanjurjo, D.; Regueiro, U.; López-López, M.; Vázquez-Vázquez, L.; Ouro, A.; Lema, I.; Sobrino, T. Alzheimer’s Disease Seen through the Eye: Ocular Alterations and Neurodegeneration. Int. J. Mol. Sci. 2022, 23, 2486. https://doi.org/10.3390/ijms23052486
Romaus-Sanjurjo D, Regueiro U, López-López M, Vázquez-Vázquez L, Ouro A, Lema I, Sobrino T. Alzheimer’s Disease Seen through the Eye: Ocular Alterations and Neurodegeneration. International Journal of Molecular Sciences. 2022; 23(5):2486. https://doi.org/10.3390/ijms23052486
Chicago/Turabian StyleRomaus-Sanjurjo, Daniel, Uxía Regueiro, Maite López-López, Laura Vázquez-Vázquez, Alberto Ouro, Isabel Lema, and Tomás Sobrino. 2022. "Alzheimer’s Disease Seen through the Eye: Ocular Alterations and Neurodegeneration" International Journal of Molecular Sciences 23, no. 5: 2486. https://doi.org/10.3390/ijms23052486