
Functionalization of Polylactide with Multiple Tetraphenyethane Inifer Groups to Form PLA Block Copolymers with Vinyl Monomers

Abstract
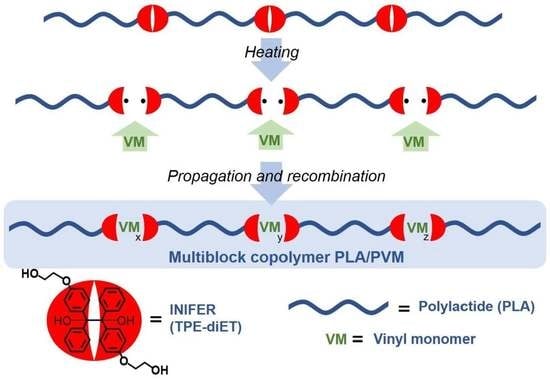
:
1. Introduction
2. Results and Discussion
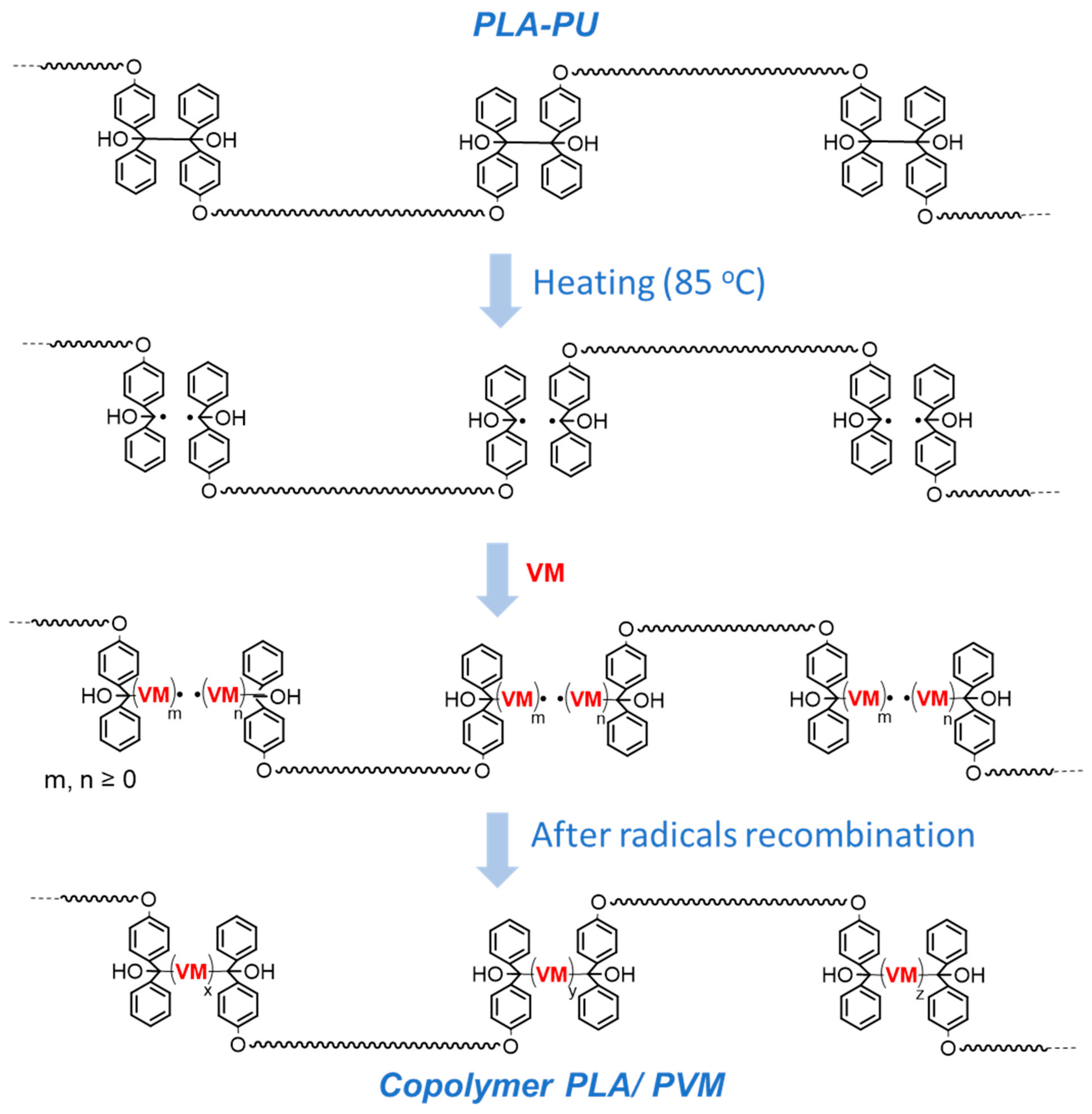
2.1. Synthesis of Polylactide-Based Polymer Containing Multiple Tetraphenylethane Groups
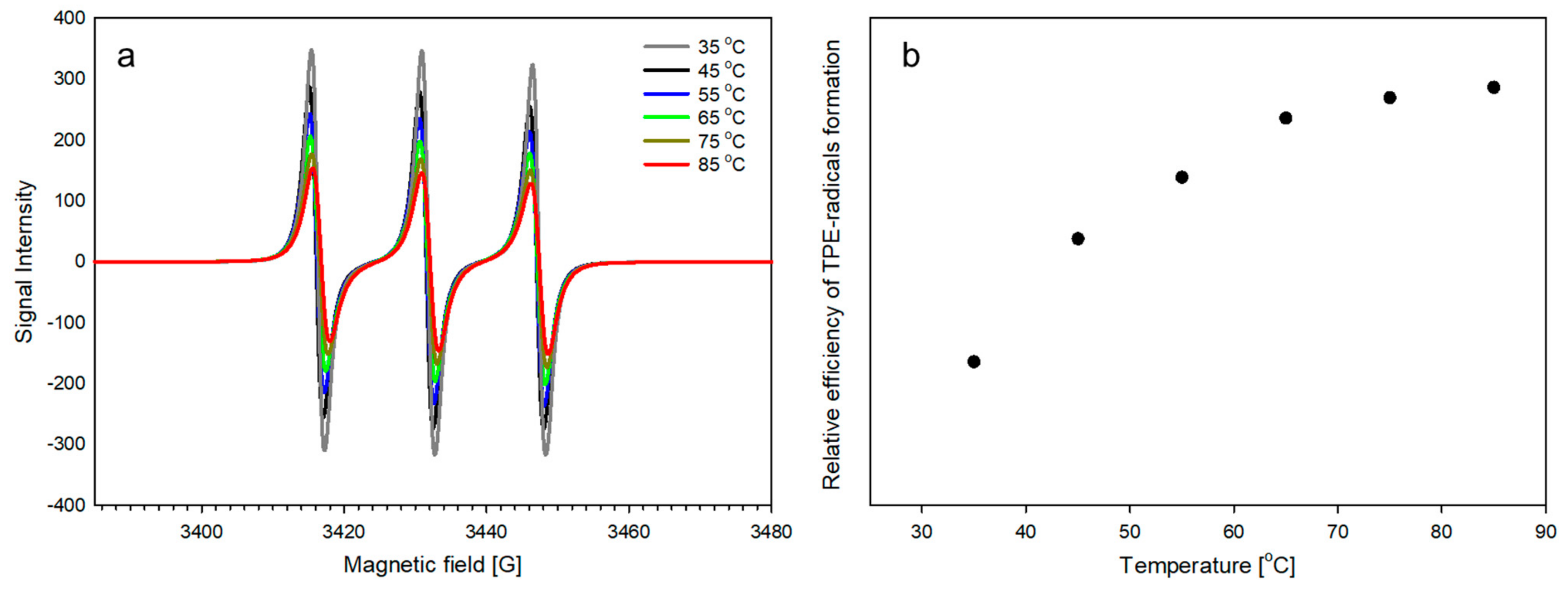
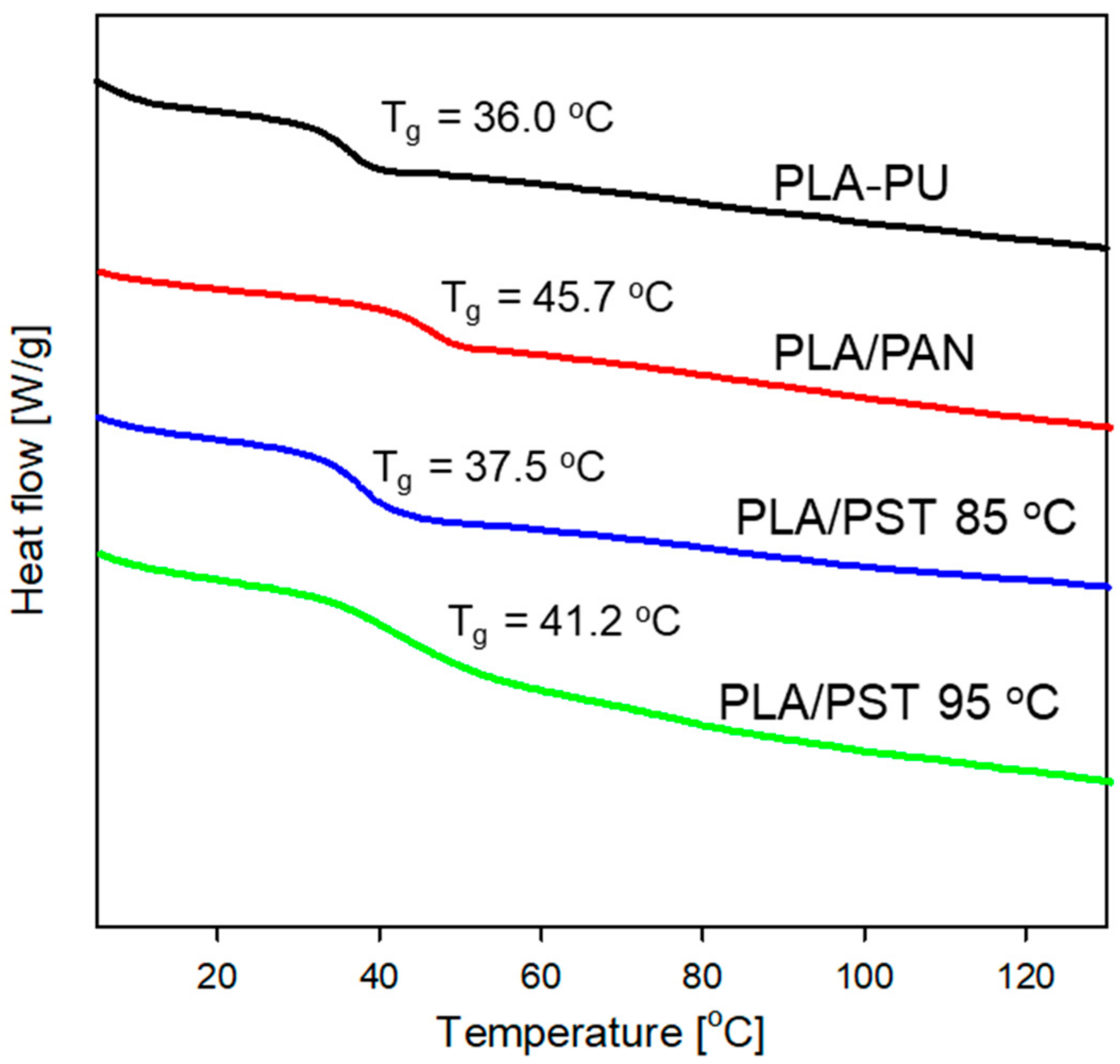
2.2. The Ability of TPE Groups inside PLA-Based Polyurethane to Form Radicals
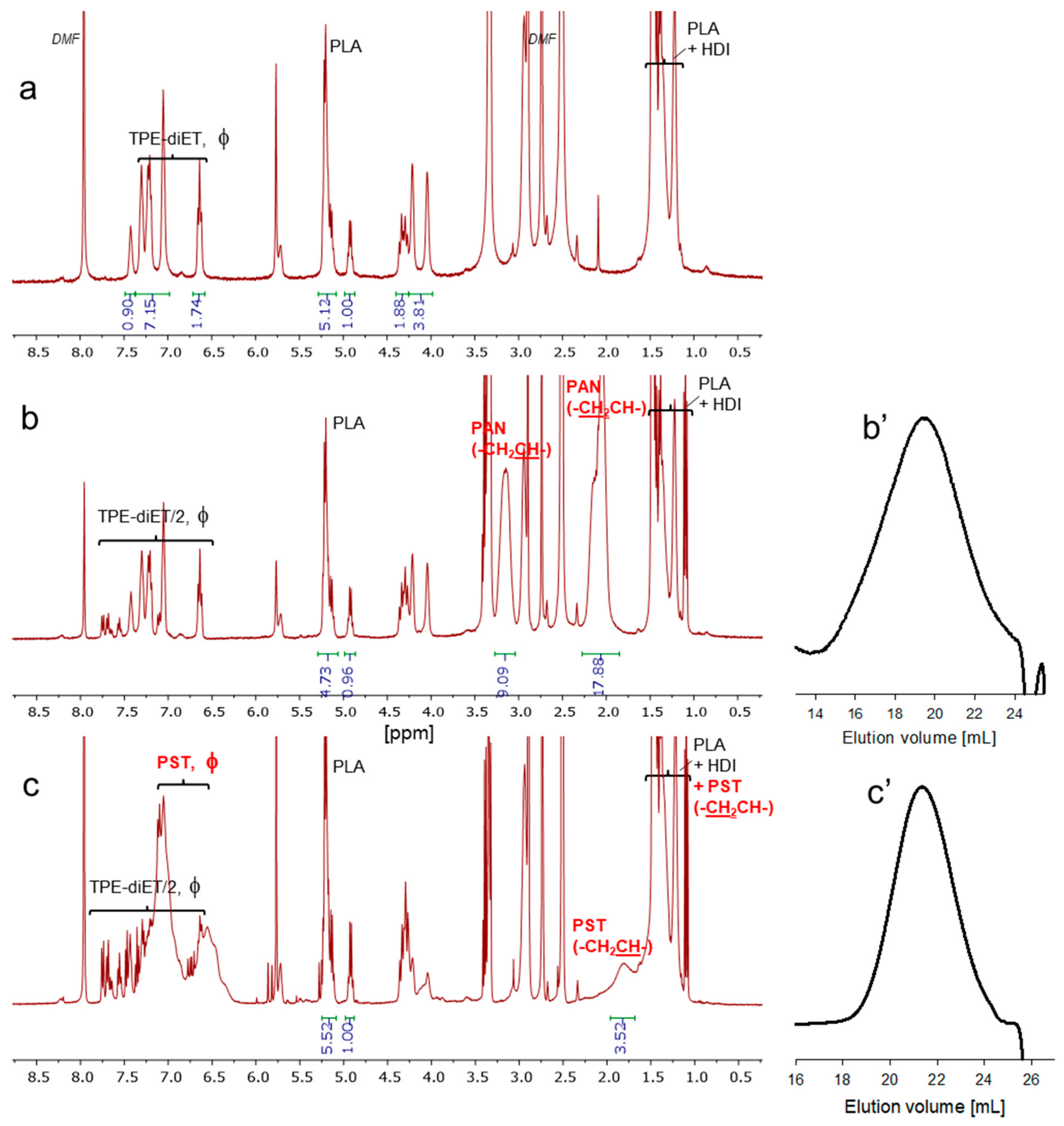
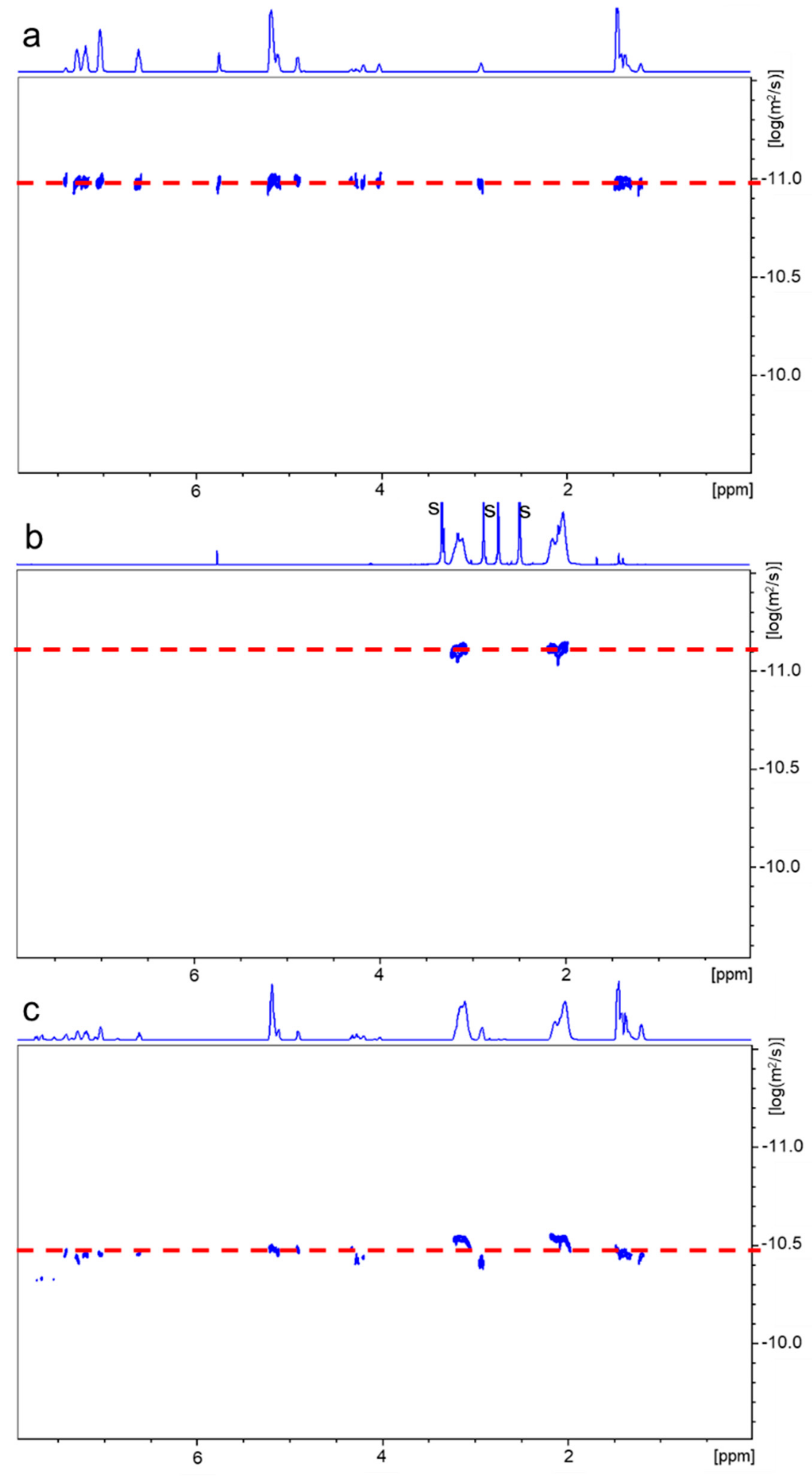
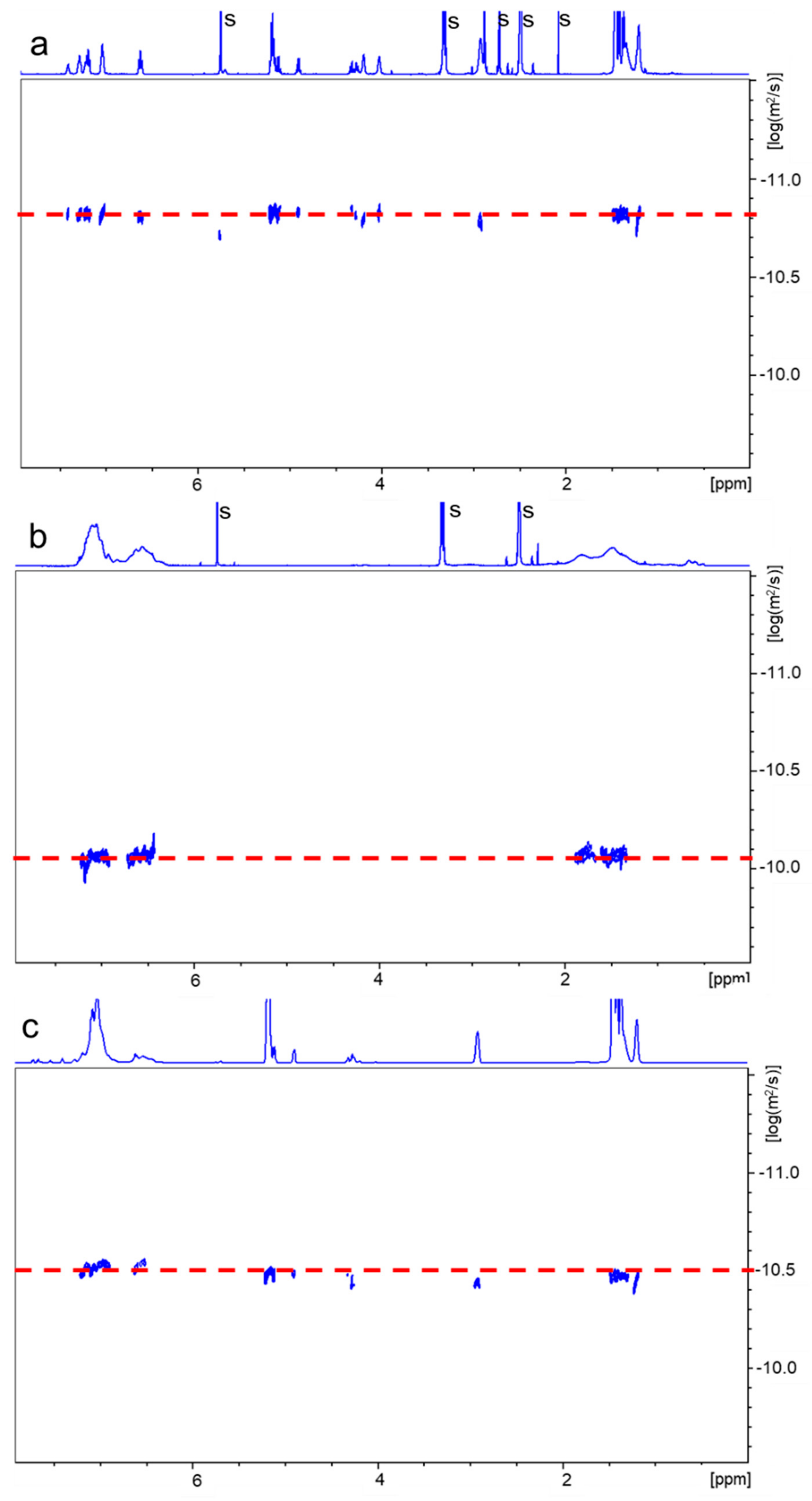
2.3. Polymerization of Acrylonitrile and Styrene Initiated with PLA-PU Containing TPE Groups
3. Materials and Methods
3.1. Materials
3.2. Synthesis of Polylactide Diol
3.3. Synthesis of TPED Derivative Containing Primary Hydroxyl Groups (TPE-diET)
3.4. Synthesis of PLA-Based Polyurethane (PLA-PU) Containing Multiple TPE-diET Groups
3.5. Polymerization of Acrylonitrile or Styrene Using PLA-PU as a Macroinifer
3.6. Instrumental Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Farah, S.; Anderson, D.G.; Langer, R. Physical and mechanical properties of PLA, and their functions in widespread applications —A comprehensive review. Adv. Drug Deliv. Rev. 2016, 107, 367–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagarajan, V.; Mohanty, A.K.; Misra, M. Perspective on Polylactic Acid (PLA) based Sustainable Materials for Durable Applications: Focus on Toughness and Heat Resistance. ACS Sustain. Chem. Eng. 2016, 4, 2899–2916. [Google Scholar] [CrossRef]
- Yildirim, I.; Weber, C.; Schubert, U.S. Old meets new: Combination of PLA and RDRP to obtain sophisticated macromolecular architectures. Prog. Polym. Sci. 2018, 76, 111–150. [Google Scholar] [CrossRef]
- Oh, J.K. Polylactide (PLA)-based amphiphilic block copolymers: Synthesis, self-assembly, and biomedical applications. Soft Matter 2011, 7, 5096–5108. [Google Scholar] [CrossRef] [Green Version]
- Dove, A.P. Controlled ring-opening polymerisation of cyclic esters: Polymer blocks in self-assembled nanostructures. Chem. Commun. 2008, 48, 6446. [Google Scholar] [CrossRef]
- Grabowski, M.; Kost, B.; Kubisa, P.; Bednarek, M. A New Approach to The Synthesis of Polylactide/Polyacrylonitrile Block Copolymers. Polymers 2022, 14, 1529. [Google Scholar] [CrossRef]
- Otsu, T. Iniferter concept and living radical polymerization. J. Polym. Sci. Part A Polym. Chem. 2000, 38, 2121–2136. [Google Scholar] [CrossRef]
- Braun, V.D.; Becker, K.H. Aromatische Pinakole als Polymerisationsinitiatoren. Angew. Makromol. Chem. 1969, 6, 186–189. [Google Scholar] [CrossRef]
- Braun, D.; Becker, K.H. Kinetik der Polymerisationsauslösung mit aromatischen pinakolen. Die Makromol. Chem. 1971, 147, 91–99. [Google Scholar] [CrossRef]
- Błedzki, A.; Braun, D.; Menzel, W.; Titzschkau, K. Polymerisationsauslösung mit substituierten ethanen, 5. Polymerisation von verschiedenen methacrylmonomeren. Die Makromol. Chem. 1983, 184, 287–294. [Google Scholar] [CrossRef]
- Otsu, T.; Matsumoto, A.; Tazaki, T. Radical polymerization of methyl methacrylate with some 1,2-disubstituted tetraphenylethanes as thermal iniferters. Polym. Bull. 1987, 17, 323–330. [Google Scholar] [CrossRef]
- Furniss, B.; Hannaford, A.; Smith, P.; Tatchell, A. Vogel’s Textbook of Practical Organic Chemistry; Longman Scientific & Technical, Longman House: London, UK, 1989. [Google Scholar]
- Grabowski, M.; Kost, B.; Gostyński, B.; Bednarek, M. Can tetraphenylethane (TPE) “iniferter” groups be introduced into polymer chains by coupling TPE diol with diisocyanates? Polymers 2022, 246, 124738. [Google Scholar] [CrossRef]
- Zhou, G.; Ma, C.; Zhang, G. Synthesis of polyurethane-g-poly(ethylene glycol) copolymers by macroiniferter and their protein resistance. Polym. Chem. 2011, 2, 1409–1414. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, L.; Zhang, Z.; Zhang, Y.; Tuo, X. Synthesis and simultaneous self-assembly of multiblock fluorinated polyurethane in iniferter polymerization. J. Polym. Sci. Part A Polym. Chem. 2013, 51, 2161–2170. [Google Scholar] [CrossRef]
- Tharanikkarasu, K.; Radhakrishnan, G. Tetraphenylethane iniferters: Polyurethane-polystyrene multiblock copolymers through “living” radical polymerization. J. Appl. Polym. Sci. 1997, 66, 1551–1560. [Google Scholar] [CrossRef]
- Sundar, S.; Tharanikkarasu, K.; Dhathathreyan, A.; Radhakrishnan, G. Aqueous dispersions of poly(urethane-co-vinyl-pyridine) synthesised from polyurethane macroiniferter. Colloid Polym. Sci. 2002, 280, 915–921. [Google Scholar] [CrossRef]
- Kim, B.K.; Tharanikkarasu, K.; Lee, J.S. Polyurethane-polymethacrylic acid multiblock copolymer dispersions through polyurethane macroiniferters. Colloid Polym. Sci. 1999, 277, 285–290. [Google Scholar] [CrossRef]
- Tharanikkarasu, K.; Kim, B.K. Modification of aqueous polyurethane dispersions via tetraphenylethane iniferters. J. Appl. Polym. Sci. 1999, 73, 2993–3000. [Google Scholar] [CrossRef]
- Ionescu, M. Chemistry and Technology of Polyols for Polyurethanes, 2nd ed.; Smithers Rapra Technology Ltd.: Shawbury, Shrewsbury, Shropshire, UK, 2016; Volume 2, ISBN 9781910242988. [Google Scholar]
- Baśko, M.; Kubisa, P. Cationic polymerization of L,L-lactide. J. Polym. Sci. Part A Polym. Chem. 2010, 48, 2650–2658. [Google Scholar] [CrossRef]
- Nim, B.; Opaprakasit, P. Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy Quantitative analyses of products from chemical recycling of polylactide (PLA) by alcoholysis with various alcohols and their applications as healable lactide-based polyurethanes. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2021, 255, 119684. [Google Scholar] [CrossRef]
- Nakamura, Y.; Yamago, S. Termination Mechanism in the Radical Polymerization of Methyl Methacrylate and Styrene Determined by the Reaction of Structurally Well-Defined Polymer End Radicals. Macromolecules 2015, 48, 6450–6456. [Google Scholar] [CrossRef]
- Ballard, N.; Hamzehlou, S.; Ruipérez, F.; Asua, J.M. On the Termination Mechanism in the Radical Polymerization of Acrylates. Macromol. Rapid Commun. 2016, 37, 1364–1368. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Ogihara, T.; Kato, T.; Nakamura, Y.; Yamago, S. Evidence for Polarity-and Viscosity-Controlled Pathways in the Termination Reaction in the Radical Polymerization of Acrylonitrile. Macromolecules 2021, 54, 4497–4506. [Google Scholar] [CrossRef]
- Reichardt, C.; Welton, T. Solvents and Solvent Effects in Organic Chemistry: Fourth Edition, 2nd ed.; Verlag Chemie: Weinheim, Germany; Basel, Switzerland; Cambridge, UK; New York, NY, USA, 2010; ISBN 9783527324736. [Google Scholar]
- Duda, A.; Penczek, S. Polymerization of ∈-Caprolactone Initiated by Aluminum Isopropoxide Trimer and/or Tetramer. Macromolecules 1995, 28, 5981–5992. [Google Scholar] [CrossRef]
- Lee, S.-H.; Kim, S.H.; Han, Y.-K.; Kim, Y.H. Synthesis and degradation of end-group-functionalized polylactide. J. Polym. Sci. Part A Polym. Chem. 2001, 39, 973–985. [Google Scholar] [CrossRef]
- Nishida, H.; Mori, T.; Hoshihara, S.; Fan, Y. Effect of tin on poly (l -lactic acid) pyrolysis. Polym. Degrad. Stab. 2003, 81, 515–523. [Google Scholar] [CrossRef]
- Wojtczak, E.; Kubisa, P.; Bednarek, M. Thermal stability of polylactide with different end-groups depending on the catalyst used for the polymerization. Polym. Degrad. Stab. 2018, 151, 100–104. [Google Scholar] [CrossRef]
- Groves, P. Diffusion ordered spectroscopy (DOSY) as applied to polymers. Polym. Chem. 2017, 8, 6700–6708. [Google Scholar] [CrossRef]
- Li, W.; Chung, H.; Daeffler, C.; Johnson, J.A.; Grubbs, R.H. Application of 1H DOSY for facile measurement of polymer molecular weights. Macromolecules 2012, 45, 9595–9603. [Google Scholar] [CrossRef] [Green Version]
- Lewinski, P.; Sosnowski, S.; Kazmierski, S.; Penczek, S. Polymer Chemistry COMMUNICATION di ff usion-ordered spectroscopy (DOSY): A “One conversion, polymer structure, M n and M w. Polym. Chem. 2015, 6, 4353–4357. [Google Scholar] [CrossRef]
- Cichoń, K.; Kost, B.; Basko, M. Synthesis and properties of ABA-triblock copolymers from polyester A-blocks and easily degradable polyacetal B-blocks. Polym. Chem. 2022, 13, 5243–5255. [Google Scholar] [CrossRef]
- Daimon, H.; Okitsu, H.; Kumanotani, J. Glass transition behaviors of random and block copolymers and polymer blends of styrene and cyclododecyl acrylate. I. Glass transition temperatures. Polym. J. 1975, 7, 460–466. [Google Scholar] [CrossRef]
- Beevers, R.B. Dependence of the glass transition temperature of polyacrylonitrile on molecular weight. J. Polym. Sci. Part A Gen. Pap. 1964, 2, 5257–5265. [Google Scholar] [CrossRef]
- Rieger, J. The glass transition temperature of polystyrene. Results of a round robin test. J. Therm. Anal. 1996, 46, 965–972. [Google Scholar] [CrossRef]
- Available online: https://www.spectroscopyonline.com/view/the-infrared-spectra-of-polymers-iii-hydrocarbon-polymers (accessed on 20 November 2022).
- Eren, O.; Ucar, N.; Onen, A.; Kizildag, N.; Karacan, I. Synergistic effect of polyaniline, nanosilver, and carbon nanotube mixtures on the structure and properties of polyacrylonitrile composite nanofiber. J. Comp. Mat. 2016, 50, 2073–2086. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substrates | Conversion of VM [%] | PLA/PVM Copolymers (a) | ||||
|---|---|---|---|---|---|---|
| Vinyl Monomer, VM | Polymeriz. No | Mol Ratio of VM/LA in Feed | Solvent | Mol Ratio of VM/LA, 1H NMR | Mn, SEC (b) | |
| AN | 1 | 4.8 | DMF | 85 | 7.0 | n.d. |
| 2 | 4.8 | Anisole | 70 | 3.8 | 16,600 | |
| ST | 3 | 2.5 | DMF | 37 | 0.4 | n.d. |
| 4 | 2.5 | DMF | 45 (95 °C) | 1.2 | 13,000 | |
| 6 | 2.5 | Anisole | 33 | 0.5 | 11,600 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grabowski, M.; Kost, B.; Bodzioch, A.; Bednarek, M. Functionalization of Polylactide with Multiple Tetraphenyethane Inifer Groups to Form PLA Block Copolymers with Vinyl Monomers. Int. J. Mol. Sci. 2023, 24, 19. https://doi.org/10.3390/ijms24010019
Grabowski M, Kost B, Bodzioch A, Bednarek M. Functionalization of Polylactide with Multiple Tetraphenyethane Inifer Groups to Form PLA Block Copolymers with Vinyl Monomers. International Journal of Molecular Sciences. 2023; 24(1):19. https://doi.org/10.3390/ijms24010019
Chicago/Turabian StyleGrabowski, Mateusz, Bartłomiej Kost, Agnieszka Bodzioch, and Melania Bednarek. 2023. "Functionalization of Polylactide with Multiple Tetraphenyethane Inifer Groups to Form PLA Block Copolymers with Vinyl Monomers" International Journal of Molecular Sciences 24, no. 1: 19. https://doi.org/10.3390/ijms24010019