
Old and Promising Markers Related to Autophagy in Traumatic Brain Injury
 , and
, and
Abstract
:1. Introduction
2. Results
2.1. Beclin 1
2.2. Lipidated Microtubule-Associated Protein Light-Chain 3 (LC3-II)

{kind=link}
{kind=link}
| Articles | Markers | Methods | Results |
|---|---|---|---|
| Diskin et al., 2005 [6] | Beclin-1 | WB, IHC, and IF in the brain of TBI mice models | Increased in the perilesional cortex and hippocampus. Increased between 4 h and 3 weeks after trauma with a peak at 1 week after trauma in neurons. Increased between 3 days and 3 weeks in astrocytes |
| Erlich et al., 2006 [12] | Beclin-1 | WB, IHC, and IF in the brain of TBI mice models | Increased in the perilesional cortex and hippocampus. Increased between 4 h and 3 weeks after trauma with a peak at 1 week after trauma in neurons. Increased between 3 days and 3 weeks in astrocytes |
| Clark et al., 2007 [7] | Beclin-1 | WB and IF in the brain of humans subjected to decompressive craniotomy after TBI | Increased in 1 of the 5 patients without correlation with LC3-II |
| LC3-II | WB and IF in the brain of humans subjected to decompressive craniotomy after TBI | Increased in 4 of the 5 patents and 3 of 5 controls without correlation with Belcin-1 | |
| Lai et al., 2008 [8] | LC3-II | WB in the brain of TBI mice models | Increased between 2 and 48 h after the trauma whit a peak at 24 h after the trauma |
| Liu et al., 2008 [9] | LC3-II | WB and IF in the brain of TBI rat models | Increased non significantly 4 h after trauma and significantly between 1 and 15 days after trauma in living neurons |
| Zhang et al., 2008 [10] | LC3-II | IHC and IF in the brain of TBI mice models | Increased between 1 h and 32 days after trauma with a peak at 8 h after trauma in the perilesional cortex. Specially in neurons until the third day, then in glial cells |
| Beclin-1 | WB, IHC, and IF in the brain of TBI mice models | Increased Between 1 h and 32 days after trauma with a peak at 8 days after trauma in perilesional cortex and hippocampus especially in neurons until the third day, then in glial cells | |
| Sadasivan et al., 2008 [11] | LC3-II | WB in the brain of TBI rat models | Increased between 2 h and 2 days in ipsilateral and contralateral brain |
| Beclin-1 | WB in the brain of TBI rat models | Increased non significantly after 2 and 6 h but the rate of beclin-1/bcl2 increase significantly at 1 and 2 days | |
| Sun et al., 2014 [15] | LC3-II | WB and IF in the brain of TBI rat models | Increased non significantly 3 h after the trauma and significantly 6, 24, and 48 h after trauma with a peak at 24 h after the trauma predominantly in living neurons of the hippocampus |
| p-CX43 | WB and IF in the brain of TBI rat models | Increased significantly between 3 and 24 h after the trauma with a peak at 6 h after the trauma in the astrocyte of the hippocampus | |
| Lin et al., 2014 [18] | LC3-II | WB in the brain of TBI rat models | Increased between 30 min and 24 h after the trauma |
| GSK-3β | WB in the brain of TBI rat models | Active form increased and inactive form decreased between 30 min and 24 h after the trauma | |
| Sarkar et al., 2014 [13] | LC3-II | WB and IF in the brain of TBI mice models | Increased between 1 h and 1 week after trauma with a peak between 1 and 3 days after traumas, at first in neurons and then in glial cells with a peak on third day. In the ipsilateral and contralateral cortex and hippocampus |
| Beclin-1 | WB in the brain of TBI mice models | No significant variation | |
| p62 | WB and IF in the brain of TBI mice models | Increased between 1 h and 7 days after the trauma in the ipsilateral cortex and hippocampus | |
| LAMP family | WB in the brain of TBI mice models | LAMP1 and LAMP2 increase between 3 and 7 days after trauma in lysosomal and cytosolic fractions | |
| CTSD | WB and IF in the brain of TBI mice models | Decreased 1 and 24 h after trauma and increased 3 and 7 days after trauma in the cortex and hippocampus | |
| Park et al., 2015 [19] | LAMP family | WB and IF in the brain of TBI rat models | LAMP2A increases between 1 and 15 days in neurons and glial cells after trauma especially in CA3 and Cx areas ipsilaterally |
| HSP70 | WB in the brain of TBI rat models | Increased between 1 and 15 days after the trauma with a peak at 3 days after the trauma | |
| Ye et al., 2017 [17] | WISP1 | WB and IF in the brain of TBI rat models | Decreased 1 day after trauma, reach the minimum at 3 days after trauma, and recovered at 5 and 7 days in neurons, in particular in the ipsilateral brain |
| LC3-II | WB and IF in the brain of TBI rat models | Increased 3 days after trauma in ipsilateral brain | |
| β-catenin | WB and IF the in brain of TBI rat models | Decreased between 1 and 7 days after trauma with a negative peak at 3 days after trauma and a partial restoration at 5 days after trauma | |
| Zhang et al., 2017 [16] | 3-MST | WB, IHC, and IF in the brain of TBI mice models | Increased between 6 h and 3 days after trauma with a peak at 1 day after trauma in vital neurons of the ipsilateral cortex |
| LC3-II | WB and IF in the brain of TBI mice models | Increased between 12 h and 2 days after trauma with a peak at 24 h in vital and non-vital neurons in the ipsilateral cortex | |
| Che et al., 2017 [20] | Cx40 | WB and IF in the brain of TBI rat models | Increased between 6 h and 6 days after trauma with a peak at 1 day after trauma in neurons of the perilesional cortex, at first in intercellular vesicles, and then diffused in the cell |
| Sebastiani et al., 2017 [14] | p62 | WB in the brain of TBI mice models | Decreased between 1 and 5 days after trauma |
| Saykally et al., 2018 [21] | TDP-43 | Radioimmunoprecipitation in the brain of TBI mice models | Increased 12 days after 1 50 g trauma. Increased 3 days after 5 30 g traumas and still increased, not significantly after 30 days in the ipsilateral cortex. Decreased in the ipsilateral hippocampus between 30 and 60 days after 5 30 g traumas |
| LC3-II | Radioimmunoprecipitation in the brain of TBI mice models | Increased 3 days after trauma | |
| Atg7 | Radioimmunoprecipitation in the brain of TBI mice models | Decreased 3 days after trauma | |
| LAMP family | Radioimmunoprecipitation in the brain of TBI mice models | LAMP2A decreases 60 days after trauma in the ipsilateral hippocampus | |
| Wiesner et al., 2018 [22] | TDP-43 | IF in the brain of TBI and stab mice models | Increased between 3 and 7 days after the trauma with a peak at 3 days after the trauma |
| Gao et al., 2018 [23] | IL-33 | WB and IF in the brain of TBI mice models | Increased 1 day after trauma in astrocyte nucleus and oligodendrocyte cytoplasm |
| ST2L | WB and IF in the brain of TBI mice models | Decreased 1 day after trauma | |
| Tominaga et al., 2019 [24] | SA-β-gal | IHC and IF in the brain of TBI mice models | Increased between 1 and 14 days after trauma with a peak at 7 days after trauma in the ipsilateral cortex |
| Ciclin-D1 | IHC and IF in the brain of TBI mice models | Increased between 1 and 14 days after trauma with a peak between 4 and 7 days in neurons, astrocytes, and microglia in the ipsilateral cortex | |
| PCNA | IHC and IF in the brain of TBI mice models | Increased between 1 and 14 days after trauma with a peak between 4 and 7 days in neurons, astrocytes, and microglia in the ipsilateral cortex | |
| p16 | IHC and IF in the brain of TBI mice models | Increased between 4 and 14 days after trauma in astrocyte of ipsilateral brain | |
| p21 | IHC and IF in the brain of TBI mice models | Increased between 4 and 14 days after trauma in neurons and microglia of the ipsilateral brain | |
| Chen et al., 2020 [25] | Chd8 | WB and IF in the brain of TBI mice models | In the perilesional cortex it decreases 3 h after trauma, partially restores at 12 h, and decreases again until the seventh day. In prefrontal cortex increases between 12 and 24 h after trauma with a peak at 12 h. In the hippocampus, it increases between 6 and 24 h with a peak at 12 h. It is localized in neurons |
| β-catenin | WB and IF in the brain of TBI mice models | In the prefrontal cortex, it increases between 6 h and 7 days after trauma with a peak at 12 h. In perilesional cortex and hippocampus it increases between 12 and 24 h with a peak at 12 h after trauma | |
| Li et al., 2021 [26] | Sec22b | WB and IF in the brain of TBI rat models | Decreased between 12 h and 7 days after trauma with a negative peak at 24 h after trauma in the perilesional cortex |
2.3. Connexins Family
2.4. Glycogen Synthase Kinase-31β (GSK-3β)
2.5. p62 or Seqeuestrsomoe-1 (SQSTM1)
2.6. Lysosomal-Associated Membrane Proteins (LAMPs) and Heat Shock Protein 70 (HSP70)
2.7. Cathepsin D (CTSD)
2.8. Wnt1 Inducible Signaling Pathway Protein 1 (WISP1)
2.9. β-Catenin
2.10. 3-Mercaptopyruvate Sulfurtransferase (3-MST) or tRNA Thiouridine Modification Protein (TUM1)
2.11. Transactivation Response Deoxyribonucleic Acid Protein 43 (TDP-43)
2.12. Autophagy Related 7 (Atg7)
2.13. Interleukin 33 (IL-33) and Interleukin 1 Receptor-like 1 (IL1RL1) or ST2
2.14. Senescence-Associated-β-Galactosidase (SA-β-gal)
2.15. Cyclin D1 and Proliferating Cell Nuclear Antigen (PCNA)
2.16. p16 and p21
2.17. Chromodomain-Helicase-DNA-Binding Protein 8 (Chd8)
2.18. Sec22b
3. Discussion
Overview of TBI Treatment Targeted on Autophagy Pathways
4. Materials and Methods
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Capizzi, A.; Woo, J.; Verduzco-Gutierrez, M. Traumatic Brain Injury. Med. Clin. N. Am. 2020, 104, 213–238. [Google Scholar] [CrossRef] [PubMed]
- Brazinova, A.; Rehorcikova, V.; Taylor, M.S.; Buckova, V.; Majdan, M.; Psota, M.; Peeters, W.; Feigin, V.; Theadom, A.; Holkovic, L.; et al. Epidemiology of Traumatic Brain Injury in Europe: A Living Systematic Review. J. Neurotrauma 2021, 38, 1411–1440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertozzi, G.; Maglietta, F.; Sessa, F.; Scoto, E.; Cipolloni, L.; Di Mizio, G.; Salerno, M.; Pomara, C. Traumatic Brain Injury: A Forensic Approach: A Literature Review. Curr. Neuropharmacol. 2020, 18, 538–550. [Google Scholar] [CrossRef]
- Zwirner, J.; Kulakofsky, R.; Fitzek, A.; Schröder, A.S.; Bohnert, S.; Franke, H.; Renné, T.; Tse, R.; Ondruschka, B. Forensic Biomarkers of Lethal Traumatic Brain Injury. Int. J. Legal Med. 2022, 136, 871–886. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Chen, X.; Xu, L.; Zhang, R.; Li, Z.; Yue, X.; Qiao, D. Traumatic Axonal Injury: Neuropathological Features, Postmortem Diagnostic Methods, and Strategies. Forensic Sci. Med. Pathol. 2022, 18, 530–544. [Google Scholar] [CrossRef] [PubMed]
- Diskin, T.; Tal-Or, P.; Erlich, S.; Mizrachy, L.; Alexandrovich, A.; Shohami, E.; Pinkas-Kramarski, R. Closed Head Injury Induces Upregulation of Beclin 1 at the Cortical Site of Injury. J. Neurotrauma 2005, 22, 750–762. [Google Scholar] [CrossRef]
- Clark, R.S.B.; Bayir, H.; Chu, C.T.; Alber, S.M.; Kochanek, P.M.; Watkins, S.C. Autophagy Is Increased in Mice after Traumatic Brain Injury and Is Detectable in Human Brain after Trauma and Critical Illness. Autophagy 2008, 4, 88–90. [Google Scholar] [CrossRef] [Green Version]
- Lai, Y.; Hickey, R.W.; Chen, Y.; Bayιr, H.; Sullivan, M.L.; Chu, C.T.; Kochanek, P.M.; Dixon, C.E.; Jenkins, L.W.; Graham, S.H.; et al. Autophagy Is Increased after Traumatic Brain Injury in Mice and Is Partially Inhibited by the Antioxidant γ-Glutamylcysteinyl Ethyl Ester. J. Cereb. Blood Flow Metab. 2008, 28, 540–550. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.L.; Chen, S.; Dietrich, D.; Hu, B.R. Changes in Autophagy after Traumatic Brain Injury. J. Cereb. Blood Flow Metab. 2008, 28, 674–683. [Google Scholar] [CrossRef]
- Zhang, Y.-B.; Li, S.-X.; Chen, X.-P.; Yang, L.; Zhang, Y.-G.; Liu, R.; Tao, L.-Y. Autophagy Is Activated and Might Protect Neurons from Degeneration after Traumatic Brain Injury. Neurosci. Bull. 2008, 24, 143–149. [Google Scholar] [CrossRef]
- Sadasivan, S.; Dunn, W.A.; Hayes, R.L.; Wang, K.K.W. Changes in Autophagy Proteins in a Rat Model of Controlled Cortical Impact Induced Brain Injury. Biochem. Biophys. Res. Commun. 2008, 373, 478–481. [Google Scholar] [CrossRef] [PubMed]
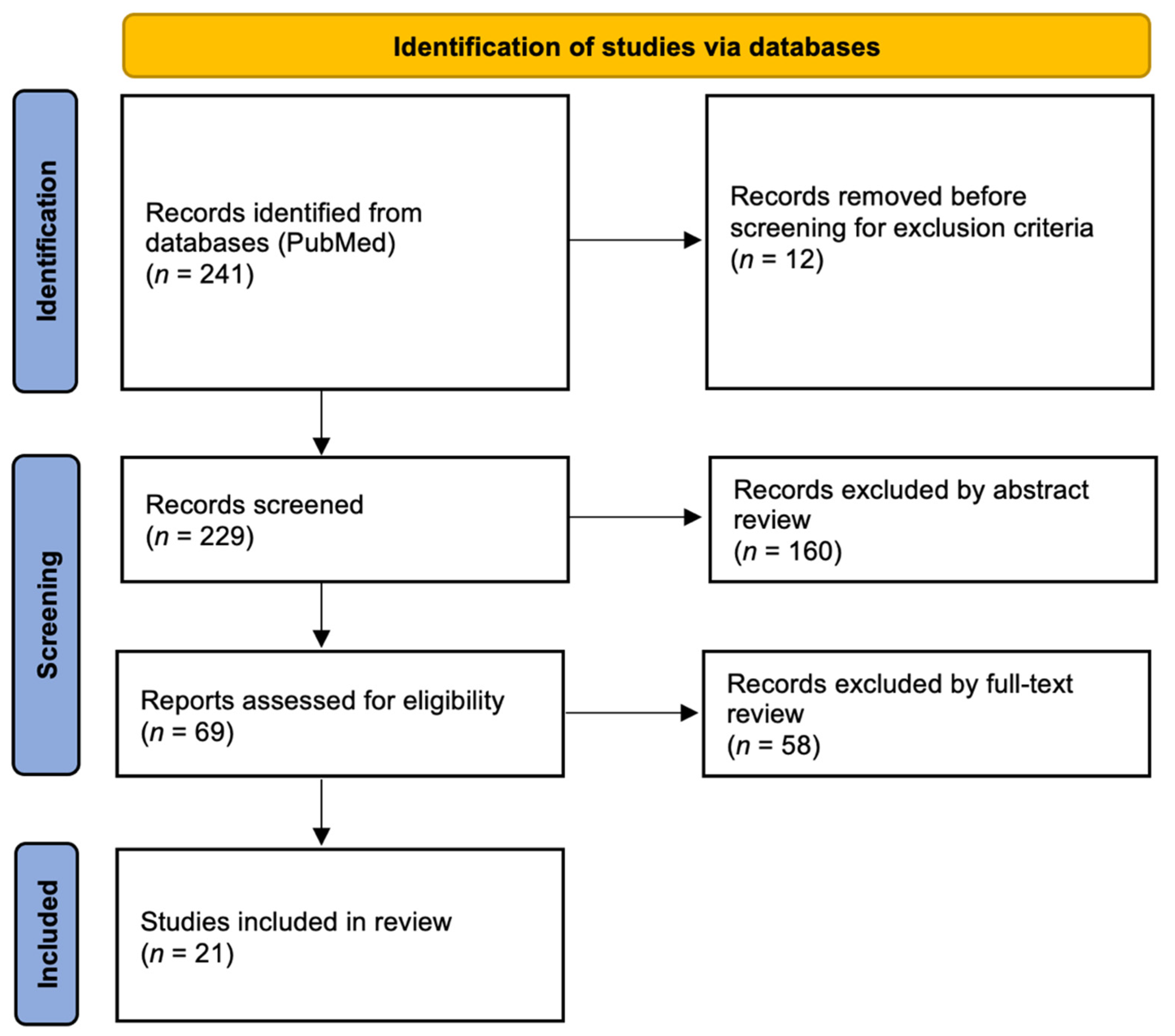
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 Statement: An Updated Guideline for Reporting Systematic Reviews. BMJ 2021, 10, 89. [Google Scholar] [CrossRef]
- Erlich, S.; Shohami, E.; Pinkas-Kramarski, R. Neurodegeneration Induces Upregulation of Beclin 1. Autophagy 2006, 2, 49–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkar, C.; Zhao, Z.; Aungst, S.; Sabirzhanov, B.; Faden, A.I.; Lipinski, M.M. Impaired Autophagy Flux Is Associated with Neuronal Cell Death after Traumatic Brain Injury. Autophagy 2014, 10, 2208–2222. [Google Scholar] [CrossRef] [Green Version]
- Sebastiani, A.; Gölz, C.; Sebastiani, P.G.; Bobkiewicz, W.; Behl, C.; Mittmann, T.; Thal, S.C.; Engelhard, K. Sequestosome 1 Deficiency Delays, but Does Not Prevent Brain Damage Formation Following Acute Brain Injury in Adult Mice. Front. Neurosci. 2017, 11, 678. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.-Q.; Gao, J.-L.; Cui, C.-M.; Cui, Y.; Jing, X.-B.; Zhao, M.-M.; Wang, Y.-C.; Tian, Y.-X.; Wang, K.-J.; Cui, J.-Z. Astrocytic P-Connexin 43 Regulates Neuronal Autophagy in the Hippocampus Following Traumatic Brain Injury in Rats. Mol. Med. Rep. 2014, 9, 77–82. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Shan, H.; Chang, P.; Ma, L.; Chu, Y.; Shen, X.; Wu, Q.; Wang, Z.; Luo, C.; Wang, T.; et al. Upregulation of 3-MST Relates to Neuronal Autophagy After Traumatic Brain Injury in Mice. Cell. Mol. Neurobiol. 2017, 37, 291–302. [Google Scholar] [CrossRef]
- Ye, Y.; Zhang, P.; Qian, Y.; Yin, B.; Yan, M. The Effect of Pyrroloquinoline Quinone on the Expression of WISP1 in Traumatic Brain Injury. Stem Cells Int. 2017, 2017, 4782820. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.-J.; Chen, T.-H.; Yang, L.-Y.; Shih, C.-M. Resveratrol Protects Astrocytes against Traumatic Brain Injury through Inhibiting Apoptotic and Autophagic Cell Death. Cell Death Dis. 2014, 5, e1147. [Google Scholar] [CrossRef] [Green Version]
- Park, Y.; Liu, C.; Luo, T.; Dietrich, W.D.; Bramlett, H.; Hu, B. Chaperone-Mediated Autophagy after Traumatic Brain Injury. J. Neurotrauma 2015, 32, 1449–1457. [Google Scholar] [CrossRef]
- Che, W.; Guo, Y.; Yang, W.; Zheng, P.; Zeng, J.; Tong, W. Involvement of Autophagy in Connexin 40 Reduction in the Late Phase of Traumatic Brain Injury in Rats. Brain Res. Bull. 2017, 131, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Saykally, J.N.; Ratliff, W.A.; Keeley, K.L.; Pick, C.G.; Mervis, R.F.; Citron, B.A. Repetitive Mild Closed Head Injury Alters Protein Expression and Dendritic Complexity in a Mouse Model. J. Neurotrauma 2018, 35, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Wiesner, D.; Tar, L.; Linkus, B.; Chandrasekar, A.; olde Heuvel, F.; Dupuis, L.; Tsao, W.; Wong, P.C.; Ludolph, A.; Roselli, F. Reversible Induction of TDP-43 Granules in Cortical Neurons after Traumatic Injury. Exp. Neurol. 2018, 299, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Zhang, M.; Wang, T.; Fan, Y.; Yu, L.; Ye, G.; Wang, Z.; Gao, C.; Wang, H.; Luo, C.; et al. IL-33/ST2L Signaling Provides Neuroprotection Through Inhibiting Autophagy, Endoplasmic Reticulum Stress, and Apoptosis in a Mouse Model of Traumatic Brain Injury. Front. Cell. Neurosci. 2018, 12, 95. [Google Scholar] [CrossRef] [PubMed]
- Tominaga, T.; Shimada, R.; Okada, Y.; Kawamata, T.; Kibayashi, K. Senescence-Associated-β-Galactosidase Staining Following Traumatic Brain Injury in the Mouse Cerebrum. PLoS ONE 2019, 14, e0213673. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wang, H.; Luo, C.; Gao, C.; Zhang, Y.; Chen, G.; Chen, W.; Chen, X.; Tao, L. Chd8 Rescued TBI-Induced Neurological Deficits by Suppressing Apoptosis and Autophagy Via Wnt Signaling Pathway. Cell. Mol. Neurobiol. 2020, 40, 1165–1184. [Google Scholar] [CrossRef]
- Li, D.; Zhang, Y.; Lu, L.; Zhang, L.; Ma, J.; Ji, J.; Li, H.; Chen, G. Upregulation of Sec22b Plays a Neuroprotective Role in a Rat Model of Traumatic Brain Injury via Inducing Protective Autophagy. Brain Res. Bull. 2021, 166, 29–36. [Google Scholar] [CrossRef]
- Bennett, M.V.L.; Contreras, J.E.; Bukauskas, F.F.; Sáez, J.C. New Roles for Astrocytes: Gap Junction Hemichannels Have Something to Communicate. Trends Neurosci. 2003, 26, 610–617. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.H.-C.; Weigel, H.; Cotrina, M.L.; Liu, S.; Bueno, E.; Hansen, A.J.; Hansen, T.W.; Goldman, S.; Nedergaard, M. Gap-Junction-Mediated Propagation and Amplification of Cell Injury. Nat. Neurosci. 1998, 1, 494–500. [Google Scholar] [CrossRef]
- Forde, J.E.; Dale, T.C. Glycogen Synthase Kinase 3: A Key Regulator of Cellular Fate. Cell. Mol. Life Sci. 2007, 64, 1930–1944. [Google Scholar] [CrossRef]
- Lamark, T.; Kirkin, V.; Dikic, I.; Johansen, T. NBR1 and P62 as Cargo Receptors for Selective Autophagy of Ubiquitinated Targets. Cell Cycle 2009, 8, 1986–1990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rusten, T.E.; Stenmark, H. P62, an Autophagy Hero or Culprit? Nat. Cell Biol. 2010, 12, 207–209. [Google Scholar] [CrossRef] [PubMed]
- Eskelinen, E.-L. Roles of LAMP-1 and LAMP-2 in Lysosome Biogenesis and Autophagy. Mol. Aspects Med. 2006, 27, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Di, Y.-Q.; Han, X.-L.; Kang, X.-L.; Wang, D.; Chen, C.-H.; Wang, J.-X.; Zhao, X.-F. Autophagy Triggers CTSD (Cathepsin D) Maturation and Localization inside Cells to Promote Apoptosis. Autophagy 2021, 17, 1170–1192. [Google Scholar] [CrossRef]
- Wang, S.; Zhong Chong, Z.; Chen Shang, Y.; Maiese, K. WISP1 (CCN4) Autoregulates Its Expression and Nuclear Trafficking of β-Catenin during Oxidant Stress with Limited Effects upon Neuronal Autophagy. Curr. Neurovasc. Res. 2012, 9, 91–101. [Google Scholar] [CrossRef]
- Valenta, T.; Hausmann, G.; Basler, K. The Many Faces and Functions of β-Catenin: β-Catenin: A Life by, beyond, and against the Wnt Canon. EMBO J. 2012, 31, 2714–2736. [Google Scholar] [CrossRef] [Green Version]
- Fräsdorf, B.; Radon, C.; Leimkühler, S. Characterization and Interaction Studies of Two Isoforms of the Dual Localized 3-Mercaptopyruvate Sulfurtransferase TUM1 from Humans. J. Biol. Chem. 2014, 289, 34543–34556. [Google Scholar] [CrossRef] [Green Version]
- Budini, M.; Buratti, E.; Morselli, E.; Criollo, A. Autophagy and Its Impact on Neurodegenerative Diseases: New Roles for TDP-43 and C9orf72. Front. Mol. Neurosci. 2017, 10, 170. [Google Scholar] [CrossRef] [Green Version]
- Bose, J.K.; Huang, C.-C.; Shen, C.-K.J. Regulation of Autophagy by Neuropathological Protein TDP-43. J. Biol. Chem. 2011, 286, 44441–44448. [Google Scholar] [CrossRef] [Green Version]
- Wicher, G.; Wallenquist, U.; Lei, Y.; Enoksson, M.; Li, X.; Fuchs, B.; Abu Hamdeh, S.; Marklund, N.; Hillered, L.; Nilsson, G.; et al. Interleukin-33 Promotes Recruitment of Microglia/Macrophages in Response to Traumatic Brain Injury. J. Neurotrauma 2017, 34, 3173–3182. [Google Scholar] [CrossRef]
- Kochanek, P.M.; Janesko, K.L.; Jenkins, L.W.; Yan, H.Q.; Kibbe, M.R.; Robichaud, P.; Wooditch, A.C.; Clark, R.S.B.; Dixon, C.E.; Marion, D.W.; et al. Adenovirus-Mediated Transfer and Expression of β-Gal in Injured Hippocampus After Traumatic Brain Injury in Mice. J. Neurotrauma 2001, 18, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Kabadi, S.V.; Stoica, B.A.; Loane, D.J.; Byrnes, K.R.; Hanscom, M.; Cabatbat, R.M.; Tan, M.T.; Faden, A.I. Cyclin D1 Gene Ablation Confers Neuroprotection in Traumatic Brain Injury. J. Neurotrauma 2012, 29, 813–827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, X.; Li, M. A New Pathway for Senescence Regulation. Genomics Proteomics Bioinformatics 2015, 13, 333–335. [Google Scholar] [CrossRef] [Green Version]
- Katano, H.; Masago, A.; Taki, H.; Nakatsuka, M.; Fuse, T.; Yamada, K. P53-Independent Transient P21WAF1/CIP1 MRNA Induction in the Rat Brain Following Experimental Traumatic Injury. NeuroReport 2000, 11, 2073–2078. [Google Scholar] [CrossRef] [PubMed]
- Weissberg, O.; Elliott, E. The Mechanisms of CHD8 in Neurodevelopment and Autism Spectrum Disorders. Genes 2021, 12, 1133. [Google Scholar] [CrossRef]
- New, J.; Thomas, S.M. Autophagy-Dependent Secretion: Mechanism, Factors Secreted, and Disease Implications. Autophagy 2019, 15, 1682–1693. [Google Scholar] [CrossRef]
- Kobayashi, S. Choose Delicately and Reuse Adequately: The Newly Revealed Process of Autophagy. Biol. Pharm. Bull. 2015, 38, 1098–1103. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of Cells and Tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Wang, W.; Li, Y. Molecular Mechanism and Regulation of Autophagy and Its Potential Role in Epilepsy. Cells 2022, 11, 2621. [Google Scholar] [CrossRef]
- Wang, P.; Shao, B.-Z.; Deng, Z.; Chen, S.; Yue, Z.; Miao, C.-Y. Autophagy in Ischemic Stroke. Prog. Neurobiol. 2018, 163–164, 98–117. [Google Scholar] [CrossRef]
- Li, H.; Wu, J.; Shen, H.; Yao, X.; Liu, C.; Pianta, S.; Han, J.; Borlongan, C.V.; Chen, G. Autophagy in Hemorrhagic Stroke: Mechanisms and Clinical Implications. Prog. Neurobiol. 2018, 163–164, 79–97. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Liu, Y.; Sun, M. Autophagy and Alzheimer’s Disease. Cell. Mol. Neurobiol. 2017, 37, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Autophagy: Biology and Diseases: Clinical Science; Le, W. (Ed.) Advances in Experimental Medicine and Biology; Springer: Singapore, 2020; Volume 1207, ISBN 9789811542718. [Google Scholar]
- Karabiyik, C.; Lee, M.J.; Rubinsztein, D.C. Autophagy Impairment in Parkinson’s Disease. Essays Biochem. 2017, 61, 711–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tripathi, M.K.; Rajput, C.; Mishra, S.; ur Rasheed, M.S.; Singh, M.P. Malfunctioning of Chaperone-Mediated Autophagy in Parkinson’s Disease: Feats, Constraints, and Flaws of Modulators. Neurotox. Res. 2019, 35, 260–270. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Watzlawik, J.O.; Fiesel, F.C.; Springer, W. Autophagy in Parkinson’s Disease. J. Mol. Biol. 2020, 432, 2651–2672. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, M.K.; Rasheed, M.S.U.; Mishra, A.K.; Patel, D.K.; Singh, M.P. Silymarin Protects Against Impaired Autophagy Associated with 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine-Induced Parkinsonism. J. Mol. Neurosci. 2020, 70, 276–283. [Google Scholar] [CrossRef]
- Zhang, L.; Li, Z.; Mao, L.; Wang, H. Circular RNA in Acute Central Nervous System Injuries: A New Target for Therapeutic Intervention. Front. Mol. Neurosci. 2022, 15, 816182. [Google Scholar] [CrossRef]
- Liu, H.; He, S.; Wang, J.; Li, C.; Liao, Y.; Zou, Q.; Chen, R. Tetrandrine Ameliorates Traumatic Brain Injury by Regulating Autophagy to Reduce Ferroptosis. Neurochem. Res. 2022, 47, 1574–1587. [Google Scholar] [CrossRef]
- Zhu, M.; Huang, X.; Shan, H.; Zhang, M. Mitophagy in Traumatic Brain Injury: A New Target for Therapeutic Intervention. Oxid. Med. Cell. Longev. 2022, 2022, 4906434. [Google Scholar] [CrossRef]
- Zhao, S.; Wang, S.; Cao, L.; Zeng, H.; Lin, S.; Lin, Z.; Chen, M.; Zhu, M.; Pang, Z.; Zhang, Y. Acupuncture Promotes Nerve Repair through the Benign Regulation of MTOR-mediated Neuronal Autophagy in Traumatic Brain Injury Rats. CNS Neurosci. Ther. 2022, cns.14018. [Google Scholar] [CrossRef]
- Farkhondeh, T.; Samarghandian, S.; Roshanravan, B.; Peivasteh-roudsari, L. Impact of Curcumin on Traumatic Brain Injury and Involved Molecular Signaling Pathways. Recent Pat. Food Nutr. Agric. 2020, 11, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Forouzanfar, F.; Read, M.I.; Barreto, G.E.; Sahebkar, A. Neuroprotective Effects of Curcumin through Autophagy Modulation. IUBMB Life 2020, 72, 652–664. [Google Scholar] [CrossRef] [PubMed]
- Unchiti, K.; Leurcharusmee, P.; Samerchua, A.; Pipanmekaporn, T.; Chattipakorn, N.; Chattipakorn, S.C. The Potential Role of Dexmedetomidine on Neuroprotection and Its Possible Mechanisms: Evidence from in Vitro and in Vivo Studies. Eur. J. Neurosci. 2021, 54, 7006–7047. [Google Scholar] [CrossRef] [PubMed]
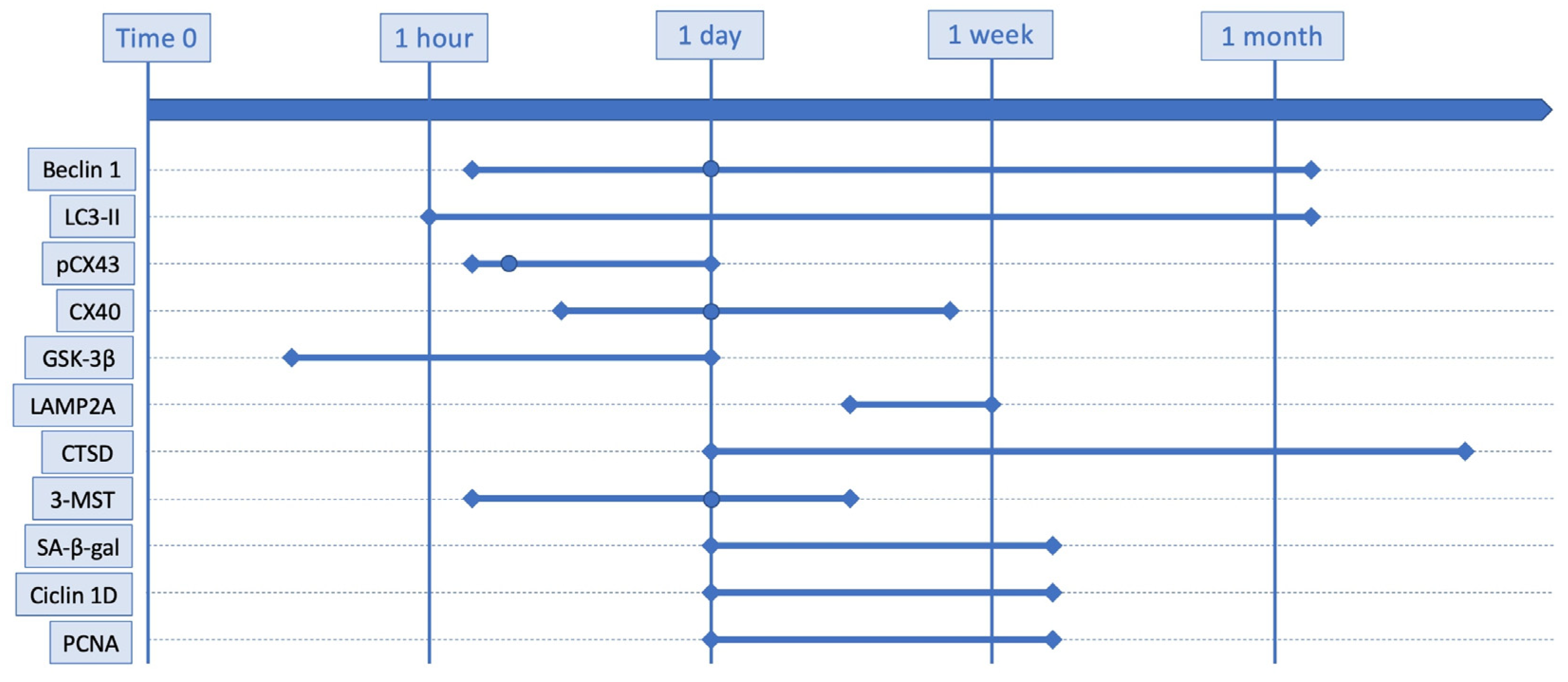
| Marker | No. of Studies | Conclusion | Models | Techniques |
|---|---|---|---|---|
| Beclin-1 | 6 | Increased between 4 h and 32 days | Mice, rats, humans | WB, IHC, IF |
| LC3-II | 11 | Increased between 1 h and 32 days with a peak at 24 h | Mice, rats, humans | WB, IHC, IF, radioimmunoprecipitation |
| pCX43 | 1 | Increased between 3 and 24 h with a peak at 6 h | Rats | WB, IF |
| CX40 | 1 | Increased between 6 h and 6 days with a peak at 24 h | Rats | WB, IF |
| GSK-3β | 1 | Increase between 30 min and 24 h | Rats | WB |
| p62 | 2 | No consensus | Mice | WB, IF |
| LAMP1 | 1 | Increased between 3 and 7 days | Mice | WB |
| LAMP2A | 3 | Increased between 1 and 15 days | Mice | WB, IHC, radioimmunoprecipitation |
| HSP70 | 1 | Increased between 1 and 15 days | Mice | WB, IHC |
| CTSD | 1 | Decreased between 1 and 24 h and increased between 3 and 7 days | Mice | WB, IF |
| WISP1 | 1 | Decreased between 1 and 7 days with a minimum at 3 days | Rats | WB, IF |
| β-catenin | 2 | No consensus | Mice, rats | WB, IF |
| 3-MST | 1 | Increased between 6 h and 3 days with a peak at 24 h | Mice | WB, IHC, IF |
| TDP-43 | 2 | Increased between 3 and 7 days | Mice | Radioimmunoprecipitation, IF |
| Atg7 | 1 | Decreased after 3 days | Mice | Radioimmunoprecipitation |
| IL-33 | 1 | Increased after 1 day | Mice | WB, IF |
| ST2L | 1 | Decreased after 1 day | Mice | WB, IF |
| SA-β-gal | 1 | Increased between 1 and 14 days with a peak at 7 days | Mice | IHC, IF |
| Cyclin-D | 1 | Increased between 1 and 14 days with a peak between 4 and 7 days | Mice | IHC, IF |
| PCNA | 1 | Increased between 1 and 14 days with a peak between 4 and 7 days | Mice | IHC, IF |
| p16 | 1 | Increased between 4 and 14 days | Mice | IHC, IF |
| p21 | 1 | Increased between 4 and 14 days | Mice | IHC, IF |
| Chd8 | 1 | Variable depending on the brain area | Mice | WB, IF |
| Sec22b | 1 | Decreased between 12 h and 7 days with a minimum at 24 h | Rats | WB, IF |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Livieri, T.; Cuttaia, C.; Vetrini, R.; Concato, M.; Peruch, M.; Neri, M.; Radaelli, D.; D’Errico, S. Old and Promising Markers Related to Autophagy in Traumatic Brain Injury. Int. J. Mol. Sci. 2023, 24, 72. https://doi.org/10.3390/ijms24010072
Livieri T, Cuttaia C, Vetrini R, Concato M, Peruch M, Neri M, Radaelli D, D’Errico S. Old and Promising Markers Related to Autophagy in Traumatic Brain Injury. International Journal of Molecular Sciences. 2023; 24(1):72. https://doi.org/10.3390/ijms24010072
Chicago/Turabian StyleLivieri, Tommaso, Calogero Cuttaia, Raffaella Vetrini, Monica Concato, Michela Peruch, Margherita Neri, Davide Radaelli, and Stefano D’Errico. 2023. "Old and Promising Markers Related to Autophagy in Traumatic Brain Injury" International Journal of Molecular Sciences 24, no. 1: 72. https://doi.org/10.3390/ijms24010072