
The Expression of FOXO3a as a Forensic Diagnostic Tool in Cases of Traumatic Brain Injury: An Immunohistochemical Study


 , , , ,
, , , ,  and
and
Abstract
1. Introduction
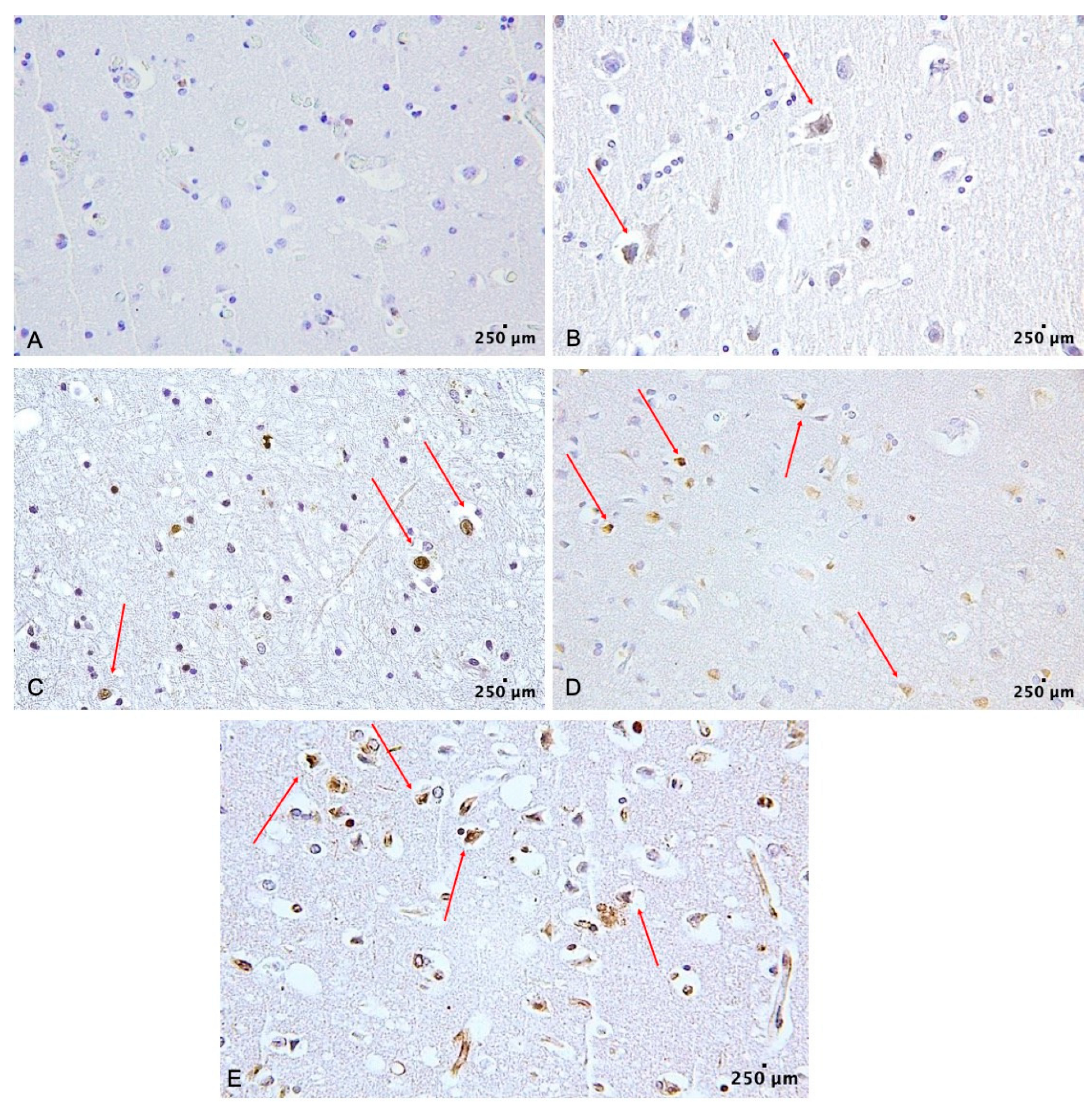
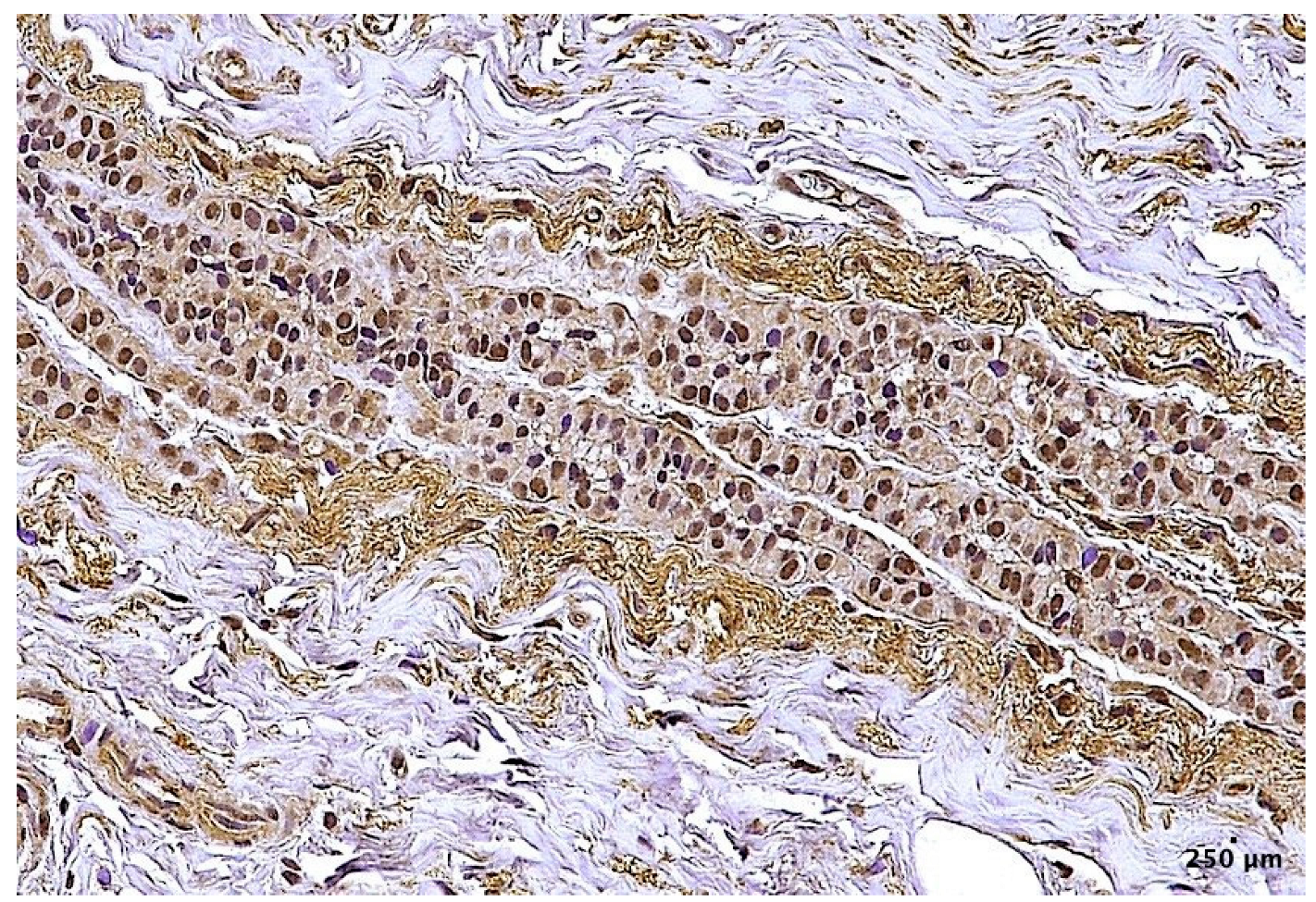
2. Results
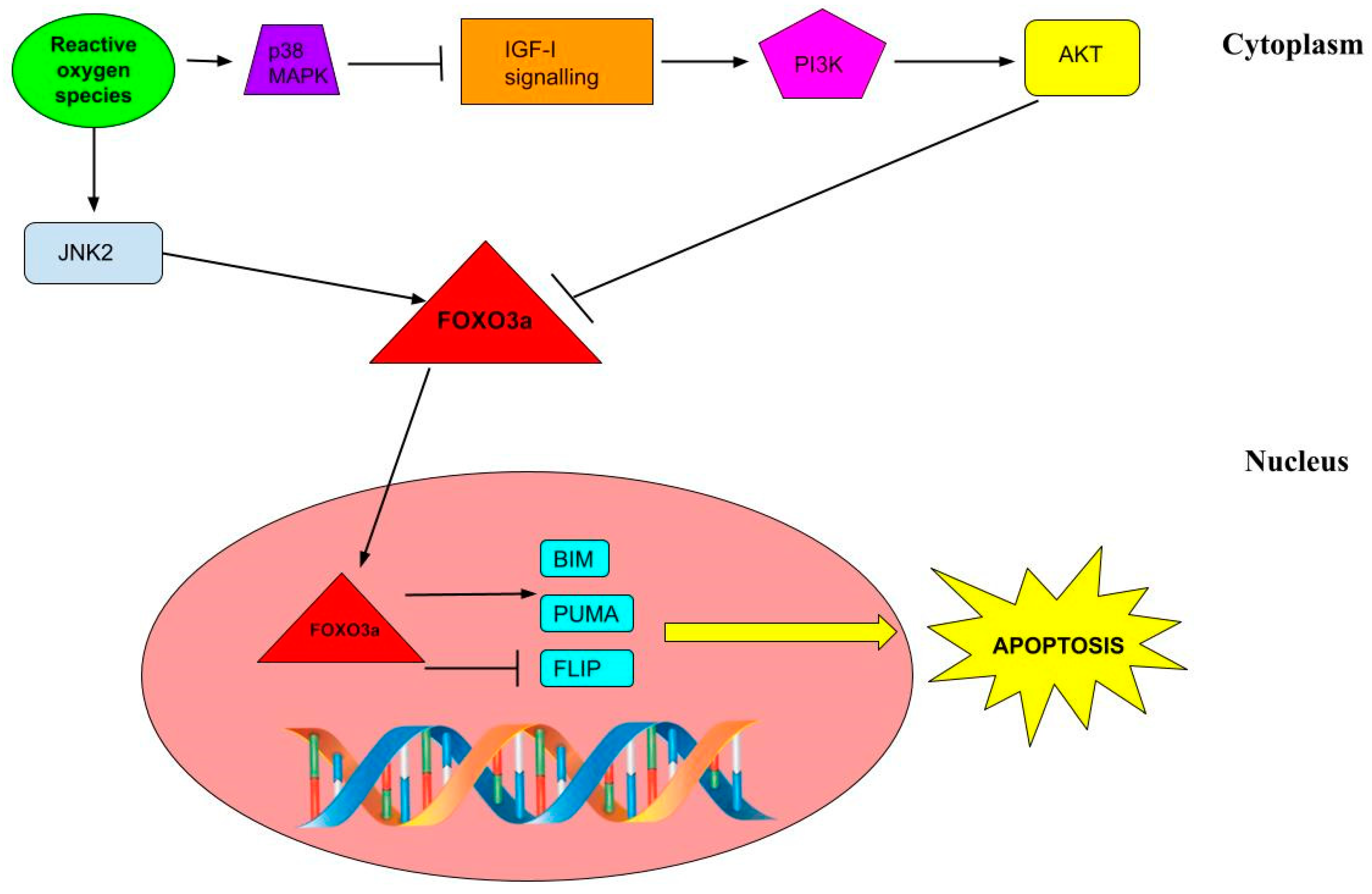
3. Discussion
4. Materials and Methods
4.1. Study Group Selection and Sample Collection
4.2. Histological and Immunohistochemical Analysis
4.3. Quantitative Analysis
4.4. Statistical Analysis
4.5. Abbreviations
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Menon, D.K.; Schwab, K.; Wright, D.W.; Maas, A.I. Position statement: Definition of traumatic brain injury. Arch. Phys. Med. Rehabil. 2010, 91, 1637–1640. [Google Scholar] [CrossRef]
- Surveillance Report of Traumatic Brain Injury-related Emergency Department Visits, Hospitalizations, and Deaths. In: Centers for Disease Control and Prevention, U.S. Department of Health and Human Services. 2014. Available online: https://www.cdc.gov/traumaticbraininjury/get_the_facts.html (accessed on 28 November 2022).
- Centers for Disease Control and Prevention. National Center for Health Statistics: Mortality Data on CDC WONDER. Available online: https://wonder.cdc.gov/mcd.html (accessed on 28 November 2022).
- Dell’Aquila, M.; Maiese, A.; De Matteis, A.; Viola, R.V.; Arcangeli, M.; La Russa, R.; Fineschi, V. Traumatic brain injury: Estimate of the age of the injury based on neuroinflammation, endothelial activation markers and adhesion molecules. Histol. Histopathol. 2021, 36, 795–806. [Google Scholar] [CrossRef]
- Simon, D.; McGeachy, M.; Bayır, H.; Clark, R.S.; Loane, D.J.; Kochanek, P.M. The far-reaching scope of neuroinflammation after traumatic brain injury. Nat. Rev. Neurol. 2017, 13, 171–191. [Google Scholar] [CrossRef]
- Zhou, J.; Liao, W.; Yang, J.; Ma, K.; Li, X.; Wang, Y.; Wang, D.; Wang, L.; Zhang, Y.; Yin, Y.; et al. FOXO3 induces FOXO1-dependent autophagy by activating the AKT1 signaling pathway. Autophagy 2012, 8, 1712–1723. [Google Scholar] [CrossRef]
- Li, Z.; He, Q.; Zhai, X.; You, Y.; Li, L.; Hou, Y.; He, F.; Zhao, Y.; Zhao, J. Foxo1-mediated inflammatory response after cerebral hemorrhage in rats. Neurosci. Lett. 2016, 26, 131–136. [Google Scholar] [CrossRef]
- Dávila, D.; Torres-Aleman, I. Neuronal death by oxidative stress involves activation of FOXO3 through a two-arm pathway that activates stress kinases and attenuates insulin-like growth factor I signaling. Mol. Biol. Cell 2008, 19, 2014–2025. [Google Scholar] [CrossRef]
- Li, D.; Qu, Y.; Mao, M.; Zhang, X.; Li, J.; Ferriero, D.; Mu, D. Involvement of the PTEN-AKT-FOXO3a pathway in neuronal apoptosis in developing rat brain after hypoxia-ischemia. J. Cereb. Blood Flow Metab. 2009, 29, 1903–1913. [Google Scholar] [CrossRef]
- Sun, L.; Zhao, M.; Liu, M.; Su, P.; Zhang, J.; Li, Y.; Yang, X.; Wu, Z. Suppression of FoxO3a attenuates neurobehavioral deficits after traumatic brain injury through inhibiting neuronal autophagy. Behav. Brain Res. 2018, 30, 271–279. [Google Scholar] [CrossRef]
- Liu, X.L.; Gao, C.C.; Qi, M.; Han, Y.L.; Zhou, M.L.; Zheng, L.R. Expression of FOXO transcription factors in the brain following traumatic brain injury. Neurosci. Lett. 2021, 14, 1358–1382. [Google Scholar] [CrossRef]
- Ng, S.Y.; Lee, A.Y.W. Traumatic Brain Injuries: Pathophysiology and Potential Therapeutic Targets. Front. Cell Neurosci. 2019, 13, 528. [Google Scholar] [CrossRef]
- Bullock, M.R.; Chesnut, R.; Ghajar, J.; Gordon, D.; Hartl, R.; Newell, D.W.; Servadei, F.; Walters, B.C.; Wilberger, J.E. Surgical Management of Traumatic Brain Injury Author Group. Surgical management of acute epidural hematomas. Neurosurgery 2006, 58, 7–15. [Google Scholar]
- Bullock, M.R.; Chesnut, R.; Ghajar, J.; Gordon, D.; Hartl, R.; Newell, D.W.; Servadei, F.; Walters, B.C.; Wilberger, J.E. Surgical Management of Traumatic Brain Injury Author Group. Surgical management of acute subdural hematomas. Neurosurgery 2006, 58, 16–24. [Google Scholar]
- Bullock, M.R.; Chesnut, R.; Ghajar, J.; Gordon, D.; Hartl, R.; Newell, D.W.; Servadei, F.; Walters, B.C.; Wilberger, J.E. Surgical Management of Traumatic Brain Injury Author Group. Surgical management of posterior fossa mass lesions. Neurosurgery 2006, 58, 47–55. [Google Scholar] [CrossRef]
- Bullock, M.R.; Chesnut, R.; Ghajar, J.; Gordon, D.; Hartl, R.; Newell, D.W.; Servadei, F.; Walters, B.C.; Wilberger, J.E. Surgical Management of Traumatic Brain Injury Author Group. Surgical management of depressed cranial fractures. Neurosurgery 2006, 58, 56–60. [Google Scholar] [CrossRef]
- Skandsen, T.; Kvistad, K.A.; Solheim, O.; Strand, I.H.; Folvik, M.; Vik, A. Prevalence and impact of diffuse axonal injury in patients with moderate and severe head injury: A cohort study of early magnetic resonance imaging findings and 1-year outcome. J. Neurosurg. 2010, 113, 556–563. [Google Scholar] [CrossRef]
- Payne, W.N.; De Jesus, O.; Payne, A.N. Contrecoup Brain Injury; StatPearls: Tampa, FL, USA, 2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK536965/ (accessed on 28 November 2022).
- Smith, D.H.; Meaney, D.F.; Shull, W.H. Diffuse axonal injury in head trauma. J. Head Trauma Rehabil. 2003, 18, 307–316. [Google Scholar] [CrossRef]
- Moen, K.G.; Skandsen, T.; Folvik, M.; Brezova, V.; Kvistad, K.A.; Rydland, J.; Manley, G.T.; Vik, A. A longitudinal MRI study of traumatic axonal injury in patients with moderate and severe traumatic brain injury. J. Neurol. Neurosurg. Psychiatry 2012, 83, 1193–1200. [Google Scholar] [CrossRef]
- Douglas, D.B.; Ro, T.; Toffoli, T.; Krawchuk, B.; Muldermans, J.; Gullo, J.; Dulberger, A.; Anderson, A.E.; Douglas, P.K.; Wintermark, M. Neuroimaging of Traumatic Brain Injury. Med. Sci. 2018, 7, 2. [Google Scholar] [CrossRef]
- Ray, S.K.; Dixon, C.E.; Banik, N.L. Molecular mechanisms in the pathogenesis of traumatic brain injury. Histol. Histopathol. 2002, 17, 1137–1152. [Google Scholar] [CrossRef]
- Schmidt, O.I.; Infanger, M.; Heyde, C.E.; Ertel, W.; Stahelet, P.F. The Role of Neuroinflammation in Traumatic Brain Injury. Eur. J. Trauma 2004, 30, 135–149. [Google Scholar] [CrossRef]
- Fukunaga, K.; Shioda, N. Pathophysiological relevance of forkhead transcription factors in brain ischemia. Adv. Exp. Med. Biol. 2009, 665, 130–142. [Google Scholar] [CrossRef]
- Martins, R.; Lithgow, G.J.; Link, W. Long live FOXO: Unraveling the role of FOXO proteins in aging and longevity. Aging Cell 2016, 15, 196–207. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Li, T.; Yang, Z.; Hu, W.; Yang, Y. Deciphering the roles of FOXO1 in human neoplasms. Int. J. Cancer 2018, 143, 1560–1568. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Ao, X.; Ding, W.; Ponnusamy, M.; Wu, W.; Hao, X.; Yu, W.; Wang, Y.; Li, P.; Wang, J. Critical role of FOXO3a in carcinogenesis. Mol. Cancer 2018, 25, 104. [Google Scholar] [CrossRef] [PubMed]
- Dharaneeswaran, H.; Abid, M.R.; Yuan, L.; Dupuis, D.; Beeler, D.; Spokes, K.C.; Janes, L.; Sciuto, T.; Kang, P.M.; Jaminet, S.S.; et al. FOXO1-mediated activation of Akt plays a critical role in vascular homeostasis. Circ. Res. 2014, 115, 238–251. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, B.T.; Lee, K.Y.; Klaus, K.; Softic, S.; Krumpoch, M.T.; Fentz, J.; Stanford, K.I.; Robinson, M.M.; Cai, W.; Kleinridders, A.; et al. Insulin and IGF-1 receptors regulate FoxO-mediated signaling in muscle proteostasis. J. Clin. Investig. 2016, 126, 3433–3446. [Google Scholar] [CrossRef]
- Hu, W.; Yang, Z.; Yang, W.; Han, M.; Xu, B.; Yu, Z.; Shen, M.; Yang, Y. Roles of forkhead box O (FoxO) transcription factors in neurodegenerative diseases: A panoramic view. Prog. Neurobiol. 2019, 181, 1016–1045. [Google Scholar] [CrossRef]
- Xin, Z.; Ma, Z.; Hu, W.; Jiang, S.; Yang, Z.; Li, T.; Chen, F.; Jia, G.; Yang, Y. FOXO1/3: Potential suppressors of fibrosis. Ageing Res. Rev. 2018, 41, 42–52. [Google Scholar] [CrossRef]
- Akhter, R.; Sanphui, P.; Biswas, S.C. The essential role of p53-up-regulated modulator of apoptosis (Puma) and its regulation by FoxO3a transcription factor in β-amyloid-induced neuron death. J. Biol. Chem. 2014, 289, 10812–10822. [Google Scholar] [CrossRef]
- Lim, S.W.; Jin, L.; Luo, K.; Jin, J.; Shin, Y.J.; Hong, S.Y.; Yang, C.W. Klotho enhances FoxO3-mediated manganese superoxide dismutase expression by negatively regulating PI3K/AKT pathway during tacrolimus-induced oxidative stress. Cell Death Dis. 2017, 8, 2972. [Google Scholar] [CrossRef]
- Palmieri, M.; Frati, A.; Santoro, A.; Frati, P.; Fineschi, V.; Pesce, A. Diffuse Axonal Injury: Clinical Prognostic Factors, Molecular Experimental Models and the Impact of the Trauma Related Oxidative Stress. An Extensive Review Concerning Milestones and Advances. Int. J. Mol. Sci. 2021, 22, 10865. [Google Scholar] [CrossRef] [PubMed]
- Willcox, B.J.; Donlon, T.A.; He, Q.; Chen, R.; Grove, J.S.; Yano, K.; Masaki, K.H.; Willcox, D.C.; Rodriguez, B.; Curb, J.D. FOXO3A genotype is strongly associated with human longevity. Proc. Natl. Acad. Sci. USA 2008, 105, 13987–13992. [Google Scholar] [CrossRef] [PubMed]
- Medema, R.H.; Kops, G.J.; Bos, J.L.; Burgering, B.M. AFX-like Forkhead transcription factors mediate cell-cycle regulation by Ras and PKB through p27kip1. Nature 2000, 404, 782–787. [Google Scholar] [CrossRef] [PubMed]
- Tran, H.; Brunet, A.; Grenier, J.M.; Datta, S.R.; Fornace, A.J.; DiStefano, P.S.; Chiang, L.W.; Greenberg, M.E. DNA repair pathway stimulated by the forkhead transcription factor FOXO3a through the Gadd45 protein. Science 2002, 296, 530–534. [Google Scholar] [CrossRef] [PubMed]
- Brunet, A.; Bonni, A.; Zigmond, M.J.; Lin, M.Z.; Juo, P.; Hu, L.S.; Anderson, M.J.; Arden, K.C.; Blenis, J.; Greenberg, M.E. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 1999, 96, 857–868. [Google Scholar] [CrossRef]
- Gilley, J.; Coffer, P.J.; Ham, J. FOXO transcription factors directly activate bim gene expression and promote apoptosis in sympathetic neurons. J. Cell Biol. 2003, 162, 613–622. [Google Scholar] [CrossRef]
- You, H.; Pellegrini, M.; Tsuchihara, K.; Yamamoto, K.; Hacker, G.; Erlacher, M.; Villunger, A.; Mak, T.W. FOXO3a-dependent regulation of Puma in response to cytokine/growth factor withdrawal. J. Exp. Med. 2006, 203, 1657–1663. [Google Scholar] [CrossRef]
- Skurk, C.; Maatz, H.; Kim, H.S.; Yang, J.; Abid, M.R.; Aird, W.C.; Walsh, K. The Akt-regulated forkhead transcription factor FOXO3a controls endothelial cell viability through modulation of the caspase-8 inhibitor FLIP. J. Biol. Chem. 2004, 279, 1513–1525. [Google Scholar] [CrossRef]
- La Russa, R.; Maiese, A.; Di Fazio, N.; Morano, A.; Di Bonaventura, C.; De Matteis, A.; Fazio, V.; Frati, P.; Fineschi, V. Post-Traumatic Meningitis Is a Diagnostic Challenging Time: A Systematic Review Focusing on Clinical and Pathological Features. Int. J. Mol. Sci. 2020, 21, 4148. [Google Scholar] [CrossRef]
- Spruiell Eldridge, S.L.; Teetsel, J.F.K.; Torres, R.A.; Ulrich, C.H.; Shah, V.V.; Singh, D.; Zamora, M.J.; Zamora, S.; Sater, A.K. A Focal Impact Model of Traumatic Brain Injury in Xenopus Tadpoles Reveals Behavioral Alterations, Neuroinflammation, and an Astroglial Response. Int. J. Mol. Sci. 2022, 23, 7578. [Google Scholar] [CrossRef]
- Neri, M.; Frati, A.; Turillazzi, E.; Cantatore, S.; Cipolloni, L.; Di Paolo, M.; Frati, P.; La Russa, R.; Maiese, A.; Scopetti, M.; et al. Immunohistochemical Evaluation of Aquaporin-4 and its Correlation with CD68, IBA-1, HIF-1α, GFAP, and CD15 Expressions in Fatal Traumatic Brain Injury. Int. J. Mol. Sci. 2018, 19, 3544. [Google Scholar] [CrossRef] [PubMed]
- Ots, H.D.; Tracz, J.A.; Vinokuroff, K.E.; Musto, A.E. CD40-CD40L in Neurological Disease. Int. J. Mol. Sci. 2022, 23, 4115. [Google Scholar] [CrossRef] [PubMed]
- Rocchi, A.; Chiti, E.; Maiese, A.; Turillazzi, E.; Spinetti, I. MicroRNAs: An Update of Applications in Forensic Science. Diagnostics 2020, 11, 32. [Google Scholar] [CrossRef] [PubMed]
- Gitto, L.; Bonaccorso, L.; Maiese, A.; dell'Aquila, M.; Arena, V.; Bolino, G. A scream from the past: A multidisciplinary approach in a concealment of a corpse found mummified. J. Forensic Leg. Med. 2015, 32, 53–58. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Case Number | Sex | Staining Intensity | Time of Death |
|---|---|---|---|
| 1 | M | 2 | Immediately |
| 2 | F | 1 | Immediately |
| 3 | M | 3 | 2 h |
| 4 | M | 2 | Immediately |
| 5 | M | 3 | 3.5 h |
| 6 | M | 1 | Immediately |
| 7 | F | 4 | 6 h |
| 8 | F | 3 | Immediately |
| 9 | F | 4 | 6 h |
| 10 | M | 2 | Immediately |
| 11 | M | 4 | 5 h |
| 12 | M | 2 | 1 h |
| 13 | M | 2 | Immediately |
| 14 | M | 4 | 4 h |
| 15 | M | 1 | Immediately |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maiese, A.; Spina, F.; Visi, G.; Del Duca, F.; De Matteis, A.; La Russa, R.; Di Paolo, M.; Frati, P.; Fineschi, V. The Expression of FOXO3a as a Forensic Diagnostic Tool in Cases of Traumatic Brain Injury: An Immunohistochemical Study. Int. J. Mol. Sci. 2023, 24, 2584. https://doi.org/10.3390/ijms24032584
Maiese A, Spina F, Visi G, Del Duca F, De Matteis A, La Russa R, Di Paolo M, Frati P, Fineschi V. The Expression of FOXO3a as a Forensic Diagnostic Tool in Cases of Traumatic Brain Injury: An Immunohistochemical Study. International Journal of Molecular Sciences. 2023; 24(3):2584. https://doi.org/10.3390/ijms24032584
Chicago/Turabian StyleMaiese, Aniello, Federica Spina, Giacomo Visi, Fabio Del Duca, Alessandra De Matteis, Raffaele La Russa, Marco Di Paolo, Paola Frati, and Vittorio Fineschi. 2023. "The Expression of FOXO3a as a Forensic Diagnostic Tool in Cases of Traumatic Brain Injury: An Immunohistochemical Study" International Journal of Molecular Sciences 24, no. 3: 2584. https://doi.org/10.3390/ijms24032584
APA StyleMaiese, A., Spina, F., Visi, G., Del Duca, F., De Matteis, A., La Russa, R., Di Paolo, M., Frati, P., & Fineschi, V. (2023). The Expression of FOXO3a as a Forensic Diagnostic Tool in Cases of Traumatic Brain Injury: An Immunohistochemical Study. International Journal of Molecular Sciences, 24(3), 2584. https://doi.org/10.3390/ijms24032584