
A Subpathway and Target Gene Cluster-Based Approach Uncovers lncRNAs Associated with Human Primordial Follicle Activation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
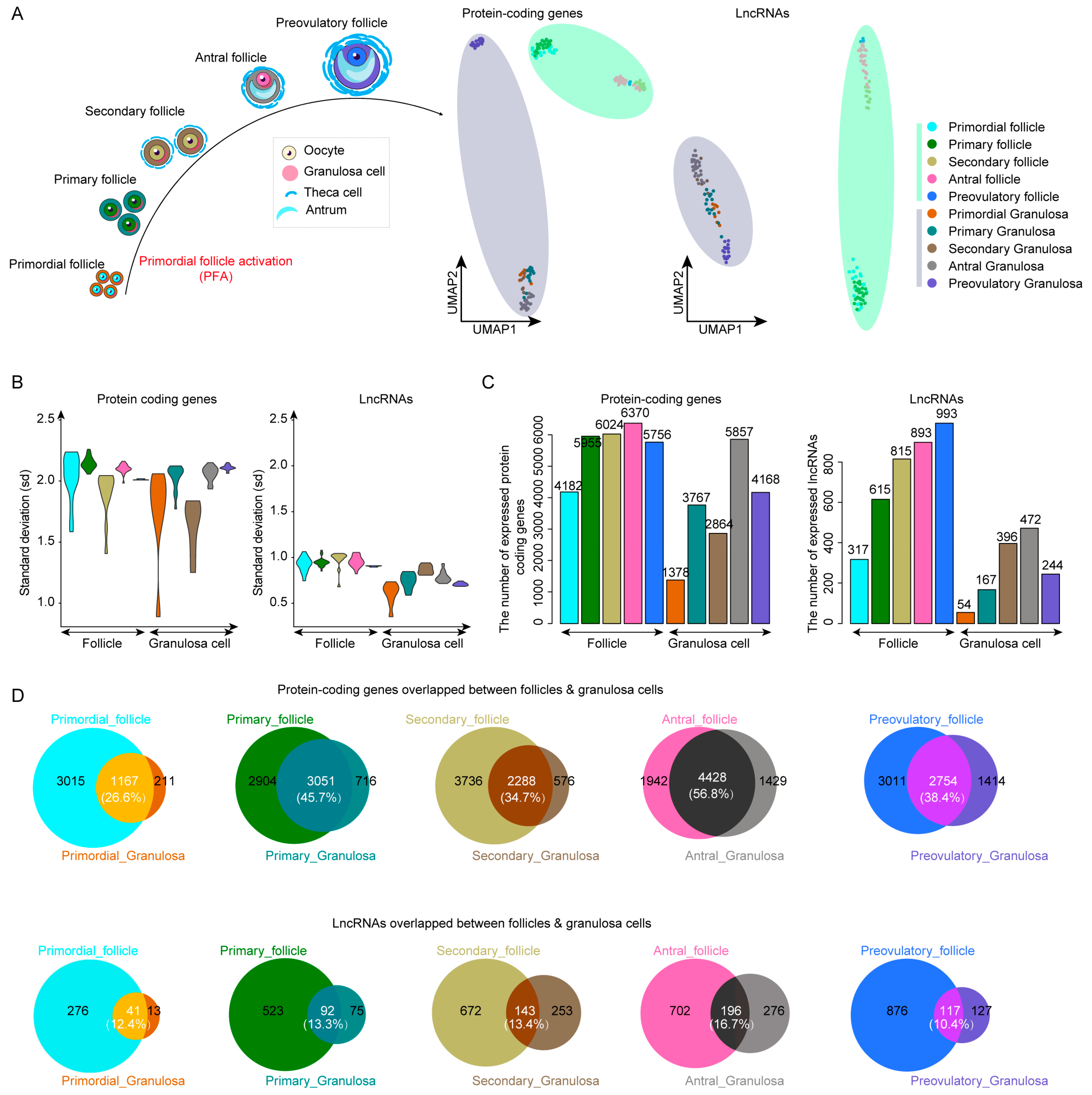
2.1. LncRNAs Improve Cell Type Classification during Human Follicle Maturation
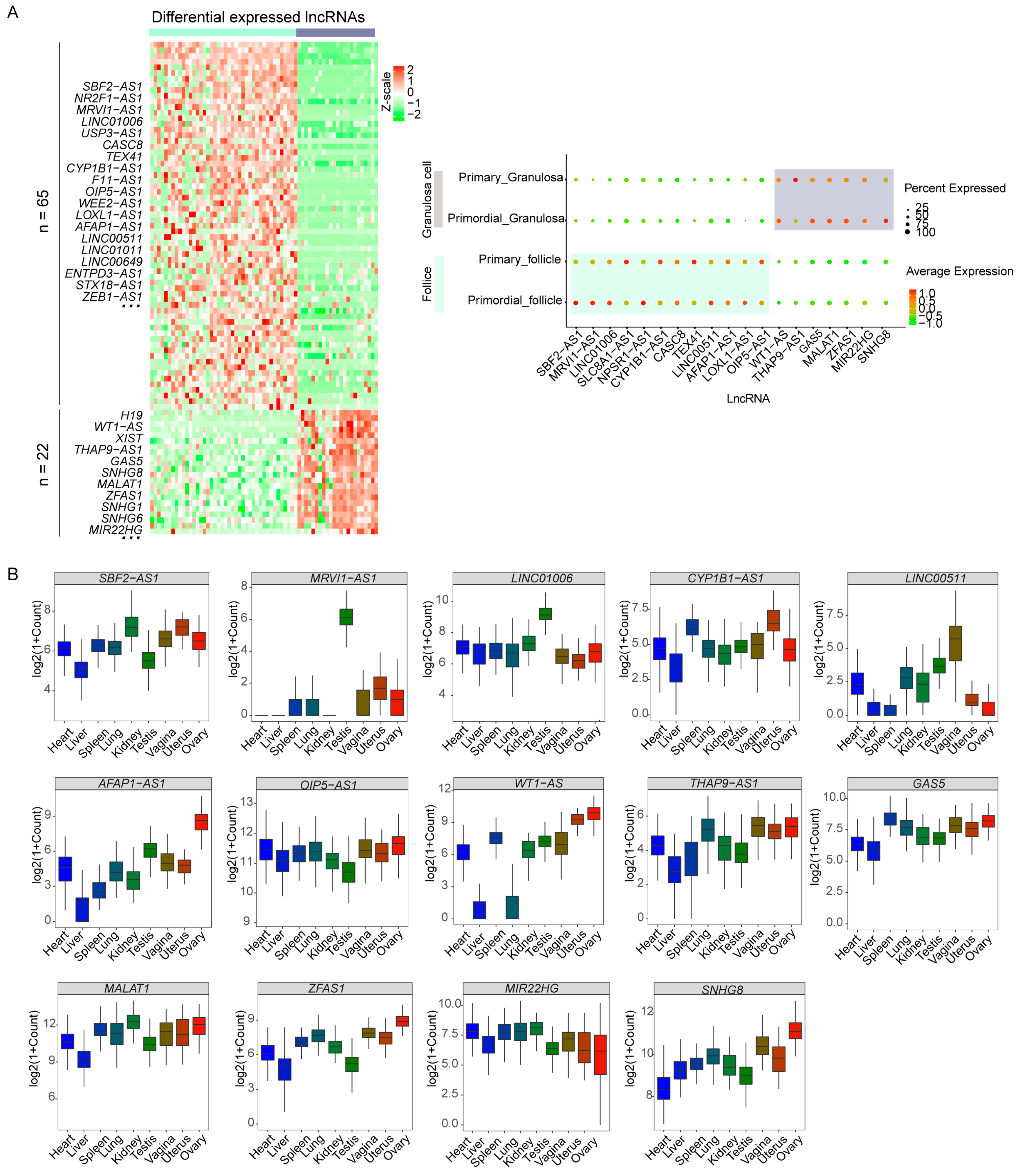
2.2. Distinct Transcriptomic Expression of lncRNAs during PFA
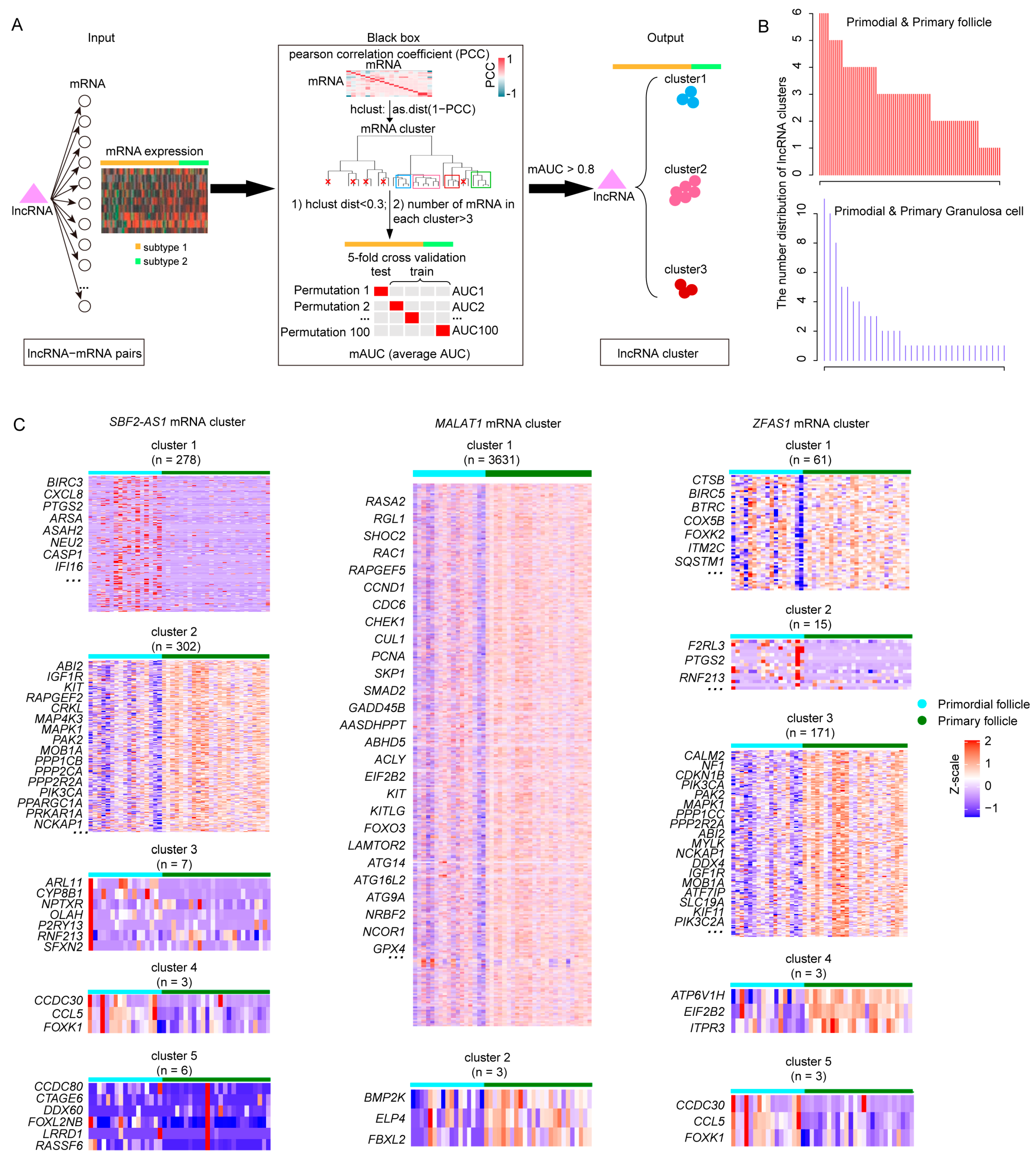
2.3. Identifying Primordial Follicle Activation Associated lncRNA Clusters
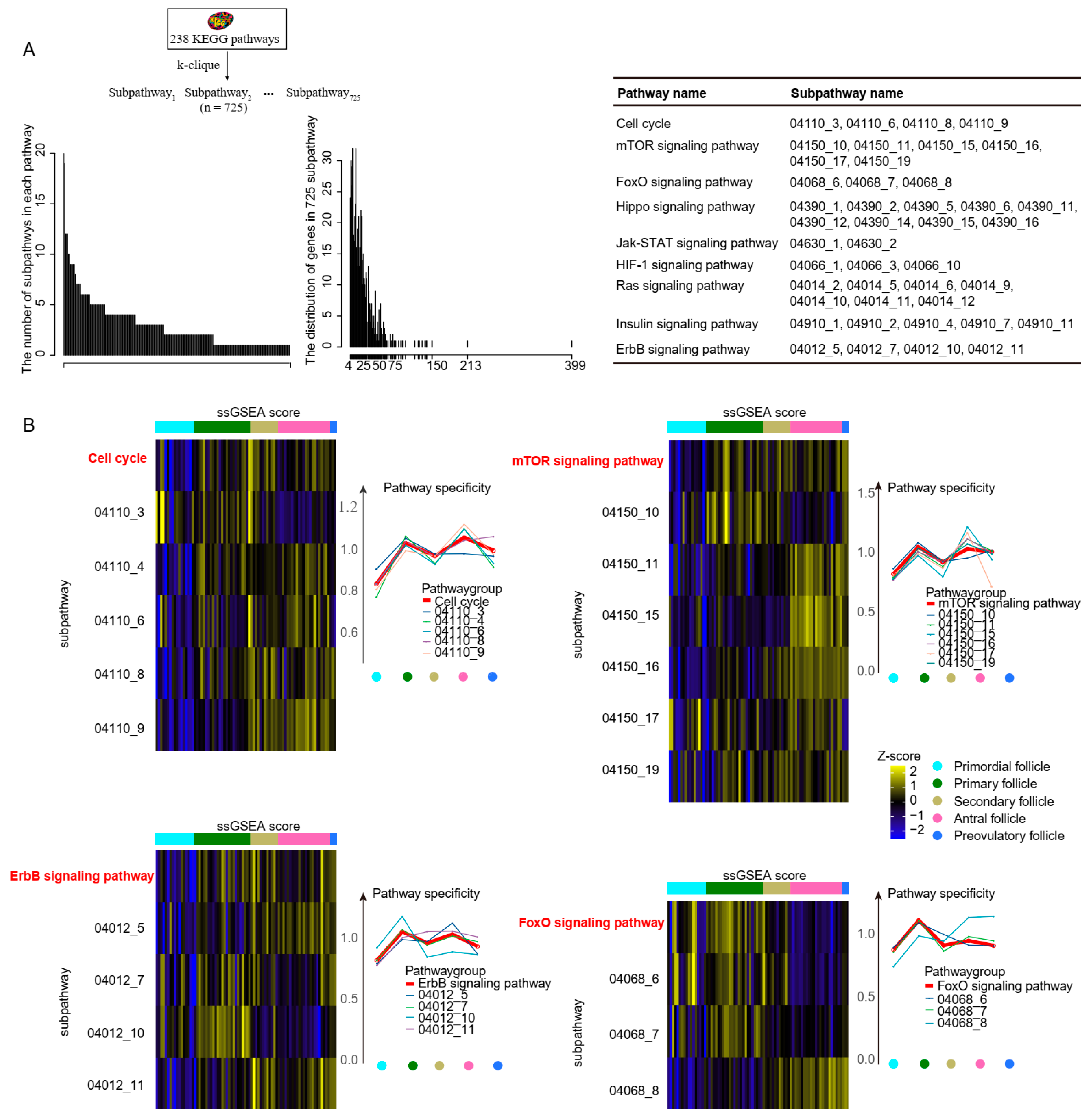
2.4. Differences between Subpathway and Pathway during Follicle Maturation
2.5. The lncRNA–Subpathway Network and Its Characteristics during Primordial Follicle Activation
2.6. Validating the Function of lncRNAs Based on Subpathway Genes after lncRNA Knockdown
3. Discussion
4. Materials and Methods
4.1. Data Collection and Pre-Processing
4.2. Cell Clustering Analysis
4.3. Identifying Follicle Maturation Associated Protein-Coding Genes and lncRNAs
4.4. Identification of lncRNA Target Gene Cluster
4.5. Obtaining Subpathway Based on k-Clique Algorithm
4.6. Subpathways Activity Score
4.7. Cell Type-Specific Pathway Specificity
4.8. LncRNA Rank
4.9. Premature Ovarian Insufficiency (POI)-Related Geneset
4.10. Download lncRNA Knockdown Datasets
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, Y.; Yan, Z.; Qin, Q.; Nisenblat, V.; Chang, H.-M.; Yu, Y.; Wang, T.; Lu, C.; Yang, M.; Yang, S.; et al. Transcriptome Landscape of Human Folliculogenesis Reveals Oocyte and Granulosa Cell Interactions. Mol. Cell 2018, 72, 1021–1034.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzumori, N.; Yan, C.; Matzuk, M.M.; Rajkovic, A. Nobox is a homeobox-encoding gene preferentially expressed in primordial and growing oocytes. Mech. Dev. 2001, 111, 137–141. [Google Scholar] [CrossRef]
- Pangas, S.; Rajkovic, A. Transcriptional regulation of early oogenesis: In search of masters. Hum. Reprod. Update 2005, 12, 65–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Feng, H.; Zhang, Y.; Zhang, J.V.; Wang, X.; Liu, D.; Wang, T.; Li, R.H.W.; Ng, E.H.Y.; Yeung, W.S.B.; et al. Current Understandings of Core Pathways for the Activation of Mammalian Primordial Follicles. Cells 2021, 10, 1491. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Yuan, D.-Z.; Wu, Y.; Yu, L.-L.; Xu, L.-Z.; Yue, L.-M.; Liu, L.; Xu, W.-M.; Qiao, X.-Y.; Zeng, R.-J.; et al. Bisphenol A Initiates Excessive Premature Activation of Primordial Follicles in Mouse Ovaries via the PTEN Signaling Pathway. Reprod. Sci. 2018, 25, 609–620. [Google Scholar] [CrossRef] [Green Version]
- Van Kasteren, Y.M.; Schoemaker, J. Premature ovarian failure: A systematic review on therapeutic interventions to restore ovarian function and achieve pregnancy. Hum. Reprod. Update 1999, 5, 483–492. [Google Scholar] [CrossRef]
- Dragojevic-Dikic, S.; Rakic, S.; Nikolic, B.; Popovac, S. Hormone replacement therapy and successful pregnancy in a patient with premature ovarian failure. Gynecol. Endocrinol. 2009, 25, 769–772. [Google Scholar] [CrossRef]
- Dekker, J.; Tr, M.; Me, D.; Js, M. Long non-coding RNAs: Insights into functions. Nat. Rev. Genet. 2009, 10, 155–159. [Google Scholar] [CrossRef]
- Fatica, A.; Bozzoni, I. Long non-coding RNAs: New players in cell differentiation and development. Nat. Rev. Genet. 2013, 15, 7–21. [Google Scholar] [CrossRef]
- Da Luz, C.M.; da Broi, M.G.; Donabela, F.C.; de Paz, C.C.P.; Meola, J.; Navarro, P.A. PTGS2 down-regulation in cumulus cells of infertile women with endometriosis. Reprod. Biomed. Online 2017, 35, 379–386. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Wang, F.; Du, L.; Li, J.; Yu, T.; Jin, Y.; Yan, Y.; Zhou, J.; Gu, J. Comprehensive Genomic Characterization Analysis of lncRNAs in Cells with Porcine Delta Coronavirus Infection. Front. Microbiol. 2020, 10, 3036. [Google Scholar] [CrossRef]
- Chen, W.; Lin, C.; Gong, L.; Chen, J.; Liang, Y.; Zeng, P.; Diao, H. Comprehensive Analysis of the mRNA-lncRNA Co-expression Profile and ceRNA Networks Patterns in Chronic Hepatitis B. Curr. Genomics 2019, 20, 231–245. [Google Scholar] [CrossRef]
- Maghsoudi, Z.; Nguyen, H.; Tavakkoli, A.; Nguyen, T. A comprehensive survey of the approaches for pathway analysis using multi-omics data integration. Brief. Bioinform. 2022, 23, bbac435. [Google Scholar] [CrossRef] [PubMed]
- Kawatsu, T. Review pathway analysis for peptide-mediated electronic coupling in the super-exchange mechanism of ET and EET. Biopolymers 2012, 100, 100–113. [Google Scholar] [CrossRef]
- Li, X.; Yan, M.L.; Yu, Q. Identification of candidate drugs for the treatment of metastatic osteosarcoma through a sub-pathway analysis method. Oncol. Lett. 2017, 13, 4378–4384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, J.; Li, Z.; Xu, M.; Ji, J.; Li, Y.; Zhang, L.; Chen, Y. Identification and Functional Analysis of Individual-Specific Sub-pathways in Lung Adenocarcinoma. Genes 2022, 13, 1122. [Google Scholar] [CrossRef]
- Huo, Y.; Shao, S.; Liu, E.; Li, J.; Tian, Z.; Wu, X.; Zhang, S.; Stover, D.; Wu, H.; Cheng, L.; et al. Subpathway Analysis of Transcriptome Profiles Reveals New Molecular Mechanisms of Acquired Chemotherapy Resistance in Breast Cancer. Cancers 2022, 14, 4878. [Google Scholar] [CrossRef] [PubMed]
- Fortune, J.; Cushman, R.; Wahl, C.; Kito, S. The primordial to primary follicle transition. Mol. Cell. Endocrinol. 2000, 163, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhou, X.; Zhu, Y.; Wang, H.; Xu, J.; Su, Y. Current mechanisms of primordial follicle activation and new strategies for fertility preservation. Mol. Hum. Reprod. 2021, 27, gaab005. [Google Scholar] [CrossRef]
- Yang, S.; Wang, S.; Luo, A.; Ding, T.; Lai, Z.; Shen, W.; Ma, X.; Cao, C.; Shi, L.; Jiang, J.; et al. Expression Patterns and Regulatory Functions of MicroRNAs During the Initiation of Primordial Follicle Development in the Neonatal Mouse Ovary. Biol. Reprod. 2013, 89, 126. [Google Scholar] [CrossRef]
- Bouckenheimer, J.; Fauque, P.; Lecellier, C.-H.; Bruno, C.; Commes, T.; Lemaître, J.-M.; De Vos, J.; Assou, S. Differential long non-coding RNA expression profiles in human oocytes and cumulus cells. Sci. Rep. 2018, 8, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Shi, Z.-M.; Chang, Y.-N.; Hu, Z.-M.; Qi, H.-X.; Hong, W. The ways of action of long non-coding RNAs in cytoplasm and nucleus. Gene 2014, 547, 1–9. [Google Scholar] [CrossRef]
- Dykes, I.M.; Emanueli, C. Transcriptional and Post-transcriptional Gene Regulation by Long Non-coding RNA. Genom. Proteom. Bioinform. 2017, 15, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Shan, Y.; Ma, Z.; Yu, M.; Gong, B. A network pharmacology approach to explore active compounds and phar-macological mechanisms of epimedium for treatment of premature ovarian insufficiency. Drug Des. Devel. Ther. 2019, 13, 2997–3007. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Han, J.; Yao, Q.; Zou, C.; Xu, Y.; Zhang, C.; Shang, D.; Zhou, L.; Zou, C.; Sun, Z.; et al. Subpathway-GM: Identification of metabolic subpathways via joint power of interesting genes and metabolites and their topologies within pathways. Nucleic Acids Res. 2013, 41, e101. [Google Scholar] [CrossRef]
- Li, C.; Li, X.; Miao, Y.; Wang, Q.; Jiang, W.; Xu, C.; Li, J.; Han, J.; Zhang, F.; Gong, B.; et al. SubpathwayMiner: A software package for flexible identification of pathways. Nucleic Acids Res. 2009, 37, e131. [Google Scholar] [CrossRef] [Green Version]
- Granados-Aparici, S.; Hardy, K.; Franks, S.; Sharum, I.B.; Waite, S.L.; Fenwick, M.A. SMAD3 directly regulates cell cycle genes to maintain arrest in granulosa cells of mouse primordial follicles. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, T.; He, M.; Zhao, L.; Qin, S.; Zhu, Z.; Du, X.; Zhou, B.; Yang, Y.; Liu, X.; Xia, G.; et al. HDAC6 regulates primordial follicle activation through mTOR signaling pathway. Cell Death Dis. 2021, 12, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Kehoe, S.; Jewgenow, K.; Johnston, P.R.; Mbedi, S.; Braun, B.C. Signalling pathways and mechanistic cues highlighted by transcriptomic analysis of primordial, primary, and secondary ovarian follicles in domestic cat. Sci. Rep. 2021, 11, 2683. [Google Scholar] [CrossRef] [PubMed]
- Ting, A.Y.; Zelinski, M.B. Characterization of FOXO1, 3 and 4 transcription factors in ovaries of fetal, prepubertal and adult rhesus macaques†. Biol. Reprod. 2017, 96, 1052–1059. [Google Scholar] [CrossRef] [Green Version]
- Ojeda, D.; Lakhal, B.; Fonseca, D.J.; Braham, R.; Landolsi, H.; Mateus, H.E.; Restrepo, C.M.; Elghezal, H.; Saâd, A.; Laissue, P. Sequence analysis of the CDKN1B gene in patients with premature ovarian failure reveals a novel mutation potentially related to the phenotype. Fertil. Steril. 2011, 95, 2658–2660.e1. [Google Scholar] [CrossRef]
- Lu, C.; Lin, L.; Tan, H.; Wu, H.; Sherman, S.L.; Gao, F.; Jin, P.; Chen, D. Fragile X premutation RNA is sufficient to cause primary ovarian insufficiency in mice. Hum. Mol. Genet. 2012, 21, 5039–5047. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.; Peng, L.; Wei, X.; Li, S. FOXO3 is a potential biomarker and therapeutic target for premature ovarian insufficiency (Review). Mol. Med. Rep. 2022, 27, 1–8. [Google Scholar] [CrossRef]
- Yao, C.; Lu, L.; Ji, Y.; Zhang, Y.; Li, W.; Shi, Y.; Liu, J.; Sun, M.; Xia, F. Hypo-Hydroxymethylation of Nobox is Associated with Ovarian Dysfunction in Rat Offspring Exposed to Prenatal Hypoxia. Reprod. Sci. 2022, 29, 1424–1436. [Google Scholar] [CrossRef] [PubMed]
- Neyroud, A.S.; Rudinger-Thirion, J.; Frugier, M.; Riley, L.G.; Bidet, M.; Akloul, L.; Simpson, A.; Gilot, D.; Christodoulou, J.; Ravel, C.; et al. LARS2 variants can present as premature ovarian insufficiency in the absence of overt hearing loss. Eur. J. Hum. Genet. 2022, 31, 453–460. [Google Scholar] [CrossRef]
- Guo, Z.-W.; Meng, Y.; Zhai, X.-M.; Xie, C.; Zhao, N.; Li, M.; Zhou, C.-L.; Li, K.; Liu, T.-C.; Yang, X.; et al. Translated Long Non-Coding Ribonucleic Acid ZFAS1 Promotes Cancer Cell Migration by Elevating Reactive Oxygen Species Production in Hepatocellular Carcinoma. Front. Genet. 2019, 10, 1111. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Wan, Q.; Zhang, L.; Li, Y.; Zhang, P.; Li, D.; Feng, C.; Yi, F.; Zhang, L.; Ding, X.; et al. Analysis of LncRNA expression in cell differentiation. RNA Biol. 2018, 15, 413–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunlop, C.E.; Anderson, R.A. The regulation and assessment of follicular growth. Scand. J. Clin. Lab. Investig. 2014, 74, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Barnett, K.; Schilling, C.; Greenfeld, C.; Tomic, D.; Flaws, J. Ovarian follicle development and transgenic mouse models. Hum. Reprod. Update 2006, 12, 537–555. [Google Scholar] [CrossRef]
- Chen, R.; Xia, W.; Wang, X.; Qiu, M.; Yin, R.; Wang, S.; Xi, X.; Wang, J.; Xu, Y.; Dong, G.; et al. Upregulated long non-coding RNA SBF2-AS1 promotes proliferation in esophageal squamous cell carcinoma. Oncol. Lett. 2018, 15, 5071–5080. [Google Scholar] [CrossRef]
- Tuo, H.; Liu, R.; Wang, Y.; Yang, W.; Liu, Q. Hypoxia-induced lncRNA MRVI1-AS1 accelerates hepatocellular carcinoma progression by recruiting RNA-binding protein CELF2 to stabilize SKA1 mRNA. World J. Surg. Oncol. 2023, 21, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.; Liu, D.; Huang, C.; Wang, M.; Xiao, X.; Zeng, F.; Wang, L.; Jiang, G. LncRNA GAS5 Inhibits Cellular Proliferation by Targeting P27Kip. Mol. Cancer Res. 2017, 15, 789–799. [Google Scholar] [CrossRef] [Green Version]
- O’brien, S.J.; Fiechter, C.; Burton, J.; Hallion, J.; Paas, M.; Patel, A.; Patel, A.; Rochet, A.; Scheurlen, K.; Gardner, S.; et al. Long non-coding RNA ZFAS1 is a major regulator of epithelial-mesenchymal transition through miR-200/ZEB1/E-cadherin, vimentin signaling in colon adenocarcinoma. Cell Death Discov. 2021, 7, 61. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-T.; Li, B.-P.; Zhang, B.; Ma, P.; Wu, Q.-L.; Ming, L.; Xie, L.-M. LncRNA SBF2-AS1 promotes hepatocellular carcinoma metastasis by regulating EMT and predicts unfavorable prognosis. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 6333–6341. [Google Scholar] [PubMed]
- Lv, J.; Qiu, M.; Xia, W.; Liu, C.; Xu, Y.; Wang, J.; Leng, X.; Huang, S.; Zhu, R.; Zhao, M.; et al. High expression of long non-coding RNA SBF2-AS1 promotes proliferation in non-small cell lung cancer. J. Exp. Clin. Cancer Res. 2016, 35, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Hussain, S.A.; Venkatesh, T. YBX1/lncRNA SBF2-AS1 interaction regulates proliferation and tamoxifen sensitivity via PI3K/AKT/MTOR signaling in breast cancer cells. Mol. Biol. Rep. 2023, 50, 3413–3428. [Google Scholar] [CrossRef]
- Sang, Q.; Zhou, Z.; Mu, J.; Wang, L. Genetic factors as potential molecular markers of human oocyte and embryo quality. J. Assist. Reprod. Genet. 2021, 38, 993–1002. [Google Scholar] [CrossRef]
- Grynberg, M.; Jacquesson, L.; Sifer, C. In vitro maturation of oocytes for preserving fertility in autoimmune premature ovarian insufficiency. Fertil. Steril. 2020, 114, 848–853. [Google Scholar] [CrossRef]
- La Marca, A.; Mastellari, E. Fertility preservation for genetic diseases leading to premature ovarian insufficiency (POI). J. Assist. Reprod. Genet. 2021, 38, 759–777. [Google Scholar] [CrossRef]
- Pinelli, S.; Artini, P.G.; Basile, S.; Obino, M.E.R.; Sergiampietri, C.; Giannarelli, D.; Simi, G.; Cela, V. Estrogen treatment in infertile women with premature ovarian insufficiency in transitional phase: A retrospective analysis. J. Assist. Reprod. Genet. 2017, 35, 475–482. [Google Scholar] [CrossRef]
- Kawamura, K.; Cheng, Y.; Suzuki, N.; Deguchi, M.; Sato, Y.; Takae, S.; Ho, C.-H.; Kawamura, N.; Tamura, M.; Hashimoto, S.; et al. Hippo signaling disruption and Akt stimulation of ovarian follicles for infertility treatment. Proc. Natl. Acad. Sci. USA 2013, 110, 17474–17479. [Google Scholar] [CrossRef] [Green Version]
- Consortium, G. Tex, The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef]
- Stuart, T.; Butler, A.; Hoffman, P.; Hafemeister, C.; Papalexi, E.; Mauck, W.M., 3rd; Hao, Y.; Stoeckius, M.; Smibert, P.; Satija, R. Comprehensive Integration of Single-Cell Data. Cell 2019, 177, 1888–1902.e21. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Zhou, G.; Soufan, O.; Xia, J. miRNet 2.0: Network-based visual analytics for miRNA functional analysis and systems biology. Nucleic Acids Res. 2020, 48, W244–W251. [Google Scholar] [CrossRef]
- Zhao, X.-M.; Liu, K.-Q.; Zhu, G.; He, F.; Duval, B.; Richer, J.-M.; Huang, D.-S.; Jiang, C.-J.; Hao, J.-K.; Chen, L. Identifying cancer-related microRNAs based on gene expression data. Bioinformatics 2014, 31, 1226–1234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, J.; Han, X.; Kong, Q.; Cheng, L. psSubpathway: A software package for flexible identification of phenotype-specific sub-pathways in cancer progression. Bioinformatics 2020, 36, 2303–2305. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, L.; Zou, J.; Wang, Z.; Li, L. A Subpathway and Target Gene Cluster-Based Approach Uncovers lncRNAs Associated with Human Primordial Follicle Activation. Int. J. Mol. Sci. 2023, 24, 10525. https://doi.org/10.3390/ijms241310525
Zhang L, Zou J, Wang Z, Li L. A Subpathway and Target Gene Cluster-Based Approach Uncovers lncRNAs Associated with Human Primordial Follicle Activation. International Journal of Molecular Sciences. 2023; 24(13):10525. https://doi.org/10.3390/ijms241310525
Chicago/Turabian StyleZhang, Li, Jiyuan Zou, Zhihao Wang, and Lin Li. 2023. "A Subpathway and Target Gene Cluster-Based Approach Uncovers lncRNAs Associated with Human Primordial Follicle Activation" International Journal of Molecular Sciences 24, no. 13: 10525. https://doi.org/10.3390/ijms241310525