


Building a Scaffold for Arteriovenous Fistula Maturation: Unravelling the Role of the Extracellular Matrix

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. AVF Maturation Failure
3. The Arterial and Venous Vessel Wall and Its ECM Components
3.1. Vascular Identity: Phenotypic Differences between Arteries and Veins
3.2. The Tunica Intima
3.3. The Tunica Media
3.4. The Tunica Adventitia
4. The Effect of Chronic Kidney Disease on the Vasculature
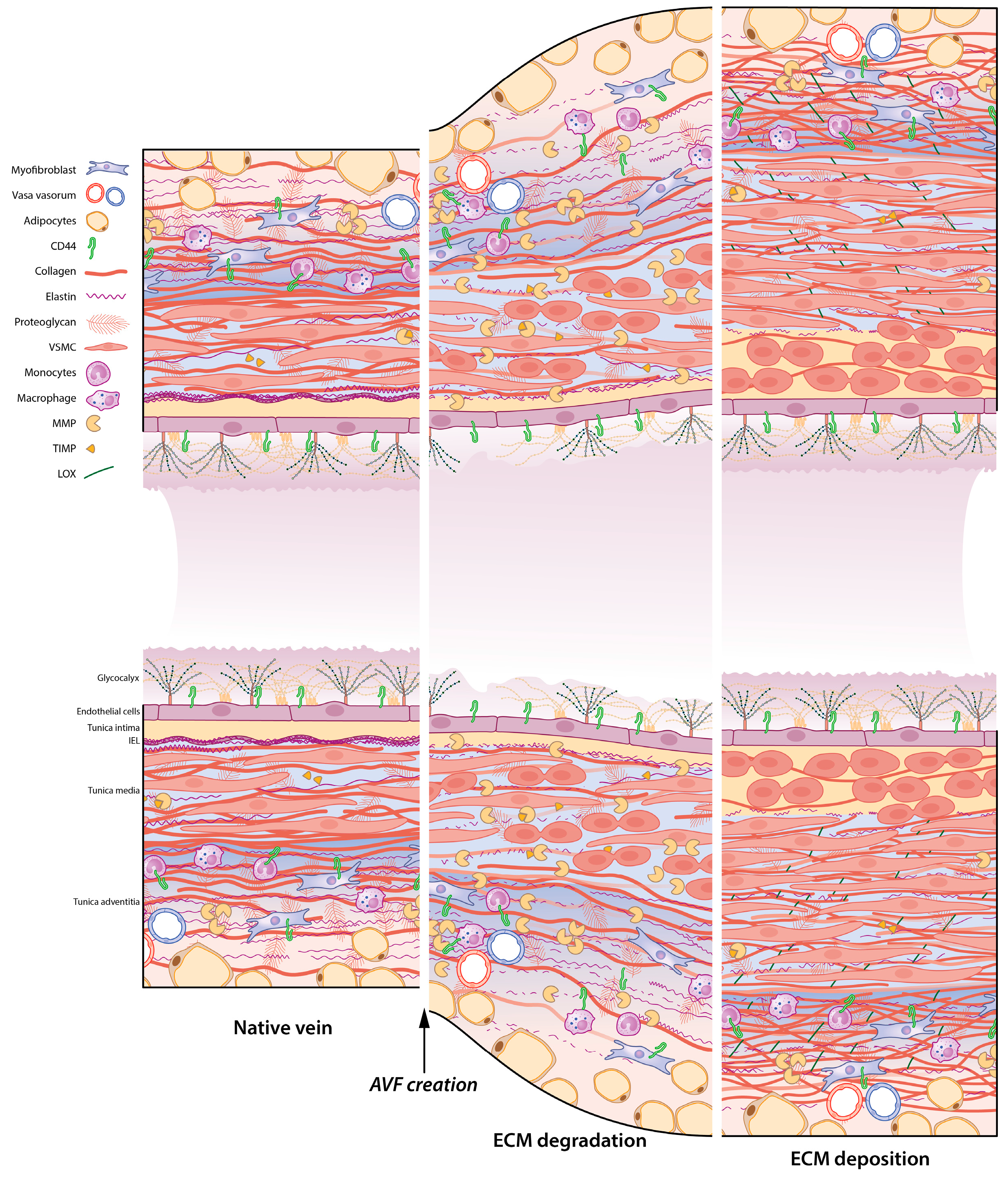
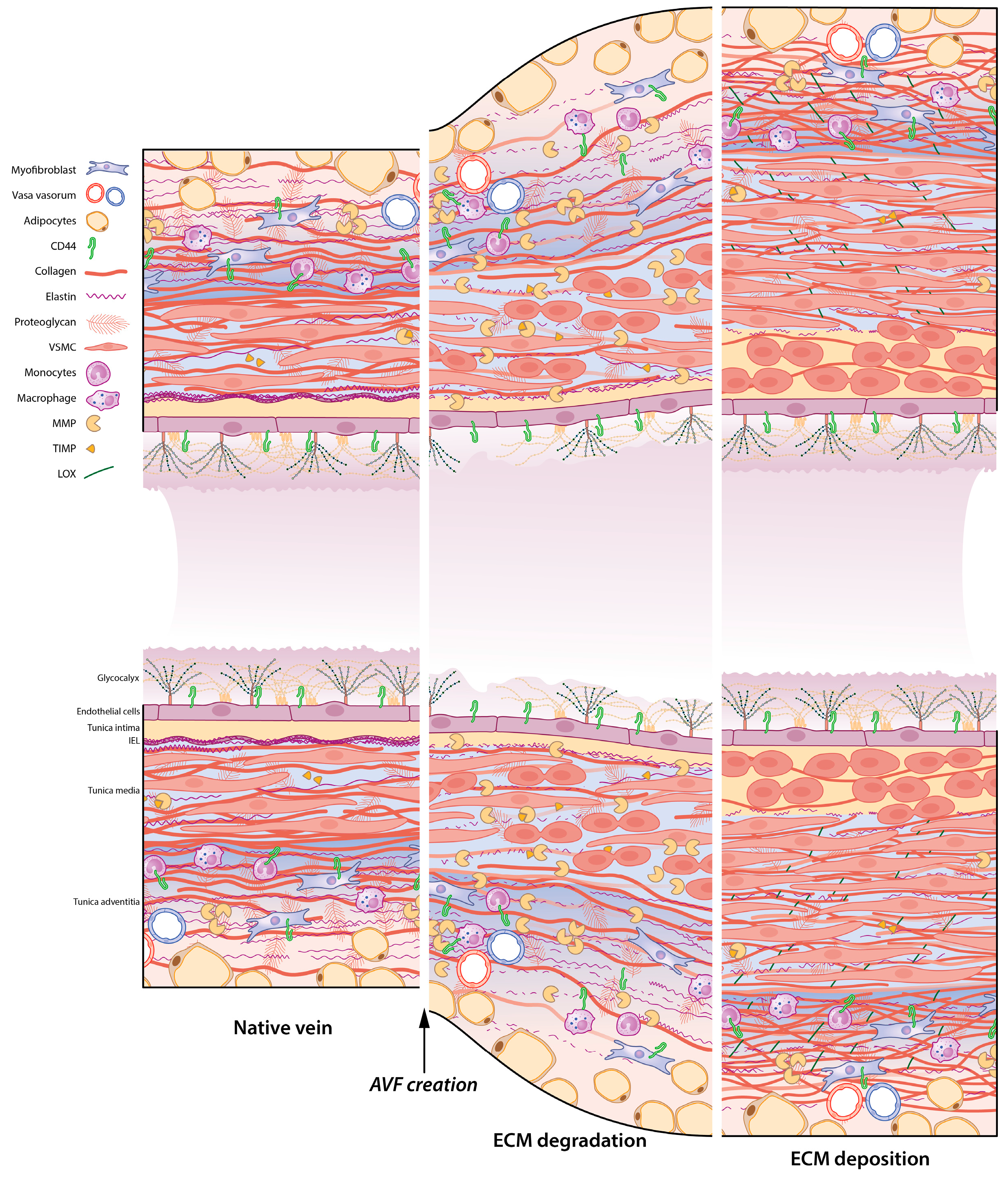
5. ECM Remodelling in the AVF: A Timely Matter
6. Rebuilding the Vascular Framework: ECM Remodelling during AVF Maturation
6.1. ECM Degradation: The Role of MMPs and TIMPs in the AVF
6.2. Strengthening the ECM Framework: Macro-Proteins Elastin and Collagen
6.3. The Effect of TGF-β on ECM Remodelling in the AVF
6.4. Inflammation Influencing ECM Remodelling
7. Interventions Creating an ECM Framework Supporting Arteriovenous Fistulas
8. Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brescia, M.J.; Cimino, J.E.; Appel, K.; Hurwich, B.J. Chronic hemodialysis using venipuncture and a surgically created arteriovenous fistula. N. Engl. J. Med. 1966, 275, 1089–1092. [Google Scholar] [CrossRef] [PubMed]
- Schmidli, J.; Widmer, M.K.; Basile, C.; de Donato, G.; Gallieni, M.; Gibbons, C.P.; Haage, P.; Hamilton, G.; Hedin, U.; Kamper, L.; et al. Editor’s Choice—Vascular Access: 2018 Clinical Practice Guidelines of the European Society for Vascular Surgery (ESVS). Eur. J. Vasc. Endovasc. Surg. 2018, 55, 757–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lok, C.E.; Huber, T.S.; Lee, T.; Shenoy, S.; Yevzlin, A.S.; Abreo, K.; Allon, M.; Asif, A.; Astor, B.C.; Glickman, M.H.; et al. KDOQI Clinical Practice Guideline for Vascular Access: 2019 Update. Am. J. Kidney Dis. 2020, 75, S1–S164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Astor, B.C.; Eustace, J.A.; Powe, N.R.; Klag, M.J.; Fink, N.E.; Coresh, J.; Josef Coresh for the CHOICE Study. Type of Vascular Access and Survival among Incident Hemodialysis Patients: The Choices for Healthy Outcomes in Caring for ESRD (CHOICE) Study. J. Am. Soc. Nephrol. 2005, 16, 1449–1455. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, T.; Kim, S.J.; Astor, B.; Shafi, T.; Coresh, J.; Powe, N.R. Vascular Access Type, Inflammatory Markers, and Mortality in Incident Hemodialysis Patients: The Choices for Healthy Outcomes in Caring for End-Stage Renal Disease (CHOICE) Study. Am. J. Kidney Dis. 2014, 64, 954–961. [Google Scholar] [CrossRef] [Green Version]
- Dhingra, R.K.; Young, E.W.; Hulbert-Shearon, T.E.; Leavey, S.F.; Port, F.K. Type of vascular access and mortality in U.S. hemodialysis patients. Kidney Int. 2001, 60, 1443–1451. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.; Ban, T.H.; Choi, B.S.; Baik, J.H.; Kim, B.S.; Kim, Y.O.; Park, C.W.; Yang, C.W.; Jin, D.C.; Park, H.S. Comparison of vascular access patency and patient survival between native arteriovenous fistula and synthetic arteriovenous graft according to age group. Hemodial Int. 2020, 24, 309–316. [Google Scholar] [CrossRef]
- Voorzaat, B.M.; van der Bogt, K.E.A.; Janmaat, C.J.; van Schaik, J.; Dekker, F.W.; Rotmans, J.I.; Voorzaat, B.M.; van der Bogt, K.E.A.; Janmaat, C.J.; van Schaik, J.; et al. Arteriovenous Fistula Maturation Failure in a Large Cohort of Hemodialysis Patients in the Netherlands. World J. Surg. 2018, 42, 1895–1903. [Google Scholar] [CrossRef] [Green Version]
- Arhuidese, I.J.; Orandi, B.J.; Nejim, B.; Malas, M. Utilization, patency, and complications associated with vascular access for hemodialysis in the United States. J. Vasc. Surg. 2018, 68, 1166–1174. [Google Scholar] [CrossRef]
- Ponticos, M.; Smith, B.D. Extracellular matrix synthesis in vascular disease: Hypertension, and atherosclerosis. J. Biomed. Res. 2014, 28, 25–39. [Google Scholar]
- Chistiakov, D.A.; Sobenin, I.A.; Orekhov, A.N. Vascular Extracellular Matrix in Atherosclerosis. Cardiol. Rev. 2013, 21, 270–288. [Google Scholar] [CrossRef]
- Stepien, K.L.; Bajdak-Rusinek, K.; Fus-Kujawa, A.; Kuczmik, W.; Gawron, K. Role of Extracellular Matrix and Inflammation in Abdominal Aortic Aneurysm. Int. J. Mol. Sci. 2022, 23, 11078. [Google Scholar] [CrossRef]
- Osol, G.; Moore, L.G. Maternal Uterine Vascular Remodeling During Pregnancy. Microcirculation 2014, 21, 38–47. [Google Scholar] [CrossRef]
- Palmer, S.K.; Zamudio, S.; Coffin, C.; Parker, S.; Stamm, E.; Moore, L.G. Quantitative estimation of human uterine artery blood flow and pelvic blood flow redistribution in pregnancy. Obstet. Gynecol. 1992, 80, 1000–1006. [Google Scholar]
- Martinez, L.; Rojas, M.G.; Tabbara, M.; Pereira-Simon, S.; Falcon, N.S.; Rauf, M.A.; Challa, A.S.; Zigmond, Z.M.; Griswold, A.J.; Duque, J.C.; et al. The Transcriptomics of the Human Vein Transformation After Arteriovenous Fistula Anastomosis Uncovers Layer-Specific Remodeling and Hallmarks of Maturation Failure. Kidney Int. Rep. 2023, 8, 837–850. [Google Scholar] [CrossRef]
- Hu, H.; Patel, S.; Hanisch, J.J.; Santana, J.M.; Hashimoto, T.; Bai, H.; Kudze, T.; Foster, T.R.; Guo, J.; Yatsula, B.; et al. Future research directions to improve fistula maturation and reduce access failure. Semin. Vasc. Surg. 2016, 29, 153–171. [Google Scholar] [CrossRef] [Green Version]
- Lu, D.Y.; Chen, E.Y.; Wong, D.J.; Yamamoto, K.; Protack, C.D.; Williams, W.T.; Assi, R.; Hall, M.R.; Sadaghianloo, N.; Dardik, A. Vein graft adaptation and fistula maturation in the arterial environment. J. Surg. Res. 2014, 188, 162–173. [Google Scholar] [CrossRef] [Green Version]
- Rothuizen, T.C.; Wong, C.; Quax, P.H.A.; van Zonneveld, A.J.; Rabelink, T.J.; Rotmans, J.I. Arteriovenous access failure: More than just intimal hyperplasia? Nephrol. Dial. Transplant. 2013, 28, 1085–1092. [Google Scholar] [CrossRef] [Green Version]
- Adams, R.H. Molecular control of arterial–venous blood vessel identity. J. Anat. 2003, 202, 105–112. [Google Scholar] [CrossRef]
- Lawson, N.D.; Scheer, N.; Pham, V.N.; Kim, C.-H.; Chitnis, A.B.; Campos-Ortega, J.A.; Weinstein, B.M. Notch signaling is required for arterial-venous differentiation during embryonic vascular development. Development 2001, 128, 3675–3683. [Google Scholar] [CrossRef]
- le Noble, F.; Moyon, D.; Pardanaud, L.; Yuan, L.; Djonov, V.; Matthijsen, R.; Bréant, C.; Fleury, V.; Eichmann, A. Flow regulates arterial-venous differentiation in the chick embryo yolk sac. Development 2004, 131, 361–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.U.; Chen, Z.F.; Anderson, D.J. Molecular distinction and angiogenic interaction between embryonic arteries and veins revealed by ephrin-B2 and its receptor Eph-B4. Cell 1998, 93, 741–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gale, N.W.; Baluk, P.; Pan, L.; Kwan, M.; Holash, J.; DeChiara, T.M.; McDonald, D.M.; Yancopoulos, G.D. Ephrin-B2 selectively marks arterial vessels and neovascularization sites in the adult, with expression in both endothelial and smooth-muscle cells. Dev. Biol. 2001, 230, 151–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinle, J.J.; Meininger, C.J.; Forough, R.; Wu, G.; Wu, M.H.; Granger, H.J. Eph B4 receptor signaling mediates endothelial cell migration and proliferation via the phosphatidylinositol 3-kinase pathway. J. Biol. Chem. 2002, 277, 43830–43835. [Google Scholar] [CrossRef] [Green Version]
- Protack, C.D.; Foster, T.R.; Hashimoto, T.; Yamamoto, K.; Lee, M.Y.; Kraehling, J.R.; Bai, H.; Hu, H.; Isaji, T.; Santana, J.M.; et al. Eph-B4 regulates adaptive venous remodeling to improve arteriovenous fistula patency. Sci. Rep. 2017, 7, 15386. [Google Scholar] [CrossRef] [Green Version]
- Rix, D.A.; Douglas, M.S.; Talbot, D.; Dark, J.H.; Kirby, J.A. Role of glycosaminoglycans (GAGs) in regulation of the immunogenicity of human vascular endothelial cells. Clin. Exp. Immunol. 1996, 104, 60–65. [Google Scholar] [CrossRef]
- Wang, G.; Tiemeier, G.L.; van den Berg, B.M.; Rabelink, T.J. Endothelial Glycocalyx Hyaluronan: Regulation and Role in Prevention of Diabetic Complications. Am. J. Pathol. 2020, 190, 781–790. [Google Scholar] [CrossRef]
- Reitsma, S.; Slaaf, D.W.; Vink, H.; van Zandvoort, M.A.M.J.; oude Egbrink, M.G.A. The endothelial glycocalyx: Composition, functions, and visualization. Pflügers Arch.—Eur. J. Physiol. 2007, 454, 345–359. [Google Scholar] [CrossRef] [Green Version]
- Tarbell, J.M.; Simon, S.I.; Curry, F.R. Mechanosensing at the vascular interface. Annu. Rev. Biomed. Eng. 2014, 16, 505–532. [Google Scholar] [CrossRef] [Green Version]
- Bartosch, A.M.W.; Mathews, R.; Tarbell, J.M. Endothelial Glycocalyx-Mediated Nitric Oxide Production in Response to Selective AFM Pulling. Biophys. J. 2017, 113, 101–108. [Google Scholar] [CrossRef] [Green Version]
- Giantsos-Adams, K.M.; Koo, A.J.-A.; Song, S.; Sakai, J.; Sankaran, J.; Shin, J.H.; Garcia-Cardena, G.; Dewey, C.F. Heparan Sulfate Regrowth Profiles Under Laminar Shear Flow Following Enzymatic Degradation. Cell. Mol. Bioeng. 2013, 6, 160–174. [Google Scholar] [CrossRef] [Green Version]
- Liew, H.; Roberts, M.A.; Pope, A.; McMahon, L.P. Endothelial glycocalyx damage in kidney disease correlates with uraemic toxins and endothelial dysfunction. BMC Nephrol. 2021, 22, 21. [Google Scholar] [CrossRef]
- Halper, J. Chapter Four—Basic Components of Vascular Connective Tissue and Extracellular Matrix. In Advances in Pharmacology; Khalil, R.A., Ed.; Academic Press: Cambridge, MA, USA, 2018; pp. 95–127. [Google Scholar]
- Kalluri, R. Basement membranes: Structure, assembly and role in tumour angiogenesis. Nat. Rev. Cancer 2003, 3, 422–433. [Google Scholar] [CrossRef]
- Tomono, Y.; Naito, I.; Ando, K.; Yonezawa, T.; Sado, Y.; Hirakawa, S.; Arata, J.; Okigaki, T.; Ninomiya, Y. Epitope-defined Monoclonal Antibodies against Multiplexin Collagens Demonstrate that Type XV and XVIII Collagens are Expressed in Specialized Basement Membranes. Cell Struct. Funct. 2002, 27, 9–20. [Google Scholar] [CrossRef] [Green Version]
- Massberg, S.; Grüner, S.; Konrad, I.; Garcia Arguinzonis, M.I.; Eigenthaler, M.; Hemler, K.; Kersting, J.; Schulz, C.; Müller, I.; Besta, F.; et al. Enhanced in vivo platelet adhesion in vasodilator-stimulated phosphoprotein (VASP)–deficient mice. Blood 2004, 103, 136–142. [Google Scholar] [CrossRef] [Green Version]
- Shi, G.; Field, D.J.; Long, X.; Mickelsen, D.; Ko, K.-a.; Ture, S.; Korshunov, V.A.; Miano, J.M.; Morrell, C.N. Platelet factor 4 mediates vascular smooth muscle cell injury responses. Blood 2013, 121, 4417–4427. [Google Scholar] [CrossRef] [Green Version]
- Fingerle, J.; Johnson, R.; Clowes, A.W.; Majesky, M.W.; Reidy, M.A. Role of platelets in smooth muscle cell proliferation and migration after vascular injury in rat carotid artery. Proc. Natl. Acad. Sci. USA 1989, 86, 8412–8416. [Google Scholar] [CrossRef] [Green Version]
- O’Connell, M.K.; Murthy, S.; Phan, S.; Xu, C.; Buchanan, J.; Spilker, R.; Dalman, R.L.; Zarins, C.K.; Denk, W.; Taylor, C.A. The three-dimensional micro- and nanostructure of the aortic medial lamellar unit measured using 3D confocal and electron microscopy imaging. Matrix Biol. 2008, 27, 171–181. [Google Scholar] [CrossRef] [Green Version]
- Bunton, T.E.; Biery, N.J.; Myers, L.; Gayraud, B.; Ramirez, F.; Dietz, H.C. Phenotypic Alteration of Vascular Smooth Muscle Cells Precedes Elastolysis in a Mouse Model of Marfan Syndrome. Circ. Res. 2001, 88, 37–43. [Google Scholar] [CrossRef] [Green Version]
- Pereira, L.; Andrikopoulos, K.; Tian, J.; Lee, S.Y.; Keene, D.R.; Ono, R.; Reinhardt, D.P.; Sakai, L.Y.; Biery, N.J.; Bunton, T.; et al. Targetting of the gene encoding fibrillin–1 recapitulates the vascular aspect of Marfan syndrome. Nat. Genet. 1997, 17, 218–222. [Google Scholar] [CrossRef]
- Davis, E.C. Smooth muscle cell to elastic lamina connections in developing mouse aorta. Role in aortic medial organization. Laboratory investigation. J. Tech. Methods Pathol. 1993, 68, 89–99. [Google Scholar]
- Niklason, L.; Dai, G. Arterial Venous Differentiation for Vascular Bioengineering. Annu. Rev. Biomed. Eng. 2018, 20, 431–447. [Google Scholar] [CrossRef] [PubMed]
- Chow, M.J.; Choi, M.; Yun, S.H.; Zhang, Y. The effect of static stretch on elastin degradation in arteries. PLoS ONE 2013, 8, e81951. [Google Scholar] [CrossRef] [PubMed]
- Basu, P.; Sen, U.; Tyagi, N.; Tyagi, S.C. Blood flow interplays with elastin: Collagen and MMP: TIMP ratios to maintain healthy vascular structure and function. Vasc. Health Risk Manag. 2010, 6, 215–228. [Google Scholar]
- Berry, C.L.; Greenwald, S.E.; Rivett, J.F. Static mechanical properties of the developing and mature rat aorta. Cardiovasc. Res. 1975, 9, 669–678. [Google Scholar] [CrossRef]
- Wanjare, M.; Agarwal, N.; Gerecht, S. Biomechanical strain induces elastin and collagen production in human pluripotent stem cell-derived vascular smooth muscle cells. Am. J. Physiol. Cell Physiol. 2015, 309, C271–C281. [Google Scholar] [CrossRef] [Green Version]
- Schwarze, U.; Schievink, W.I.; Petty, E.; Jaff, M.R.; Babovic-Vuksanovic, D.; Cherry, K.J.; Pepin, M.; Byers, P.H. Haploinsufficiency for One COL3A1 Allele of Type III Procollagen Results in a Phenotype Similar to the Vascular Form of Ehlers-Danlos Syndrome, Ehlers-Danlos Syndrome Type IV. Am. J. Hum. Genet. 2001, 69, 989–1001. [Google Scholar] [CrossRef] [Green Version]
- Krady, M.M.; Zeng, J.; Yu, J.; MacLauchlan, S.; Skokos, E.A.; Tian, W.; Bornstein, P.; Sessa, W.C.; Kyriakides, T.R. Thrombospondin-2 modulates extracellular matrix remodeling during physiological angiogenesis. Am. J. Pathol. 2008, 173, 879–891. [Google Scholar] [CrossRef] [Green Version]
- Chiang, H.Y.; Korshunov, V.A.; Serour, A.; Shi, F.; Sottile, J. Fibronectin is an important regulator of flow-induced vascular remodeling. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1074–1079. [Google Scholar] [CrossRef] [Green Version]
- Nagase, H.; Woessner, J.F. Matrix Metalloproteinases. J. Biol. Chem. 1999, 274, 21491–21494. [Google Scholar] [CrossRef] [Green Version]
- Páramo, J.A. New mechanisms of vascular fibrosis: Role of lysyl oxidase. Clínica Investig. Arterioscler. Engl. Ed. 2017, 29, 166–167. [Google Scholar] [CrossRef]
- Pinnell, S.R.; Martin, G.R. The cross-linking of collagen and elastin: Enzymatic conversion of lysine in peptide linkage to alpha-aminoadipic-delta-semialdehyde (allysine) by an extract from bone. Proc. Natl. Acad. Sci. USA 1968, 61, 708–716. [Google Scholar] [CrossRef] [Green Version]
- Csiszar, K. Lysyl oxidases: A novel multifunctional amine oxidase family. In Progress in Nucleic Acid Research and Molecular Biology; Academic Press: Cambridge, MA, USA, 2001; pp. 1–32. [Google Scholar]
- Takaoka, M.; Nagata, D.; Kihara, S.; Shimomura, I.; Kimura, Y.; Tabata, Y.; Saito, Y.; Nagai, R.; Sata, M. Periadventitial Adipose Tissue Plays a Critical Role in Vascular Remodeling. Circ. Res. 2009, 105, 906–911. [Google Scholar] [CrossRef]
- Dashwood, M.R.; Anand, R.; Loesch, A.; Souza, D.S.R. Hypothesis: A Potential Role for the Vasa Vasorum in the Maintenance of Vein Graft Patency. Angiology 2004, 55, 385–395. [Google Scholar] [CrossRef]
- Duque, J.C.; Martinez, L.; Tabbara, M.; Parikh, P.; Paez, A.; Selman, G.; Salman, L.H.; Velazquez, O.C.; Vazquez-Padron, R.I. Vascularization of the arteriovenous fistula wall and association with maturation outcomes. J. Vasc. Access 2020, 21, 161–168. [Google Scholar] [CrossRef]
- Hellström, M.; Engström-Laurent, A.; Hellström, S. Expression of the CD44 Receptor in the Blood Vessel System: An Experimental Study in Rat. Cells Tissues Organs 2005, 179, 102–108. [Google Scholar] [CrossRef]
- Goodison, S.; Urquidi, V.; Tarin, D. CD44 cell adhesion molecules. Mol. Pathol. 1999, 52, 189–196. [Google Scholar] [CrossRef] [Green Version]
- Stenmark, K.R.; Yeager, M.E.; El Kasmi, K.C.; Nozik-Grayck, E.; Gerasimovskaya, E.V.; Li, M.; Riddle, S.R.; Frid, M.G. The adventitia: Essential regulator of vascular wall structure and function. Annu. Rev. Physiol. 2013, 75, 23–47. [Google Scholar] [CrossRef] [Green Version]
- Go, A.S.; Chertow, G.M.; Fan, D.; McCulloch, C.E.; Hsu, C.-y. Chronic Kidney Disease and the Risks of Death, Cardiovascular Events, and Hospitalization. N. Engl. J. Med. 2004, 351, 1296–1305. [Google Scholar] [CrossRef]
- Brunet, P.; Gondouin, B.; Duval-Sabatier, A.; Dou, L.; Cerini, C.; Dignat-George, F.; Jourde-Chiche, N.; Argiles, A.; Burtey, S. Does Uremia Cause Vascular Dysfunction. Kidney Blood Press. Res. 2011, 34, 284–290. [Google Scholar] [CrossRef]
- Wali, M.A.; Eid, R.A.; Al-Homrany, M.A. Smooth Muscle Changes in the Cephalic Vein of Renal Failure Patients before Use as an Arteriovenous Fistula (AVF). J. Smooth Muscle Res. 2002, 38, 75–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alpers, C.E.; Imrey, P.B.; Hudkins, K.L.; Wietecha, T.A.; Radeva, M.; Allon, M.; Cheung, A.K.; Dember, L.M.; Roy-Chaudhury, P.; Shiu, Y.-T.; et al. Histopathology of Veins Obtained at Hemodialysis Arteriovenous Fistula Creation Surgery. J. Am. Soc. Nephrol. 2017, 28, 3076–3088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martens, C.R.; Edwards, D.G. Peripheral vascular dysfunction in chronic kidney disease. Cardiol. Res. Pract. 2011, 2011, 267257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Günthner, T.; Jankowski, V.; Kretschmer, A.; Nierhaus, M.; Van Der Giet, M.; Zidek, W.; Jankowski, J. Progress in Uremic Toxin Research: Endothelium and Vascular Smooth Muscle Cells in the Context of Uremia. Semin. Dial. 2009, 22, 428–432. [Google Scholar] [CrossRef]
- Moody, W.E.; Edwards, N.C.; Madhani, M.; Chue, C.D.; Steeds, R.P.; Ferro, C.J.; Townend, J.N. Endothelial dysfunction and cardiovascular disease in early-stage chronic kidney disease: Cause or association? Atherosclerosis 2012, 223, 86–94. [Google Scholar] [CrossRef]
- Geenen, I.L.; Kolk, F.F.; Molin, D.G.; Wagenaar, A.; Compeer, M.G.; Tordoir, J.H.; Schurink, G.W.; De Mey, J.G.; Post, M.J. Nitric Oxide Resistance Reduces Arteriovenous Fistula Maturation in Chronic Kidney Disease in Rats. PLoS ONE 2016, 11, e0146212. [Google Scholar] [CrossRef] [Green Version]
- Stockler-Pinto, M.B.; Mafra, D.; Moraes, C.; Lobo, J.; Boaventura, G.T.; Farage, N.E.; Silva, W.S.; Cozzolino, S.F.; Malm, O. Brazil Nut (Bertholletia excelsa, H.B.K.) Improves Oxidative Stress and Inflammation Biomarkers in Hemodialysis Patients. Biol. Trace Elem. Res. 2014, 158, 105–112. [Google Scholar] [CrossRef]
- Hall, M.R.; Yamamoto, K.; Protack, C.D.; Tsuneki, M.; Kuwahara, G.; Assi, R.; Brownson, K.E.; Bai, H.; Madri, J.A.; Dardik, A. Temporal regulation of venous extracellular matrix components during arteriovenous fistula maturation. J. Vasc. Access 2015, 16, 93–106. [Google Scholar] [CrossRef] [Green Version]
- Fanjul-Fernández, M.; Folgueras, A.R.; Cabrera, S.; López-Otín, C. Matrix metalloproteinases: Evolution, gene regulation and functional analysis in mouse models. Biochim. Biophys. Acta BBA—Mol. Cell Res. 2010, 1803, 3–19. [Google Scholar] [CrossRef] [Green Version]
- Chan, C.-Y.; Chen, Y.-S.; Ma, M.-C.; Chen, C.-F. Remodeling of experimental arteriovenous fistula with increased matrix metalloproteinase expression in rats. J. Vasc. Surg. 2007, 45, 804–811. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.S.; Shen, Q.; Pitts, R.L.; Guo, M.; Wu, M.H.; Sun, S.C.; Yuan, S.Y. Serum metalloproteinases MMP-2, MMP-9, and metalloproteinase tissue inhibitors in patients are associated with arteriovenous fistula maturation. J. Vasc. Surg. 2011, 54, 454–459. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.S.; Shen, Q.; Pitts, R.L.; Guo, M.; Wu, M.H.; Yuan, S.Y. Vein tissue expression of matrix metalloproteinase as biomarker for hemodialysis arteriovenous fistula maturation. Vasc. Endovasc. Surg. 2010, 44, 674–679. [Google Scholar] [CrossRef]
- Misra, S.; Fu, A.A.; Anderson, J.L.; Sethi, S.; Glockner, J.F.; McKusick, M.A.; Bjarnason, H.; Woodrum, D.A.; Mukhopadhyay, D. The rat femoral arteriovenous fistula model: Increased expression of matrix metalloproteinase-2 and -9 at the venous stenosis. J. Vasc. Interv. Radiol. 2008, 19, 587–594. [Google Scholar] [CrossRef]
- Ruan, L.; Yao, X.; Li, W.; Zhang, L.; Yang, H.; Sun, J.; Li, A. Effect of galectin-3 in the pathogenesis of arteriovenous fistula stenosis formation. Ren. Fail. 2021, 43, 566–576. [Google Scholar] [CrossRef]
- Misra, S.; Fu, A.A.; Rajan, D.K.; Juncos, L.A.; McKusick, M.A.; Bjarnason, H.; Mukhopadhyay, D. Expression of Hypoxia Inducible Factor–1α, Macrophage Migration Inhibition Factor, Matrix Metalloproteinase–2 and −9, and Their Inhibitors in Hemodialysis Grafts and Arteriovenous Fistulas. J. Vasc. Interv. Radiol. 2008, 19, 252–259. [Google Scholar] [CrossRef]
- Diskin, C.; Stokes, T.J.; Dansby, L.M.; Radcliff, L.; Carter, T.B. Doxycycline may reduce the incidence of aneurysms in haemodialysis vascular accesses. Nephrol. Dial. Transplant. 2005, 20, 959–961. [Google Scholar] [CrossRef] [Green Version]
- Nath, K.A.; Grande, J.P.; Kang, L.; Juncos, J.P.; Ackerman, A.W.; Croatt, A.J.; Katusic, Z.S. ß-Catenin is markedly induced in a murine model of an arteriovenous fistula: The effect of metalloproteinase inhibition: The effect of metalloproteinase inhibition. Am. J. Physiol. Renal Physiol. 2010, 299, F1270–F1277. [Google Scholar] [CrossRef]
- Lardenoye, J.H.; de Vries, M.R.; Deckers, M.; van Lent, N.; Hanemaaijer, R.; van Bockel, J.H.; Quax, P.H. Inhibition of intimal hyperplasia by the tetracycline derived mmp inhibitor doxycycline in vein graft disease in vitro and in vivo. EuroIntervention 2005, 1, 236–243. [Google Scholar]
- Shih, Y.-C.; Chen, P.-Y.; Ko, T.-M.; Huang, P.-H.; Ma, H.; Tarng, D.-C. MMP-9 Deletion Attenuates Arteriovenous Fistula Neointima through Reduced Perioperative Vascular Inflammation. Int. J. Mol. Sci. 2021, 22, 5448. [Google Scholar] [CrossRef]
- Guo, L.; Ning, W.; Tan, Z.; Gong, Z.; Li, X. Mechanism of matrix metalloproteinase axis-induced neointimal growth. J. Mol. Cell. Cardiol. 2014, 66, 116–125. [Google Scholar] [CrossRef]
- Lin, C.-C.; Yang, W.-C.; Chung, M.-Y.; Lee, P.-C. Functional Polymorphisms in Matrix Metalloproteinases-1, -3, -9 are Associated with Arteriovenous Fistula Patency in Hemodialysis Patients. Clin. J. Am. Soc. Nephrol. 2010, 5, 1805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okamura, H.; Pisani, L.J.; Dalal, A.R.; Emrich, F.; Dake, B.A.; Arakawa, M.; Onthank, D.C.; Cesati, R.R.; Robinson, S.P.; Milanesi, M.; et al. Assessment of Elastin Deficit in a Marfan Mouse Aneurysm Model Using an Elastin-Specific Magnetic Resonance Imaging Contrast Agent. Circ. Cardiovasc. Imaging 2014, 7, 690–696. [Google Scholar] [CrossRef] [Green Version]
- Vouyouka, A.G.; Pfeiffer, B.J.; Liem, T.K.; Taylor, T.A.; Mudaliar, J.; Phillips, C.L. The role of type I collagen in aortic wall strength with a homotrimeric [α1(I)]3 collagen mouse model. J. Vasc. Surg. 2001, 33, 1263–1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wynn, T.A. Cellular and molecular mechanisms of fibrosis. J. Pathol. 2008, 214, 199–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suki, B.; Jesudason, R.; Sato, S.; Parameswaran, H.; Araujo, A.D.; Majumdar, A.; Allen, P.G.; Bartolák-Suki, E. Mechanical failure, stress redistribution, elastase activity and binding site availability on elastin during the progression of emphysema. Pulm. Pharmacol. Ther. 2012, 25, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Davis, P.F.; Ryan, P.A.; Osipowicz, J.; Anderson, M.J.; Sweeney, A.; Stehbens, W.E. The biochemical composition of hemodynamically stressed vascular tissue: The insoluble elastin of experimental arteriovenous fistulae. Exp. Mol. Pathol. 1989, 51, 103–110. [Google Scholar] [CrossRef]
- Chang, C.-J.; Chen, C.-C.; Hsu, L.-A.; Chang, G.-J.; Ko, Y.-H.; Chen, C.-F.; Chen, M.-Y.; Yang, S.-H.; Pang, J.-H.S. Degradation of the Internal Elastic Laminae in Vein Grafts of Rats with Aortocaval Fistulae: Potential Impact on Graft Vasculopathy. Am. J. Pathol. 2009, 174, 1837–1846. [Google Scholar] [CrossRef] [Green Version]
- Masuda, H.; Zhuang, Y.-J.; Singh, T.M.; Kawamura, K.; Murakami, M.; Zarins, C.K.; Glagov, S. Adaptive Remodeling of Internal Elastic Lamina and Endothelial Lining During Flow-Induced Arterial Enlargement. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 2298–2307. [Google Scholar] [CrossRef] [Green Version]
- Greenhill, N.S.; Stehbens, W.E. Scanning electron microscopic investigation of the afferent arteries of experimental femoral arteriovenous fistulae in rabbits. Pathology 1987, 19, 22–27. [Google Scholar] [CrossRef]
- Sho, E.; Sho, M.; Singh, T.M.; Nanjo, H.; Komatsu, M.; Xu, C.; Masuda, H.; Zarins, C.K. Arterial Enlargement in Response to High Flow Requires Early Expression of Matrix Metalloproteinases to Degrade Extracellular Matrix. Exp. Mol. Pathol. 2002, 73, 142–153. [Google Scholar] [CrossRef] [Green Version]
- Tronc, F.; Wassef, M.; Esposito, B.; Henrion, D.; Glagov, S.; Tedgui, A. Role of NO in Flow-Induced Remodeling of the Rabbit Common Carotid Artery. Arterioscler. Thromb. Vasc. Biol. 1996, 16, 1256–1262. [Google Scholar] [CrossRef]
- Jones, G.T.; Stehbens, W.E.; Martin, B.J. Ultrastructural changes in arteries proximal to short-term experimental carotid-jugular arteriovenous fistulae in rabbits. Int. J. Exp. Pathol. 1994, 75, 225–232. [Google Scholar]
- Wong, C.Y.; Rothuizen, T.C.; de Vries, M.R.; Rabelink, T.J.; Hamming, J.F.; van Zonneveld, A.J.; Quax, P.H.; Rotmans, J.I. Elastin is a key regulator of outward remodeling in arteriovenous fistulas. Eur. J. Vasc. Endovasc. Surg. 2015, 49, 480–486. [Google Scholar] [CrossRef] [Green Version]
- Bezhaeva, T.; de Vries, M.R.; Geelhoed, W.J.; van der Veer, E.P.; Versteeg, S.; van Alem, C.M.A.; Voorzaat, B.M.; Eijkelkamp, N.; van der Bogt, K.E.; Agoulnik, A.I.; et al. Relaxin receptor deficiency promotes vascular inflammation and impairs outward remodeling in arteriovenous fistulas. FASEB J. 2018, 32, 6293–6304. [Google Scholar] [CrossRef] [Green Version]
- Peden, E.K.; Lucas, J.F.; Browne, B.J.; Settle, S.M.; Scavo, V.A.; Bleyer, A.J.; Ozaki, C.K.; Teruya, T.H.; Wilson, S.E.; Mishler, R.E.; et al. PATENCY-2 trial of vonapanitase to promote radiocephalic fistula use for hemodialysis and secondary patency. J. Vasc. Access 2022, 23, 265–274. [Google Scholar] [CrossRef]
- Bleyer, A.J.; Scavo, V.A.; Wilson, S.E.; Browne, B.J.; Ferris, B.L.; Ozaki, C.K.; Lee, T.; Peden, E.K.; Dixon, B.S.; Mishler, R.; et al. A randomized trial of vonapanitase (PATENCY-1) to promote radiocephalic fistula patency and use for hemodialysis. J. Vasc. Surg. 2019, 69, 507–515. [Google Scholar] [CrossRef] [Green Version]
- Dwivedi, A.J.; Roy-Chaudhury, P.; Peden, E.K.; Browne, B.J.; Ladenheim, E.D.; Scavo, V.A.; Gustafson, P.N.; Wong, M.D.; Magill, M.; Lindow, F.; et al. Application of Human Type I Pancreatic Elastase (PRT-201) to the venous Anastomosis of Arteriovenous Grafts in Patients with Chronic Kidney Disease. J. Vasc. Access 2014, 15, 376–384. [Google Scholar] [CrossRef]
- Hye, R.J.; Peden, E.K.; O’Connor, T.P.; Browne, B.J.; Dixon, B.S.; Schanzer, A.S.; Jensik, S.C.; Dember, L.M.; Jaff, M.R.; Burke, S.K. Human type I pancreatic elastase treatment of arteriovenous fistulas in patients with chronic kidney disease. J. Vasc. Surg. 2014, 60, 454–461.e1. [Google Scholar] [CrossRef] [Green Version]
- Andraska, E.; Skirtich, N.; McCreary, D.; Kulkarni, R.; Tzeng, E.; McEnaney, R. Simultaneous Upregulation of Elastolytic and Elastogenic Factors Are Necessary for Regulated Collateral Diameter Expansion. Front. Cardiovasc. Med. 2022, 8, 762094. [Google Scholar] [CrossRef]
- Field, M.A.; McGrogan, D.G.; Tullet, K.; Inston, N.G. Arteriovenous fistula aneurysms in patients with Alport’s. J. Vasc. Access 2013, 14, 397–399. [Google Scholar] [CrossRef]
- Klüsch, V.; Aper, T.; Sonnenschein, K.; Becker, L.S.; Umminger, J.; Haverich, A.; Rustum, S. A Hyperdynamic Arteriovenous Fistula Aneurysm After Long Time Renal Transplantation. Vasc. Endovasc. Surg. 2022, 57, 182–185. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.A.; Stehbens, W.E.; Weber, P. Hemodynamically-induced increase in soluble collagen in the anastomosed veins of experimental arteriovenous fistulae. Atherosclerosis 1976, 23, 429–436. [Google Scholar] [CrossRef]
- Shiu, Y.T.; Litovsky, S.H.; Cheung, A.K.; Pike, D.B.; Tey, J.C.; Zhang, Y.; Young, C.J.; Robbin, M.; Hoyt, K.; Allon, M. Preoperative Vascular Medial Fibrosis and Arteriovenous Fistula Development. Clin. J. Am. Soc. Nephrol. 2016, 11, 1615–1623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, L.; Duque, J.C.; Tabbara, M.; Paez, A.; Selman, G.; Hernandez, D.R.; Sundberg, C.A.; Tey, J.C.S.; Shiu, Y.T.; Cheung, A.K.; et al. Fibrotic Venous Remodeling and Nonmaturation of Arteriovenous Fistulas. J. Am. Soc. Nephrol. 2018, 29, 1030–1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.C. Lysyl Oxidase. In International Review of Connective Tissue Research; Hall, D.A., Jackson, D.S., Eds.; Elsevier: Amsterdam, The Netherlands, 1979; pp. 73–118. [Google Scholar]
- Hernandez, D.R.; Applewhite, B.; Martinez, L.; Laurito, T.; Tabbara, M.; Rojas, M.G.; Wei, Y.; Selman, G.; Knysheva, M.; Velazquez, O.C.; et al. Inhibition of Lysyl Oxidase with β-aminopropionitrile Improves Venous Adaptation after Arteriovenous Fistula Creation. Kidney360 2021, 2, 270–278. [Google Scholar] [CrossRef]
- Applewhite, B.; Gupta, A.; Wei, Y.; Yang, X.; Martinez, L.; Rojas, M.G.; Andreopoulos, F.; Vazquez-Padron, R.I. Periadventitial β-aminopropionitrile-loaded nanofibers reduce fibrosis and improve arteriovenous fistula remodeling in rats. Front. Cardiovasc. Med. 2023, 10, 1124106. [Google Scholar] [CrossRef]
- Wilmarth, K.R.; Froines, J.R. In vitro and in vivo inhibition of lysyl oxidase byaminopropionitriles. J. Toxicol. Environ. Health 1992, 37, 411–423. [Google Scholar] [CrossRef]
- Meng, X.-M.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-β: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef]
- Suwanabol, P.A.; Seedial, S.M.; Shi, X.; Zhang, F.; Yamanouchi, D.; Roenneburg, D.; Liu, B.; Kent, K.C. Transforming growth factor-β increases vascular smooth muscle cell proliferation through the Smad3 and extracellular signal-regulated kinase mitogen-activated protein kinases pathways. J. Vasc. Surg. 2012, 56, 446–454.e1. [Google Scholar] [CrossRef] [Green Version]
- Atsawasuwan, P.; Mochida, Y.; Katafuchi, M.; Kaku, M.; Fong, K.S.; Csiszar, K.; Yamauchi, M. Lysyl oxidase binds transforming growth factor-beta and regulates its signaling via amine oxidase activity. J. Biol. Chem. 2008, 283, 34229–34240. [Google Scholar] [CrossRef] [Green Version]
- Gacheru, S.N.; Thomas, K.M.; Murray, S.A.; Csiszar, K.; Smith-Mungo, L.I.; Kagan, H.M. Transcriptional and post-transcriptional control of lysyl oxidase expression in vascular smooth muscle cells: Effects of TGF-beta 1 and serum deprivation. J. Cell. Biochem. 1997, 65, 395–407. [Google Scholar] [CrossRef]
- Taniguchi, R.; Ohashi, Y.; Lee, J.S.; Hu, H.; Gonzalez, L.; Zhang, W.; Langford, J.; Matsubara, Y.; Yatsula, B.; Tellides, G.; et al. Endothelial Cell TGF- β (Transforming Growth Factor-Beta) Signaling Regulates Venous Adaptive Remodeling to Improve Arteriovenous Fistula Patency. Arterioscler. Thromb. Vasc. Biol. 2022, 42, 868–883. [Google Scholar] [CrossRef]
- Hu, H.; Lee, S.R.; Bai, H.; Guo, J.; Hashimoto, T.; Isaji, T.; Guo, X.; Wang, T.; Wolf, K.; Liu, S.; et al. TGFβ (Transforming Growth Factor-Beta)-Activated Kinase 1 Regulates Arteriovenous Fistula Maturation. Arterioscler. Thromb. Vasc. Biol. 2020, 40, e203–e213. [Google Scholar] [CrossRef]
- Bashar, K.; Zafar, A.; Elsheikh, S.; Healy, D.A.; Clarke-Moloney, M.; Casserly, L.; Burke, P.E.; Kavanagh, E.G.; Walsh, S.R. Predictive Parameters of Arteriovenous Fistula Functional Maturation in a Population of Patients with End-Stage Renal Disease. PLoS ONE 2015, 10, e0119958. [Google Scholar] [CrossRef]
- Peterson, W.J.; Barker, J.; Allon, M. Disparities in Fistula Maturation Persist Despite Preoperative Vascular Mapping. Clin. J. Am. Soc. Nephrol. 2008, 3, 437–441. [Google Scholar] [CrossRef] [Green Version]
- Cai, C.; Kilari, S.; Singh, A.K.; Zhao, C.; Simeon, M.L.; Misra, A.; Li, Y.; Misra, S. Differences in Transforming Growth Factor-β1/BMP7 Signaling and Venous Fibrosis Contribute to Female Sex Differences in Arteriovenous Fistulas. J. Am. Heart Assoc. 2020, 9, e017420. [Google Scholar] [CrossRef]
- Cai, C.; Zhao, C.; Kilari, S.; Sharma, A.; Singh, A.K.; Simeon, M.L.; Misra, A.; Li, Y.; Misra, S. Effect of sex differences in treatment response to angioplasty in a murine arteriovenous fistula model. Am. J. Physiol.-Ren. Physiol. 2020, 318, F565–F575. [Google Scholar] [CrossRef] [Green Version]
- Heine, G.H.; Ulrich, C.; Sester, U.; Sester, M.; Köhler, H.; Girndt, M. Transforming growth factor β1 genotype polymorphisms determine AV fistula patency in hemodialysis patients. Kidney Int. 2003, 64, 1101–1107. [Google Scholar] [CrossRef] [Green Version]
- Stirbu, O.; Gadalean, F.; Pitea, I.V.; Ciobanu, G.; Schiller, A.; Grosu, I.; Nes, A.; Bratescu, R.; Olariu, N.; Timar, B.; et al. C-reactive protein as a prognostic risk factor for loss of arteriovenous fistula patency in hemodialyzed patients. J. Vasc. Surg. 2019, 70, 208–215. [Google Scholar] [CrossRef]
- Kaller, R.; Arbănași, E.M.; Mureșan, A.V.; Voidăzan, S.; Arbănași, E.M.; Horváth, E.; Suciu, B.A.; Hosu, I.; Halmaciu, I.; Brinzaniuc, K.; et al. The Predictive Value of Systemic Inflammatory Markers, the Prognostic Nutritional Index, and Measured Vessels’ Diameters in Arteriovenous Fistula Maturation Failure. Life 2022, 12, 1447. [Google Scholar] [CrossRef]
- Chang, C.-J.; Ko, Y.-S.; Ko, P.-J.; Hsu, L.-A.; Chen, C.-F.; Yang, C.-W.; Hsu, T.-S.; Pang, J.-H.S. Thrombosed arteriovenous fistula for hemodialysis access is characterized by a marked inflammatory activity. Kidney Int. 2005, 68, 1312–1319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sener, E.F.; Taheri, S.; Korkmaz, K.; Zararsiz, G.; Serhatlioglu, F.; Unal, A.; Emirogullari, O.N.; Ozkul, Y. Association of TNF-α −308 G > A and ACE I/D gene polymorphisms in hemodialysis patients with arteriovenous fistula thrombosis. Int. Urol. Nephrol. 2014, 46, 1419–1425. [Google Scholar] [CrossRef] [PubMed]
- Juncos, J.P.; Grande, J.P.; Kang, L.; Ackerman, A.W.; Croatt, A.J.; Katusic, Z.S.; Nath, K.A. MCP-1 Contributes to Arteriovenous Fistula Failure. J. Am. Soc. Nephrol. 2011, 22, 43–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marrone, D.; Pertosa, G.; Simone, S.; Loverre, A.; Capobianco, C.; Cifarelli, M.; Memoli, B.; Schena, F.P.; Grandaliano, G. Local Activation of Interleukin 6 Signaling Is Associated with Arteriovenous Fistula Stenosis in Hemodialysis Patients. Am. J. Kidney Dis. 2007, 49, 664–673. [Google Scholar] [CrossRef]
- Wong, C.; Bezhaeva, T.; Rothuizen, T.C.; Metselaar, J.M.; de Vries, M.R.; Verbeek, F.P.R.; Vahrmeijer, A.L.; Wezel, A.; van Zonneveld, A.-J.; Rabelink, T.J.; et al. Liposomal prednisolone inhibits vascular inflammation and enhances venous outward remodeling in a murine arteriovenous fistula model. Sci. Rep. 2016, 6, 30439. [Google Scholar] [CrossRef] [Green Version]
- Voorzaat, B.M.; van der Bogt, K.E.A.; Bezhaeva, T.; van Schaik, J.; Eefting, D.; van der Putten, K.; van Nieuwenhuizen, R.C.; Groeneveld, J.O.; Hoogeveen, E.K.; van der Meer, I.M.; et al. A Randomized Trial of Liposomal Prednisolone (LIPMAT) to Enhance Radiocephalic Fistula Maturation: A Pilot Study. Kidney Int. Rep. 2020, 5, 1327–1332. [Google Scholar] [CrossRef]
- Penn, D.L.; Witte, S.R.; Komotar, R.J.; Sander Connolly, E. The role of vascular remodeling and inflammation in the pathogenesis of intracranial aneurysms. J. Clin. Neurosci. 2014, 21, 28–32. [Google Scholar] [CrossRef]
- Nguyen, M.; Thankam, F.G.; Agrawal, D.K. Sterile inflammation in the pathogenesis of maturation failure of arteriovenous fistula. J. Mol. Med. 2021, 99, 729–741. [Google Scholar] [CrossRef]
- Samra, G.; Rai, V.; Agrawal, D.K. Innate and adaptive immune cells associate with arteriovenous fistula maturation and failure. Can. J. Physiol. Pharmacol. 2022, 100, 716–727. [Google Scholar] [CrossRef]
- Satish, M.; Gunasekar, P.; Agrawal, D.K. Pro-inflammatory and pro-resolving mechanisms in the immunopathology of arteriovenous fistula maturation. Expert Rev. Cardiovasc. Ther. 2019, 17, 369–376. [Google Scholar] [CrossRef]
- Matsubara, Y.; Kiwan, G.; Fereydooni, A.; Langford, J.; Dardik, A. Distinct subsets of T cells and macrophages impact venous remodeling during arteriovenous fistula maturation. JVS Vasc. Sci. 2020, 1, 207–218. [Google Scholar] [CrossRef]
- Chan, S.M.; Weininger, G.; Langford, J.; Jane-Wit, D.; Dardik, A. Sex Differences in Inflammation During Venous Remodeling of Arteriovenous Fistulae. Front. Cardiovasc. Med. 2021, 8, 715114. [Google Scholar] [CrossRef]
- Zhao, X.; Chen, J.; Sun, H.; Zhang, Y.; Zou, D. New insights into fibrosis from the ECM degradation perspective: The macrophage-MMP-ECM interaction. Cell Biosci. 2022, 12, 117. [Google Scholar] [CrossRef]
- Sukhova, G.K.; Schönbeck, U.; Rabkin, E.; Schoen, F.J.; Poole, A.R.; Billinghurst, R.C.; Libby, P. Evidence for Increased Collagenolysis by Interstitial Collagenases-1 and -3 in Vulnerable Human Atheromatous Plaques. Circulation 1999, 99, 2503–2509. [Google Scholar] [CrossRef]
- Galis, Z.S.; Muszynski, M.; Sukhova, G.K.; Simon-Morrissey, E.; Unemori, E.N.; Lark, M.W.; Amento, E.; Libby, P. Cytokine-stimulated human vascular smooth muscle cells synthesize a complement of enzymes required for extracellular matrix digestion. Circ. Res. 1994, 75, 181–189. [Google Scholar] [CrossRef] [Green Version]
- Alexander, M.R.; Moehle, C.W.; Johnson, J.L.; Yang, Z.; Lee, J.K.; Jackson, C.L.; Owens, G.K. Genetic inactivation of IL-1 signaling enhances atherosclerotic plaque instability and reduces outward vessel remodeling in advanced atherosclerosis in mice. J. Clin. Investig. 2012, 122, 70–79. [Google Scholar] [CrossRef] [Green Version]
- Kuwahara, G.; Hashimoto, T.; Tsuneki, M.; Yamamoto, K.; Assi, R.; Foster, T.R.; Hanisch, J.J.; Bai, H.; Hu, H.; Protack, C.D.; et al. CD44 Promotes Inflammation and Extracellular Matrix Production During Arteriovenous Fistula Maturation. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1147–1156. [Google Scholar] [CrossRef] [Green Version]
- Matsubara, Y.; Kiwan, G.; Liu, J.; Gonzalez, L.; Langford, J.; Gao, M.; Gao, X.; Taniguchi, R.; Yatsula, B.; Furuyama, T.; et al. Inhibition of T-Cells by Cyclosporine A Reduces Macrophage Accumulation to Regulate Venous Adaptive Remodeling and Increase Arteriovenous Fistula Maturation. Arterioscler. Thromb. Vasc. Biol. 2021, 41, e160–e174. [Google Scholar] [CrossRef]
- Yang, B.; Kilari, S.; Brahmbhatt, A.; McCall, D.L.; Torres, E.N.; Leof, E.B.; Mukhopadhyay, D.; Misra, S. CorMatrix Wrapped Around the Adventitia of the Arteriovenous Fistula Outflow Vein Attenuates Venous Neointimal Hyperplasia. Sci. Rep. 2017, 7, 14298. [Google Scholar] [CrossRef] [Green Version]
- Leskovar, B.; Furlan, T.; Poznic, S.; Hrastelj, M.; Adamlje, A. Using CorMatrix for partial and complete (re)construction of arteriovenous fistulas in haemodialysis patients: (Re)construction of arteriovenous fistulas with CorMatrix. J. Vasc. Access 2019, 20, 597–603. [Google Scholar] [CrossRef]
- DuBose, J.J.; Azizzadeh, A. Utilization of a Tubularized CorMatrix Extracellular Matrix for Repair of an Arteriovenous Fistula Aneurysm. Ann. Vasc. Surg. 2015, 29, e1–e366. [Google Scholar] [CrossRef] [PubMed]
- Shiu, Y.T.; He, Y.; Tey, J.C.S.; Knysheva, M.; Anderson, B.; Kauser, K. Natural Vascular Scaffolding Treatment Promotes Outward Remodeling During Arteriovenous Fistula Development in Rats. Front. Bioeng. Biotechnol. 2021, 9, 622617. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Anderson, B.; Hu, Q.; Hayes, R.; Huff, K.; Isaacson, J.; Warner, K.S.; Hauser, H.; Greenberg, M.; Chandra, V.; et al. Photochemically Aided Arteriovenous Fistula Creation to Accelerate Fistula Maturation. Int. J. Mol. Sci. 2023, 24, 7571. [Google Scholar] [CrossRef] [PubMed]
- Protti, A.; Lavin, B.; Dong, X.; Lorrio, S.; Robinson, S.; Onthank, D.; Shah, A.M.; Botnar, R.M. Assessment of Myocardial Remodeling Using an Elastin/Tropoelastin Specific Agent with High Field Magnetic Resonance Imaging (MRI). J. Am. Heart Assoc. 2015, 4, e001851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brangsch, J.; Reimann, C.; Kaufmann, J.O.; Adams, L.C.; Onthank, D.C.; Thöne-Reineke, C.; Robinson, S.P.; Buchholz, R.; Karst, U.; Botnar, R.M.; et al. Concurrent Molecular Magnetic Resonance Imaging of Inflammatory Activity and Extracellular Matrix Degradation for the Prediction of Aneurysm Rupture. Circ. Cardiovasc. Imaging 2019, 12, e008707. [Google Scholar] [CrossRef] [Green Version]
- Laboyrie, S.L.; Vries, M.R.d.; Jong, A.d.; Boer, H.C.d.; Lalai, R.A.; Martinez, L.; Vazquez-Padron, R.I.; Rotmans, J.I. von Willebrand Factor: A Central Regulator of Arteriovenous Fistula Maturation Through Smooth Muscle Cell Proliferation and Outward Remodeling. J. Am. Heart Assoc. 2022, 11, e024581. [Google Scholar] [CrossRef]
- Murphy, G.J.; Angelini, G.D. Insights into the pathogenesis of vein graft disease: Lessons from intravascular ultrasound. Cardiovasc. Ultrasound 2004, 2, 8. [Google Scholar] [CrossRef] [Green Version]
- de Jong, A.; Sier, V.Q.; Peters, H.A.B.; Schilder, N.K.M.; Jukema, J.W.; Goumans, M.J.T.H.; Quax, P.H.A.; de Vries, M.R. Interfering in the ALK1 Pathway Results in Macrophage-Driven Outward Remodeling of Murine Vein Grafts. Front. Cardiovasc. Med. 2022, 8, 784980. [Google Scholar] [CrossRef]
- Wu, M.; Awasthi, N.; Rad, N.M.; Pluim, J.P.W.; Lopata, R.G.P. Advanced Ultrasound and Photoacoustic Imaging in Cardiology. Sensors 2021, 21, 7947. [Google Scholar] [CrossRef]
- Muzard, J.; Sarda-Mantel, L.; Loyau, S.; Meulemans, A.; Louedec, L.; Bantsimba-Malanda, C.; Hervatin, F.; Marchal-Somme, J.; Michel, J.B.; Le Guludec, D.; et al. Non-invasive molecular imaging of fibrosis using a collagen-targeted peptidomimetic of the platelet collagen receptor glycoprotein VI. PLoS ONE 2009, 4, e5585. [Google Scholar] [CrossRef] [Green Version]
- De Jong, S.; van Middendorp, L.B.; Hermans, R.H.A.; de Bakker, J.M.T.; Bierhuizen, M.F.A.; Prinzen, F.W.; van Rijen, H.V.M.; Losen, M.; Vos, M.A.; van Zandvoort, M.A.M.J. Ex Vivo and In Vivo Administration of Fluorescent CNA35 Specifically Marks Cardiac Fibrosis. Mol. Imaging 2014, 13, 1–9. [Google Scholar] [CrossRef]
- Adams, L.C.; Brangsch, J.; Reimann, C.; Kaufmann, J.O.; Buchholz, R.; Karst, U.; Botnar, R.M.; Hamm, B.; Makowski, M.R. Simultaneous molecular MRI of extracellular matrix collagen and inflammatory activity to predict abdominal aortic aneurysm rupture. Sci. Rep. 2020, 10, 15206. [Google Scholar] [CrossRef]
- Bezhaeva, T.; Wong, C.; de Vries, M.R.; van der Veer, E.P.; van Alem, C.M.A.; Que, I.; Lalai, R.A.; van Zonneveld, A.-J.; Rotmans, J.I.; Quax, P.H.A. Deficiency of TLR4 homologue RP105 aggravates outward remodeling in a murine model of arteriovenous fistula failure. Sci. Rep. 2017, 7, 10269. [Google Scholar] [CrossRef]
- Ding, X.; Chen, J.; Wu, C.; Wang, G.; Zhou, C.; Chen, S.; Wang, K.; Zhang, A.; Ye, P.; Wu, J.; et al. Nucleotide-Binding Oligomerization Domain-Like Receptor Protein 3 Deficiency in Vascular Smooth Muscle Cells Prevents Arteriovenous Fistula Failure Despite Chronic Kidney Disease. J. Am. Heart Assoc. 2019, 8, e011211. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.K.; Cai, C.; Kilari, S.; Zhao, C.; Simeon, M.L.; Takahashi, E.; Edelman, E.R.; Kong, H.; Macedo, T.; Singh, R.J.; et al. 1α,25-Dihydroxyvitamin D3 Encapsulated in Nanoparticles Prevents Venous Neointimal Hyperplasia and Stenosis in Porcine Arteriovenous Fistulas. J. Am. Soc. Nephrol. 2021, 32, 866–885. [Google Scholar] [CrossRef]
- Tanaka, L.Y.; Laurindo, F.R.M. Vascular remodeling: A redox-modulated mechanism of vessel caliber regulation. Free. Radic. Biol. Med. 2017, 109, 11–21. [Google Scholar] [CrossRef]
- George, S.J.; Wan, S.; Hu, J.; MacDonald, R.; Johnson, J.L.; Baker, A.H. Sustained Reduction of Vein Graft Neointima Formation by Ex Vivo TIMP-3 Gene Therapy. Circulation 2011, 124 (Suppl. 1), S135–S142. [Google Scholar] [CrossRef] [Green Version]
- Ballmann, M.Z.; Raus, S.; Engelhart, R.; Kaján, G.L.; Beqqali, A.; Hadoke, P.W.F.; van der Zalm, C.; Papp, T.; John, L.; Khan, S.; et al. Human AdV-20-42-42, a Promising Novel Adenoviral Vector for Gene Therapy and Vaccine Product Development. J. Virol. 2021, 95, e0038721. [Google Scholar] [CrossRef]
- Rotmans, J.I.; Verhagen, H.J.; Velema, E.; de Kleijn, D.P.; van den Heuvel, M.; Kastelein, J.J.; Pasterkamp, G.; Stroes, E.S. Local overexpression of C-type natriuretic peptide ameliorates vascular adaptation of porcine hemodialysis grafts. Kidney Int. 2004, 65, 1897–1905. [Google Scholar] [CrossRef] [Green Version]
- van Rooij, E.; Sutherland, L.B.; Thatcher, J.E.; DiMaio, J.M.; Naseem, R.H.; Marshall, W.S.; Hill, J.A.; Olson, E.N. Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13027–13032. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Huang, A.; Ferruzzi, J.; Mecham, R.P.; Starcher, B.C.; Tellides, G.; Humphrey, J.D.; Giordano, F.J.; Niklason, L.E.; Sessa, W.C. Inhibition of MicroRNA-29 Enhances Elastin Levels in Cells Haploinsufficient for Elastin and in Bioengineered Vessels—Brief Report. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 756–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothuizen, T.C.; Kemp, R.; Duijs, J.M.; de Boer, H.C.; Bijkerk, R.; van der Veer, E.P.; Moroni, L.; van Zonneveld, A.J.; Weiss, A.S.; Rabelink, T.J.; et al. Promoting Tropoelastin Expression in Arterial and Venous Vascular Smooth Muscle Cells and Fibroblasts for Vascular Tissue Engineering. Tissue Eng. Part C Methods 2016, 22, 923–931. [Google Scholar] [CrossRef] [PubMed]
- Sudo, R.; Sato, F.; Azechi, T.; Wachi, H. MiR-29-mediated elastin down-regulation contributes to inorganic phosphorus-induced osteoblastic differentiation in vascular smooth muscle cells. Genes Cells 2015, 20, 1077–1087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kilari, S.; Cai, C.; Zhao, C.; Sharma, A.; Chernogubova, E.; Simeon, M.; Wu, C.C.; Song, H.L.; Maegdefessel, L.; Misra, S. The Role of MicroRNA-21 in Venous Neointimal Hyperplasia: Implications for Targeting miR-21 for VNH Treatment. Mol. Ther. 2019, 27, 1681–1693. [Google Scholar] [CrossRef] [Green Version]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laboyrie, S.L.; de Vries, M.R.; Bijkerk, R.; Rotmans, J.I. Building a Scaffold for Arteriovenous Fistula Maturation: Unravelling the Role of the Extracellular Matrix. Int. J. Mol. Sci. 2023, 24, 10825. https://doi.org/10.3390/ijms241310825
Laboyrie SL, de Vries MR, Bijkerk R, Rotmans JI. Building a Scaffold for Arteriovenous Fistula Maturation: Unravelling the Role of the Extracellular Matrix. International Journal of Molecular Sciences. 2023; 24(13):10825. https://doi.org/10.3390/ijms241310825
Chicago/Turabian StyleLaboyrie, Suzanne L., Margreet R. de Vries, Roel Bijkerk, and Joris I. Rotmans. 2023. "Building a Scaffold for Arteriovenous Fistula Maturation: Unravelling the Role of the Extracellular Matrix" International Journal of Molecular Sciences 24, no. 13: 10825. https://doi.org/10.3390/ijms241310825