
CSB Regulates Pathway Choice in Response to DNA Replication Stress Induced by Camptothecin

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
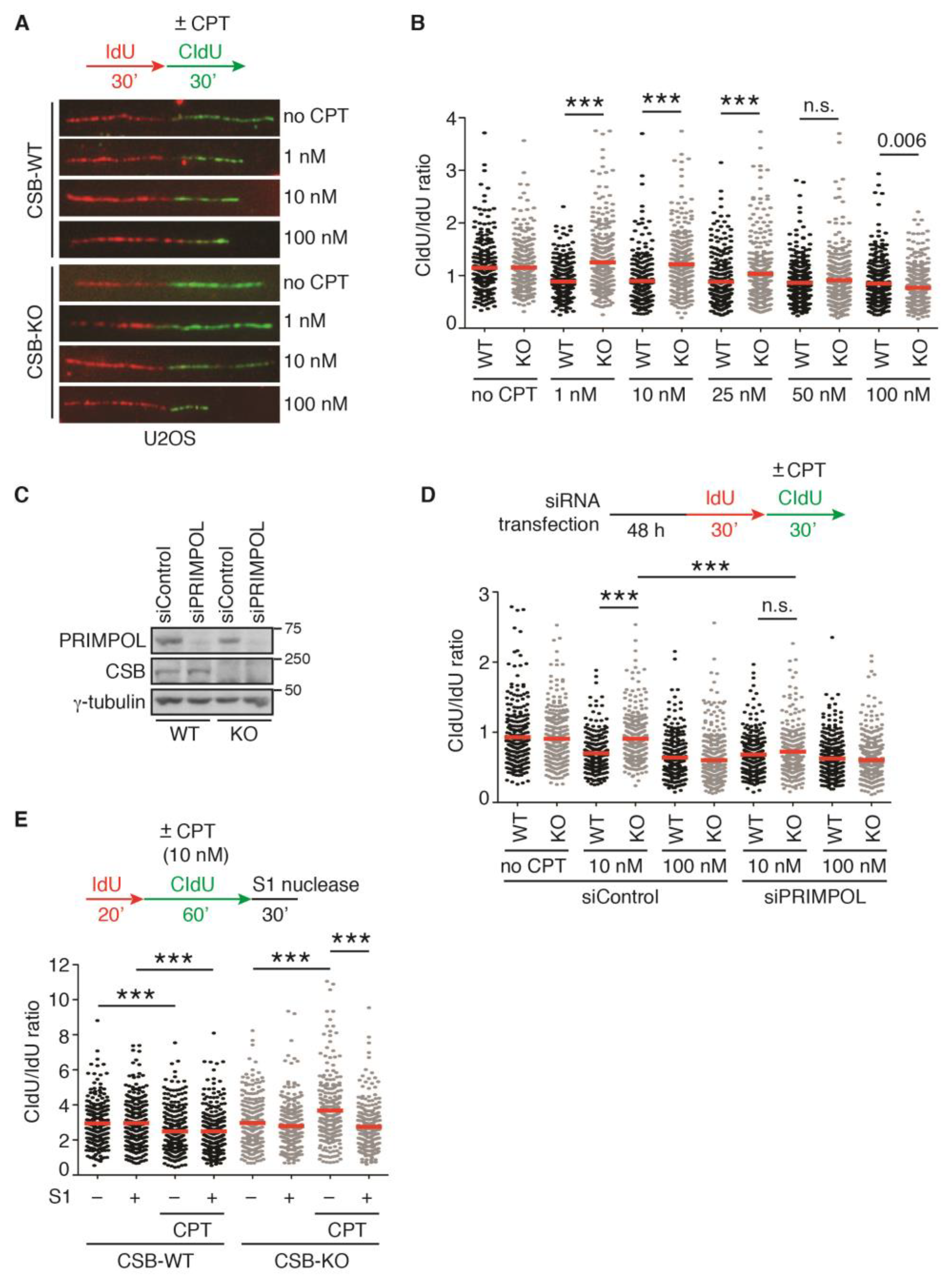
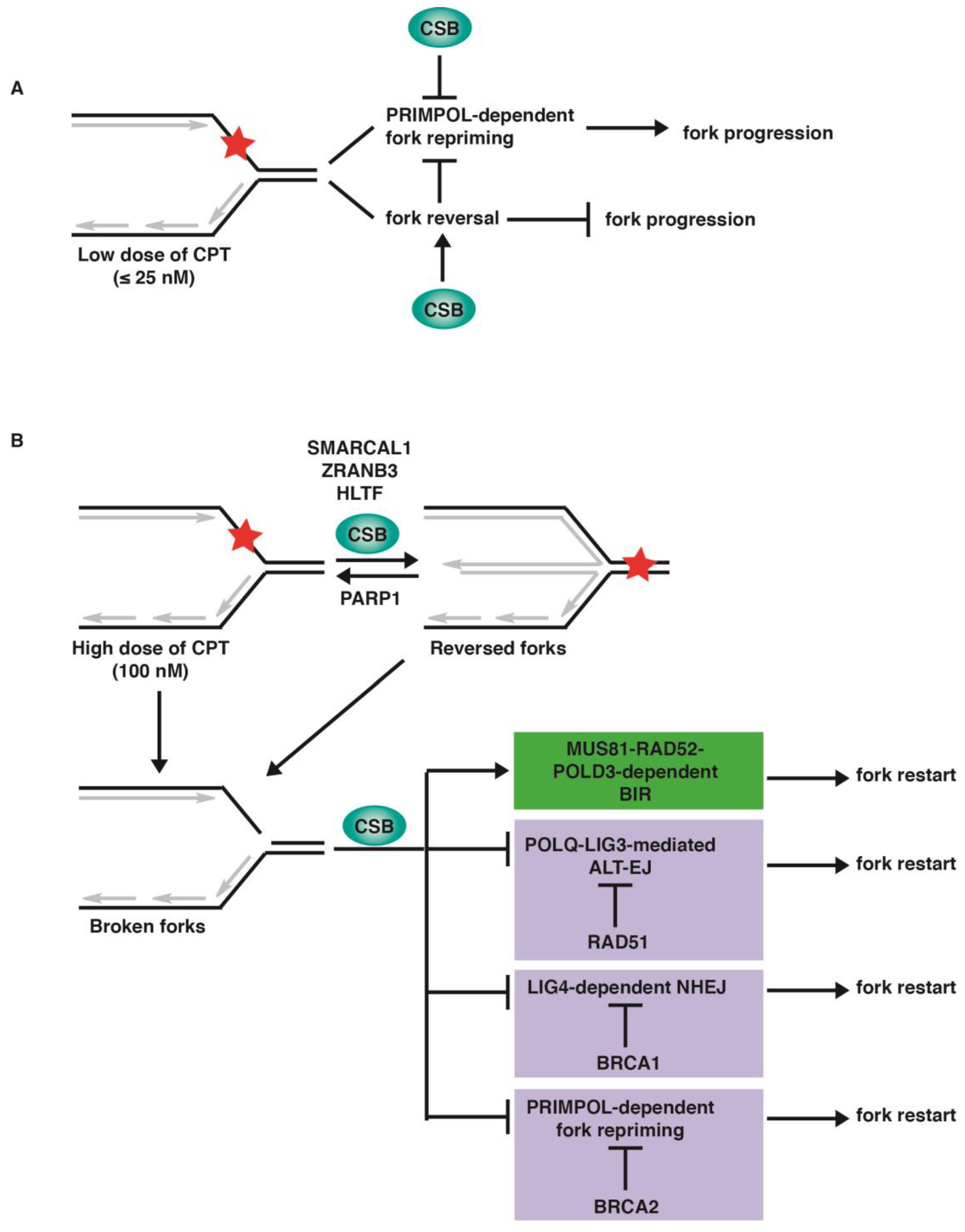
2.1. Loss of CSB Leads to Unrestrained Fork Progression in Response to CPT at Low Concentrations
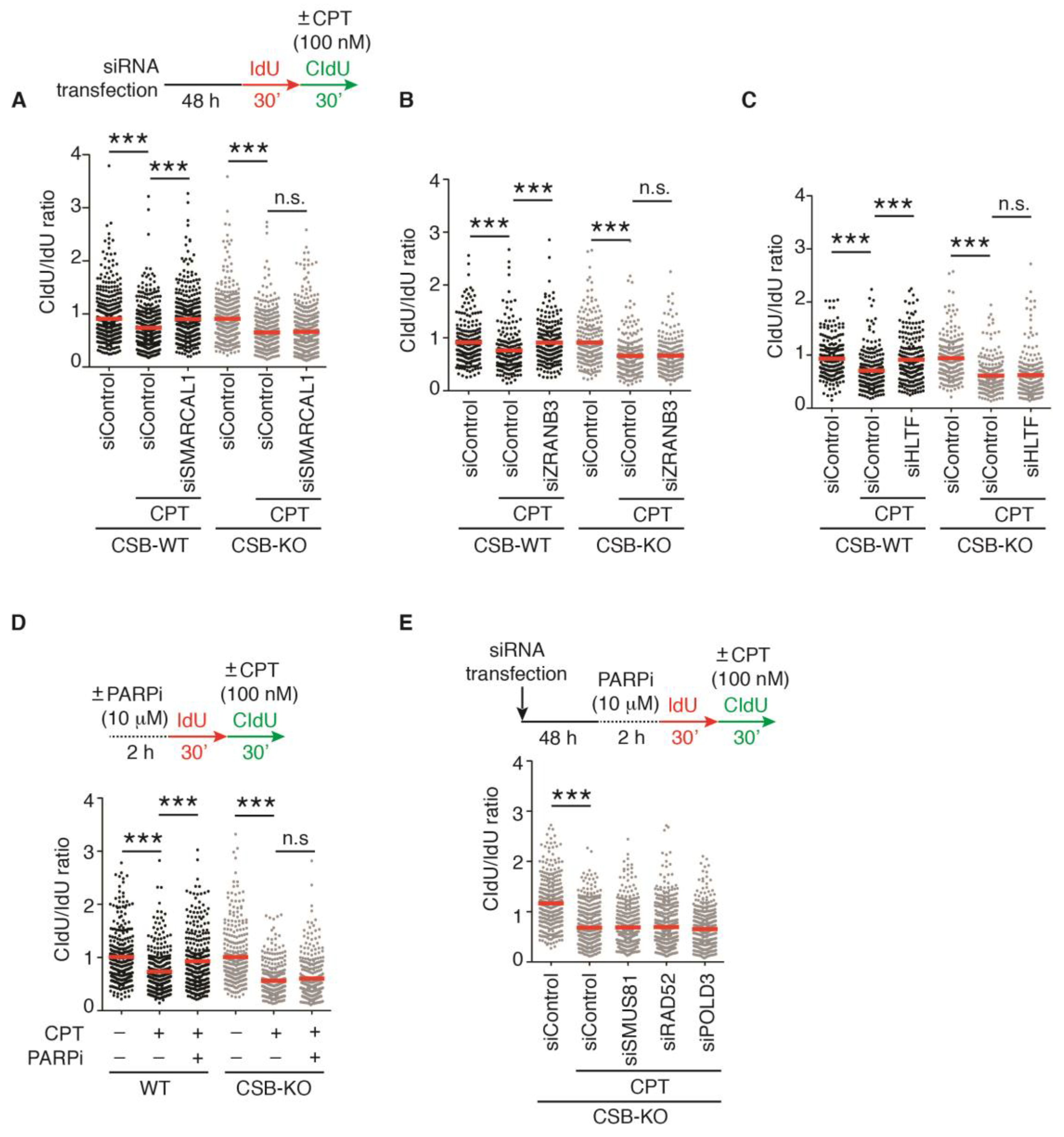
2.2. Loss of Fork Reversal Is Insufficient to Allow Restart of DNA Synthesis in CSB-KO Cells following 100 nM CPT-Induced Fork Stalling
2.3. CSB Is Epistatic to MUS81/RAD52/POLD3 to Restart DNA Replication in Response to a High Dose of CPT
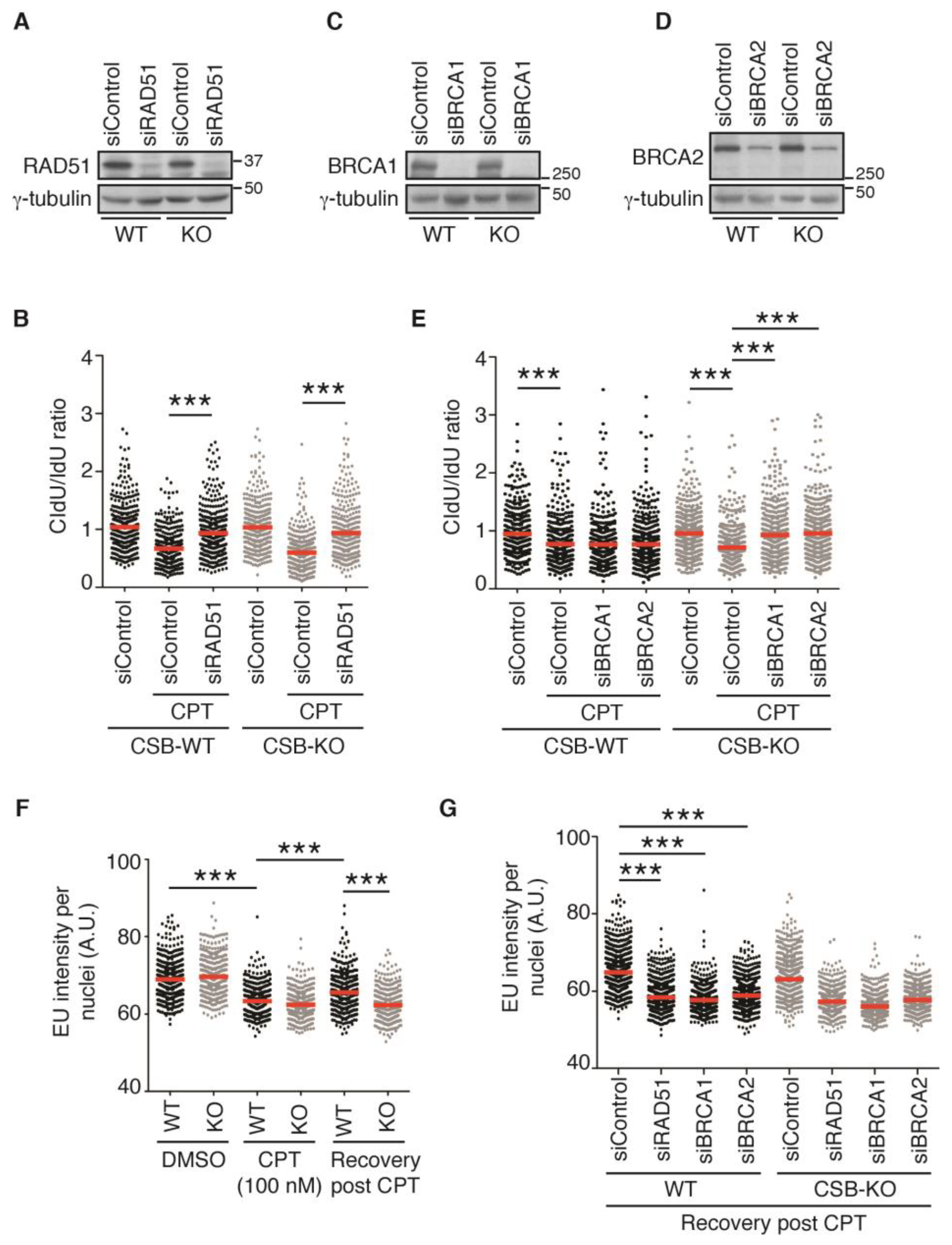
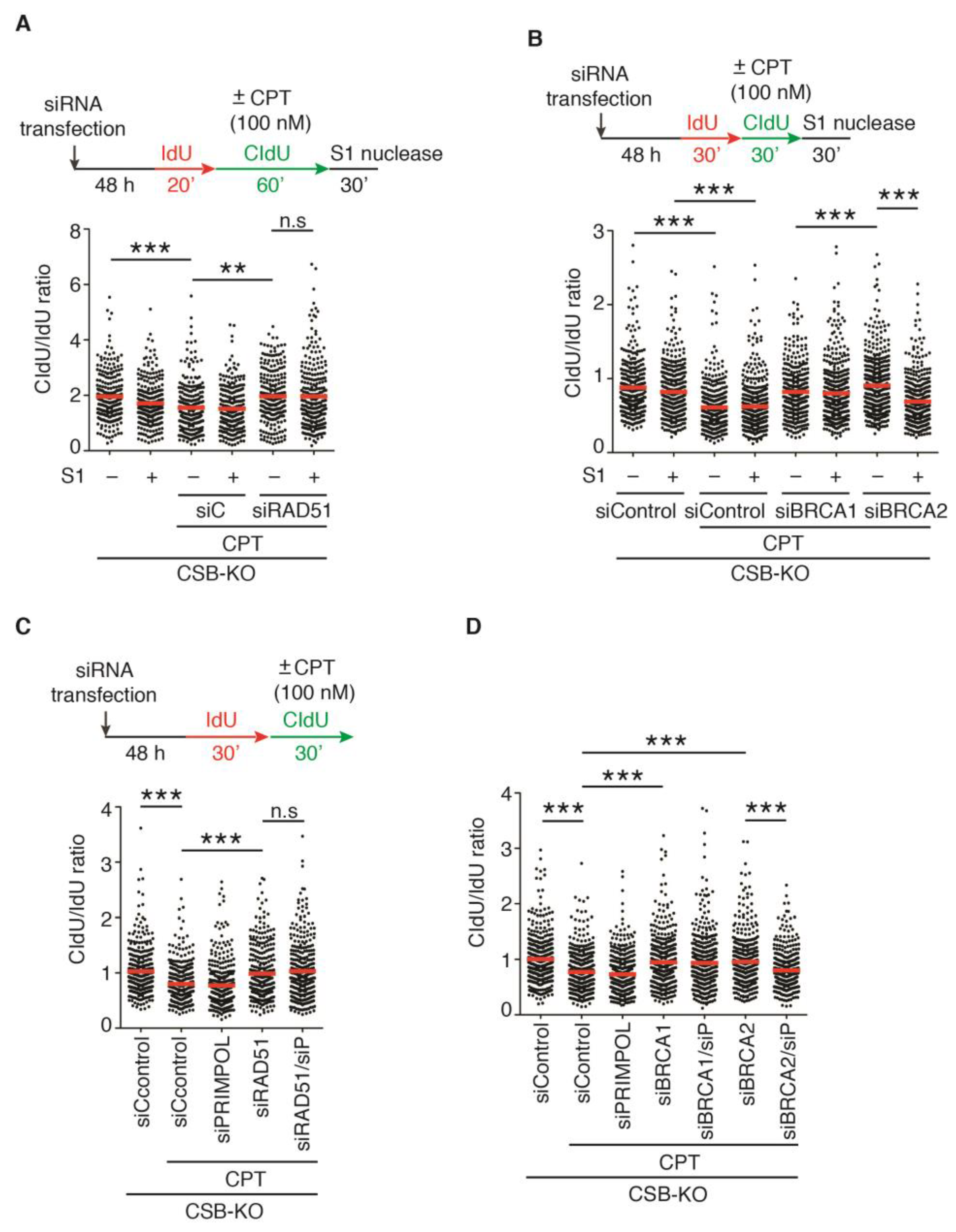
2.4. Depletion of RAD51, BRCA1, or BRCA2 Restarts DNA Replication upon Treatment with 100 nM CPT in the Absence of CSB
2.5. Transcription Recovery Is Not Required for Restart of DNA Replication upon Treatment with 100 nM CPT in CSB-KO Cells Lacking RAD51, BRCA1, or BRCA2
2.6. PRIMPOL Mediates Restart of DNA Replication upon Treatment with 100 nM CPT in CSB-KO Cells Depleted for BRCA2 but Not RAD51 or BRCA1
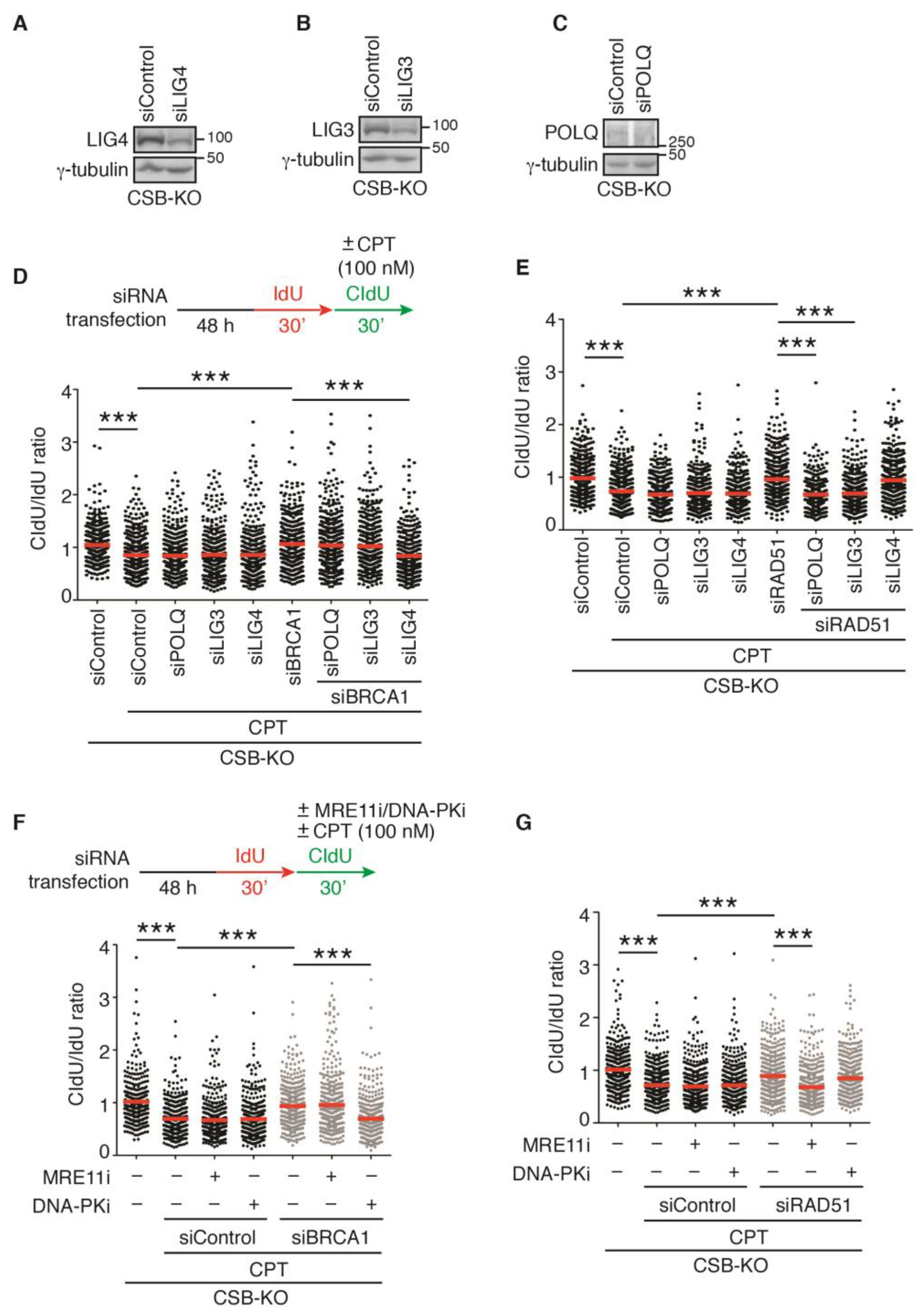
2.7. LIG3 and LIG4, Respectively, Mediate Restart of DNA Replication upon Treatment with 100 nM CPT in RAD51- and BRCA1-Depleted CSB-KO Cells
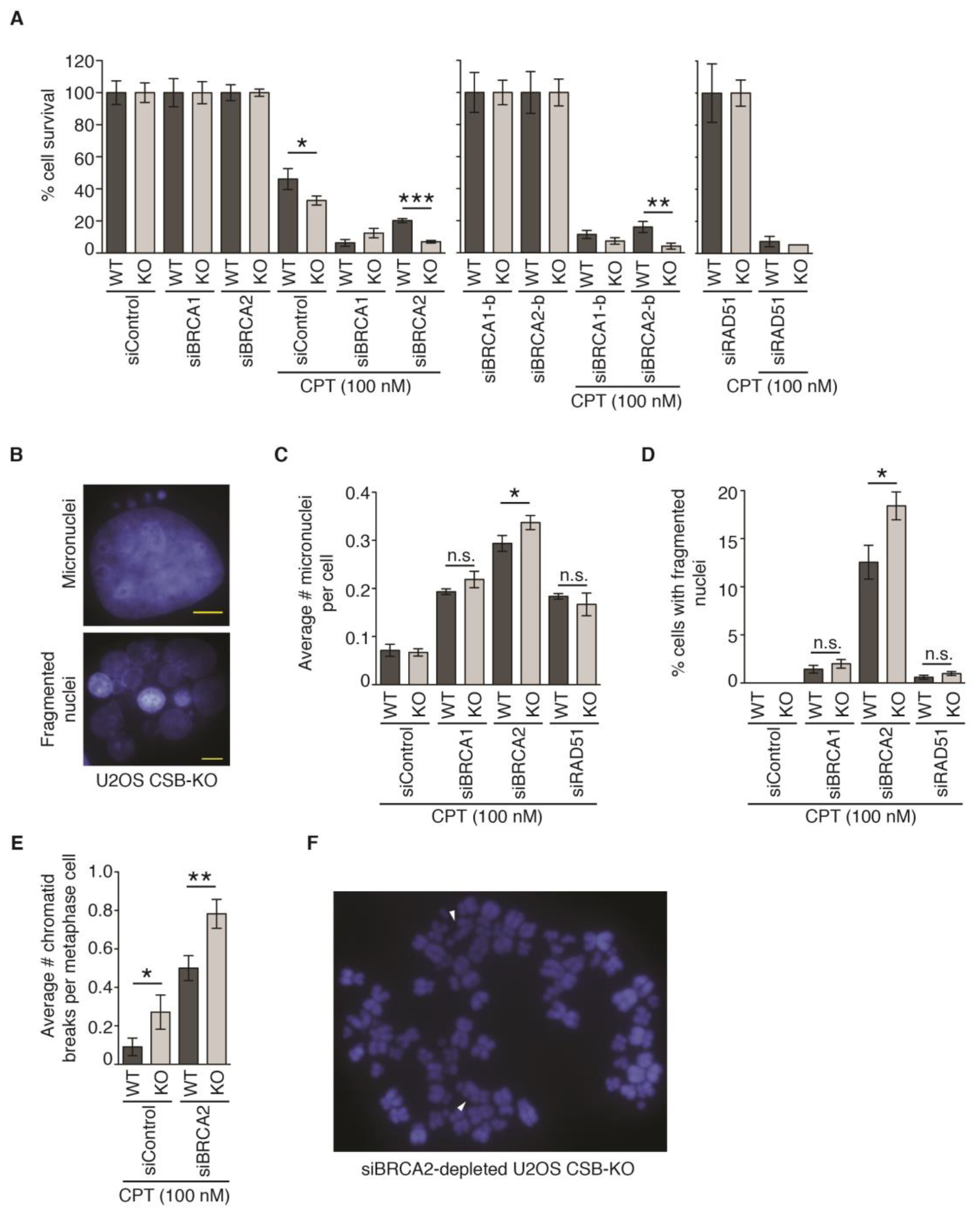
2.8. CSB and BRCA2 Are a Toxic Combination to Genomic Stability and Cell Survival in Response to Treatment with 100 nM CPT
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Transfection
4.2. Small Interfering RNAs, Antibodies, and Drugs
4.3. DNA Fiber Assay
4.4. S1 Nuclease Assay
4.5. Transcriptional Recovery Assay (EU Incorporation)
4.6. Metaphase Chromosome Spreads
4.7. Immunoblotting
4.8. Clonogenic Survival Assays
4.9. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Champoux, J.J. DNA topoisomerases: Structure, function, and mechanism. Annu. Rev. Biochem. 2001, 70, 369–413. [Google Scholar] [CrossRef]
- Wang, J.C. Cellular roles of DNA topoisomerases: A molecular perspective. Nat. Rev. Mol. Cell Biol. 2002, 3, 430–440. [Google Scholar] [CrossRef]
- Liu, L.F.; Desai, S.D.; Li, T.K.; Mao, Y.; Sun, M.; Sim, S.P. Mechanism of action of camptothecin. Ann. N. Y. Acad. Sci. 2000, 922, 1–10. [Google Scholar] [CrossRef]
- Pommier, Y. Topoisomerase I inhibitors: Camptothecins and beyond. Nat. Rev. Cancer 2006, 6, 789–802. [Google Scholar] [CrossRef] [PubMed]
- Hsiang, Y.H.; Lihou, M.G.; Liu, L.F. Arrest of replication forks by drug-stabilized topoisomerase I-DNA cleavage complexes as a mechanism of cell killing by camptothecin. Cancer Res. 1989, 49, 5077–5082. [Google Scholar] [PubMed]
- Beretta, G.L.; Gatti, L.; Perego, P.; Zaffaroni, N. Camptothecin resistance in cancer: Insights into the molecular mechanisms of a DNA-damaging drug. Curr. Med. Chem. 2013, 20, 1541–1565. [Google Scholar] [CrossRef]
- Ray Chaudhuri, A.; Hashimoto, Y.; Herrador, R.; Neelsen, K.J.; Fachinetti, D.; Bermejo, R.; Cocito, A.; Costanzo, V.; Lopes, M. Topoisomerase I poisoning results in PARP-mediated replication fork reversal. Nat. Struct. Mol. Biol. 2012, 19, 417–423. [Google Scholar] [CrossRef]
- Berti, M.; Ray Chaudhuri, A.; Thangavel, S.; Gomathinayagam, S.; Kenig, S.; Vujanovic, M.; Odreman, F.; Glatter, T.; Graziano, S.; Mendoza-Maldonado, R.; et al. Human RECQ1 promotes restart of replication forks reversed by DNA topoisomerase I inhibition. Nat. Struct. Mol. Biol. 2013, 20, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, J.; McGlynn, P. Replication fork reversal and the maintenance of genome stability. Nucleic Acids Res. 2009, 37, 3475–3492. [Google Scholar] [CrossRef]
- Neelsen, K.J.; Lopes, M. Replication fork reversal in eukaryotes: From dead end to dynamic response. Nat. Rev. Mol. Cell Biol. 2015, 16, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Quinet, A.; Tirman, S.; Cybulla, E.; Meroni, A.; Vindigni, A. To skip or not to skip: Choosing repriming to tolerate DNA damage. Mol. Cell 2021, 81, 649–658. [Google Scholar] [CrossRef]
- Walker, J.R.; Zhu, X.-D. Role of Cockayne Syndrome Group B Protein in Replication Stress: Implications for Cancer Therapy. Int. J. Mol. Sci. 2022, 23, 10212. [Google Scholar] [CrossRef] [PubMed]
- Joseph, S.A.; Taglialatela, A.; Leuzzi, G.; Huang, J.W.; Cuella-Martin, R.; Ciccia, A. Time for remodeling: SNF2-family DNA translocases in replication fork metabolism and human disease. DNA Repair. 2020, 95, 102943. [Google Scholar] [CrossRef] [PubMed]
- Zellweger, R.; Dalcher, D.; Mutreja, K.; Berti, M.; Schmid, J.A.; Herrandor, R.; Vindigni, A.; Lopes, M. Rad51-mediated replication fork reversal is a global response to genotoxic treatments in human cells. J. Cell Biol. 2015, 208, 563–579. [Google Scholar] [CrossRef] [PubMed]
- Bétous, R.; Mason, A.C.; Rambo, R.P.; Bansbach, C.E.; Badu-Nkansah, A.; Sirbu, B.M.; Eichman, B.F.; Cortez, D. SMARCAL1 catalyzes fork regression and Holliday junction migration to maintain genome stability during DNA replication. Genes Dev. 2012, 26, 151–162. [Google Scholar] [CrossRef]
- Bai, G.; Kermi, C.; Stoy, H.; Schiltz, C.J.; Bacal, J.; Zaino, A.M.; Hadden, M.K.; Eichman, B.F.; Lopes, M.; Cimprich, K.A. HLTF Promotes Fork Reversal, Limiting Replication Stress Resistance and Preventing Multiple Mechanisms of Unrestrained DNA Synthesis. Mol. Cell 2020, 78, 1237–1251.e7. [Google Scholar] [CrossRef]
- Vujanovic, M.; Krietsch, J.; Raso, M.C.; Terraneo, N.; Zellweger, R.; Schmid, J.A.; Taglialatela, A.; Huang, J.W.; Holland, C.L.; Zwicky, K.; et al. Replication Fork Slowing and Reversal upon DNA Damage Require PCNA Polyubiquitination and ZRANB3 DNA Translocase Activity. Mol. Cell 2017, 67, 882–890.e5. [Google Scholar] [CrossRef]
- Batenburg, N.L.; Mersaoui, S.Y.; Walker, J.R.; Coulombe, Y.; Hammond-Martel, I.; Wurtele, H.; Masson, J.Y.; Zhu, X.D. Cockayne syndrome group B protein regulates fork restart, fork progression, and MRE11-dependent fork degradation in BRCA1/2-deficient cells. Nucleic Acid. Res. 2021, 49, 12836–12854. [Google Scholar] [CrossRef]
- Chappidi, N.; Nascakova, Z.; Boleslavska, B.; Zellweger, R.; Isik, E.; Andrs, M.; Menon, S.; Dobrovolna, J.; Balbo Pogliano, C.; Matos, J.; et al. Fork Cleavage-Religation Cycle and Active Transcription Mediate Replication Restart after Fork Stalling at Co-transcriptional R-Loops. Mol. Cell 2020, 77, 528–541. [Google Scholar] [CrossRef]
- Tiwari, V.; Baptiste, B.A.; Okur, M.N.; Bohr, V.A. Current and emerging roles of Cockayne syndrome group B (CSB) protein. Nucleic Acids Res. 2021, 49, 2418–2434. [Google Scholar] [CrossRef]
- Troelstra, C.; van Gool, A.; de Wit, J.; Vermeulen, W.; Bootsma, D.; Hoeijmakers, J.H. ERCC6, a member of a subfamily of putative helicases, is involved in Cockayne’s syndrome and preferential repair of active genes. Cell 1992, 71, 939–953. [Google Scholar] [CrossRef] [PubMed]
- Batenburg, N.L.; Walker, J.R.; Noordermeer, S.M.; Moatti, N.; Durocher, D.; Zhu, X.D. ATM and CDK2 control chromatin remodeler CSB to inhibit RIF1 in DSB repair pathway choice. Nat. Commun. 2017, 8, 1921. [Google Scholar] [CrossRef] [PubMed]
- Batenburg, N.L.; Walker, J.R.; Coulombe, Y.; Sherker, A.; Masson, J.Y.; Zhu, X.D. CSB interacts with BRCA1 in late S/G2 to promote MRN- and CtIP-mediated DNA end resection. Nucleic Acids Res. 2019, 47, 10678–10692. [Google Scholar] [CrossRef] [PubMed]
- Batenburg, N.L.; Thompson, E.L.; Hendrickson, E.A.; Zhu, X.D. Cockayne syndrome group B protein regulates DNA double-strand break repair and checkpoint activation. EMBO J. 2015, 34, 1399–1416. [Google Scholar] [CrossRef]
- Taglialatela, A.; Alvarez, S.; Leuzzi, G.; Sannino, V.; Ranjha, L.; Huang, J.W.; Madubata, C.; Anand, R.; Levy, B.; Rabadan, R.; et al. Restoration of Replication Fork Stability in BRCA1- and BRCA2-Deficient Cells by Inactivation of SNF2-Family Fork Remodelers. Mol. Cell 2017, 68, 414–430. [Google Scholar] [CrossRef]
- Mijic, S.; Zellweger, R.; Chappidi, N.; Berti, M.; Jacobs, K.; Mutreja, K.; Ursich, S.; Ray Chaudhuri, A.; Nussenzweig, A.; Janscak, P.; et al. Replication fork reversal triggers fork degradation in BRCA2-defective cells. Nat. Commun. 2017, 8, 859. [Google Scholar] [CrossRef] [PubMed]
- Kolinjivadi, A.M.; Sannino, V.; De Antoni, A.; Zadorozhny, K.; Kilkenny, M.; Técher, H.; Baldi, G.; Shen, R.; Ciccia, A.; Pellegrini, L.; et al. Smarcal1-Mediated Fork Reversal Triggers Mre11-Dependent Degradation of Nascent DNA in the Absence of Brca2 and Stable Rad51 nucleofilaments. Mol. Cell 2017, 67, 867–881.e7. [Google Scholar] [CrossRef]
- Bhowmick, R.; Minocherhomji, S.; Hickson, I.D. RAD52 Facilitates Mitotic DNA Synthesis Following Replication Stress. Mol. Cell 2016, 64, 1117–1126. [Google Scholar] [CrossRef]
- Cui, S.; Walker, J.R.; Batenburg, N.L.; Zhu, X.-D. Cockayne syndrome group B protein uses its DNA translocase activity to promote mitotic DNA synthesis. DNA Repair. 2022, 116, 103354. [Google Scholar] [CrossRef] [PubMed]
- Taglialatela, A.; Leuzzi, G.; Sannino, V.; Cuella-Martin, R.; Huang, J.W.; Wu-Baer, F.; Baer, R.; Costanzo, V.; Ciccia, A. REV1-Polζ maintains the viability of homologous recombination-deficient cancer cells through mutagenic repair of RPIMOL-dependent ssDNA gaps. Mol. Cell 2021, 81, 4008–4025.e7. [Google Scholar] [CrossRef]
- Tirman, S.; Quinet, A.; Wood, M.; Meroni, A.; Cybulla, E.; Jackson, J.; Pegoraro, S.; Simoneau, A.; Zou, L.; Vindigni, A. Temporally distinct post-replicative repair mechanisms fill PRIMPOL-dependent ssDNA gaps in human cells. Mol. Cell 2021, 81, 4026–4040.e8. [Google Scholar] [CrossRef]
- Piberger, A.L.; Bowry, A.; Kelly, R.D.W.; Walker, A.K.; González-Acosta, D.; Bailey, L.J.; Doherty, A.J.; Méndez, J.; Morris, J.R.; Bryant, H.E.; et al. PrimPol-dependent single-stranded gap formation mediates homologous recombination at bulky DNA adducts. Nat. Commun. 2020, 11, 5863. [Google Scholar] [CrossRef] [PubMed]
- Tye, S.; Ronson, G.E.; Morris, J.R. A fork in the road: Where homologous recombination and stalled replication fork protection part ways. Semin. Cell Dev. Biol. 2021, 113, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Velez-Cruz, R.; Egly, J.M. Cockayne syndrome group B (CSB) protein: At the crossroads of transcriptional networks. Mech. Ageing Dev. 2013, 134, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Kang, Z.; Fu, P.; Alcivar, A.L.; Fu, H.; Redon, C.; Foo, T.K.; Zuo, Y.; Ye, C.; Baxley, R.; Madireddy, A.; et al. BRCA2 associates with MCM10 to suppress PRIMPOL-mediated repriming and single-stranded gap formation after DNA damage. Nat. Commun. 2021, 12, 5966. [Google Scholar] [CrossRef]
- Stok, C.; Kok, Y.P.; van den Tempel, N.; van Vugt, M.A.T.M. Shaping the BRCAness mutational landscape by alternative double-strand break repair, replication stress and mitotic aberrancies. Nucleic Acid. Res. 2021, 49, 4239–4257. [Google Scholar] [CrossRef]
- Audoynaud, C.; Vagner, S.; Lambert, S. Non-homologous end-joining at challenged replication forks: An RNA connection? Trends Genet. 2021, 37, 973–985. [Google Scholar] [CrossRef]
- Ho, Y.C.; Ku, C.S.; Tsai, S.S.; Shiu, J.L.; Jiang, Y.Z.; Mirian, H.E.; Zhang, H.W.; Chen, Y.T.; Chiu, W.T.; Chang, S.B.; et al. PARP1 recruits DNA translocases to restrain DNA replication and facilitate DNA repair. PLoS Genet. 2022, 18, e1010545. [Google Scholar] [CrossRef]
- Quinet, A.; Tirman, S.; Jackson, J.; Šviković, S.; Lemaçon, D.; Carvajal-Maldonado, D.; González-Acosta, D.; Vessoni, A.T.; Cybulla, E.; Wood, M.; et al. PRIMPOL-Mediated Adaptive Response Suppresses Replication Fork Reversal in BRCA-Deficient Cells. Mol. Cell 2020, 77, 461–474.e9. [Google Scholar] [CrossRef]
- Vallerga, M.B.; Mansilla, S.F.; Federico, M.B.; Bertolin, A.P.; Gottifredi, V. Rad51 recombinase prevents Mre11 nuclease-dependent degradation and excessive PrimPol-mediated elongation of nascent DNA after UV irradiation. Proc. Natl. Acad. Sci. USA 2015, 112, E6624–E6633. [Google Scholar] [CrossRef]
- Duan, H.; Mansour, S.; Reed, R.; Gillis, M.K.; Parent, B.; Liu, B.; Sztupinszki, Z.; Birkbak, N.; Szallasi, Z.; Elia, A.E.H.; et al. E3 ligase RFWD3 is a novel modulator of stalled fork stability in BRCA2-deficient cells. J. Cell Biol. 2020, 219, e201908192. [Google Scholar] [CrossRef] [PubMed]
- Cong, K.; Peng, M.; Kousholt, A.N.; Lee, W.T.C.; Lee, S.; Nayak, S.; Krais, J.; VanderVere-Carozza, P.S.; Pawelczak, K.S.; Calvo, J.; et al. Replication gaps are a key determinant of PARP inhibitor synthetic lethality with BRCA deficiency. Mol. Cell 2021, 81, 3128–3144.e7. [Google Scholar] [CrossRef] [PubMed]
- Proietti-De-Santis, L.; Balzerano, A.; Prantera, G. CSB: An Emerging Actionable Target for Cancer Therapy. Trends Cancer 2018, 4, 172–175. [Google Scholar] [CrossRef]
- Feng, E.; Batenburg, N.L.; Walker, J.R.; Ho, A.; Mitchell, T.R.H.; Qin, J.; Zhu, X.D. CSB cooperates with SMARCAL1 to maintain telomere stability in ALT cells. J. Cell Sci. 2020, 133, jcs234914. [Google Scholar] [CrossRef]
- Zhu, X.D.; Kuster, B.; Mann, M.; Petrini, J.H.; Lange, T. Cell-cycle-regulated association of RAD50/MRE11/NBS1 with TRF2 and human telomeres. Nat. Genet. 2000, 25, 347–352. [Google Scholar]
- Mitchell, T.R.; Glenfield, K.; Jeyanthan, K.; Zhu, X.D. Arginine methylation regulates telomere length and stability. Mol. Cell Biol. 2009, 29, 4918–4934. [Google Scholar] [CrossRef] [PubMed]
- Batenburg, N.L.; Qin, J.; Walker, J.R.; Zhu, X.D. Efficient UV repair requires disengagement of the CSB winged helix domain from the CSB ATPase domain. DNA Repair. 2018, 68, 58–67. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Batenburg, N.L.; Walker, J.R.; Zhu, X.-D. CSB Regulates Pathway Choice in Response to DNA Replication Stress Induced by Camptothecin. Int. J. Mol. Sci. 2023, 24, 12419. https://doi.org/10.3390/ijms241512419
Batenburg NL, Walker JR, Zhu X-D. CSB Regulates Pathway Choice in Response to DNA Replication Stress Induced by Camptothecin. International Journal of Molecular Sciences. 2023; 24(15):12419. https://doi.org/10.3390/ijms241512419
Chicago/Turabian StyleBatenburg, Nicole L., John R. Walker, and Xu-Dong Zhu. 2023. "CSB Regulates Pathway Choice in Response to DNA Replication Stress Induced by Camptothecin" International Journal of Molecular Sciences 24, no. 15: 12419. https://doi.org/10.3390/ijms241512419
APA StyleBatenburg, N. L., Walker, J. R., & Zhu, X.-D. (2023). CSB Regulates Pathway Choice in Response to DNA Replication Stress Induced by Camptothecin. International Journal of Molecular Sciences, 24(15), 12419. https://doi.org/10.3390/ijms241512419