
Fighting Pseudomonas aeruginosa Infections: Antibacterial and Antibiofilm Activity of D-Q53 CecB, a Synthetic Analog of a Silkworm Natural Cecropin B Variant
 , , , , and
, , , , and
Abstract
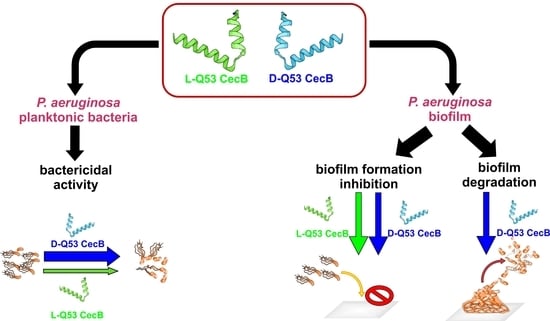
:
1. Introduction
2. Results
2.1. L- and D-Q53 CecB Sensitivity to Degradation by Human and P. aeruginosa Elastases
2.2. L- and D-Q53 CecB Antimicrobial Activity against P. aeruginosa Planktonic Bacteria
2.3. Effects of Cations, Serum, and Heat Treatments on the L- and D-Q53 CecB Anti-Pseudomonas Activity
2.4. L- and D-Q53 CecB Inhibitory Effects on P. aeruginosa Biofilm Formation
2.5. L- and D-Q53 CecB Degradation Activities on Pre-Formed P. aeruginosa Biofilm
2.6. Exploring L- and D-Q53 CecB Mechanisms Inhibiting P. aeruginosa Biofilm Formation
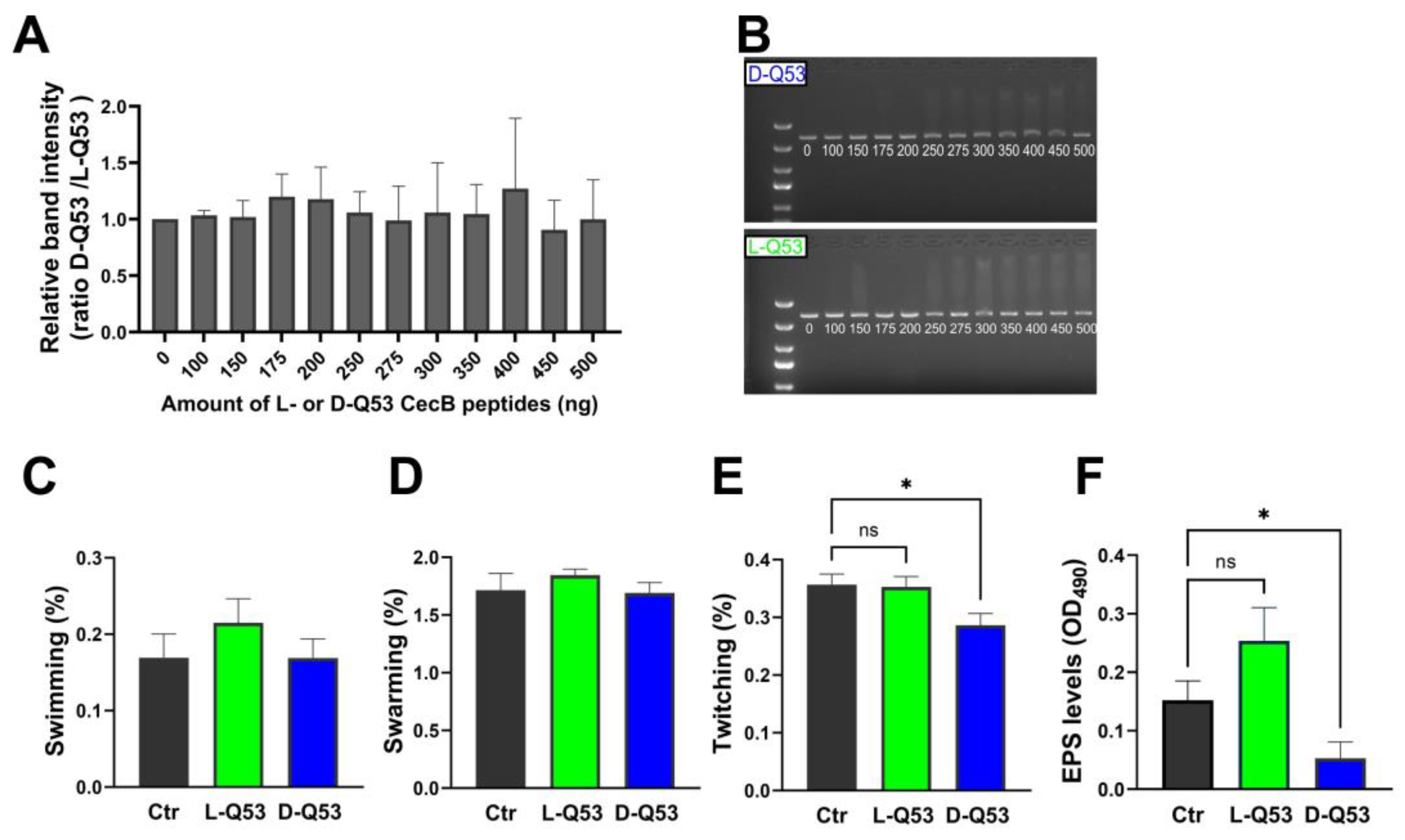
2.6.1. In Vitro Interaction with DNA
2.6.2. Peptides’ Effects on Bacterial Motility and EPS Production
2.6.3. L- and D-Q53 CecB Effects on Transcription of Representative Genes Involved in Biofilm Formation
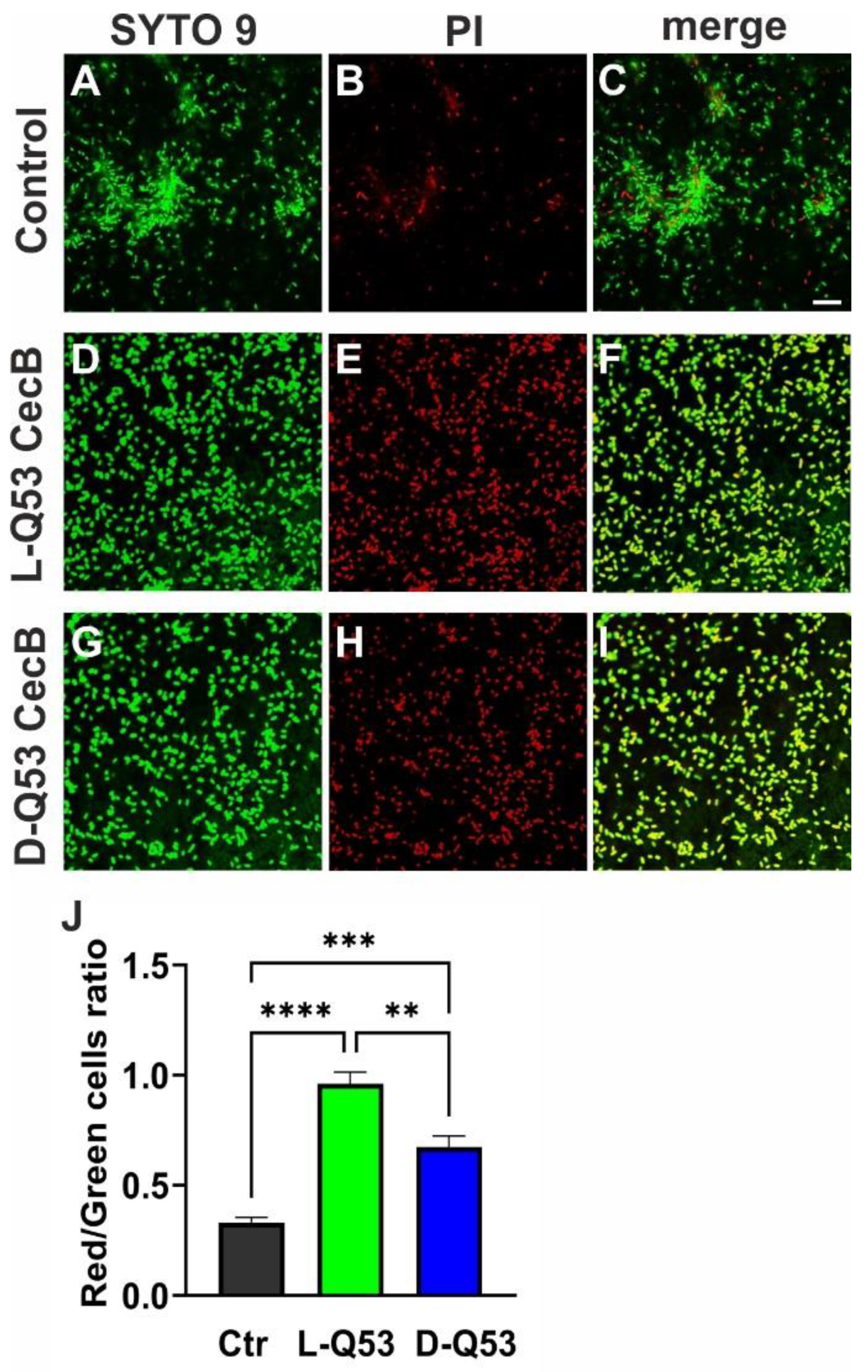
2.7. Exploring L- and D-Q53 CecB Effects on Pre-Formed P. aeruginosa Biofilm
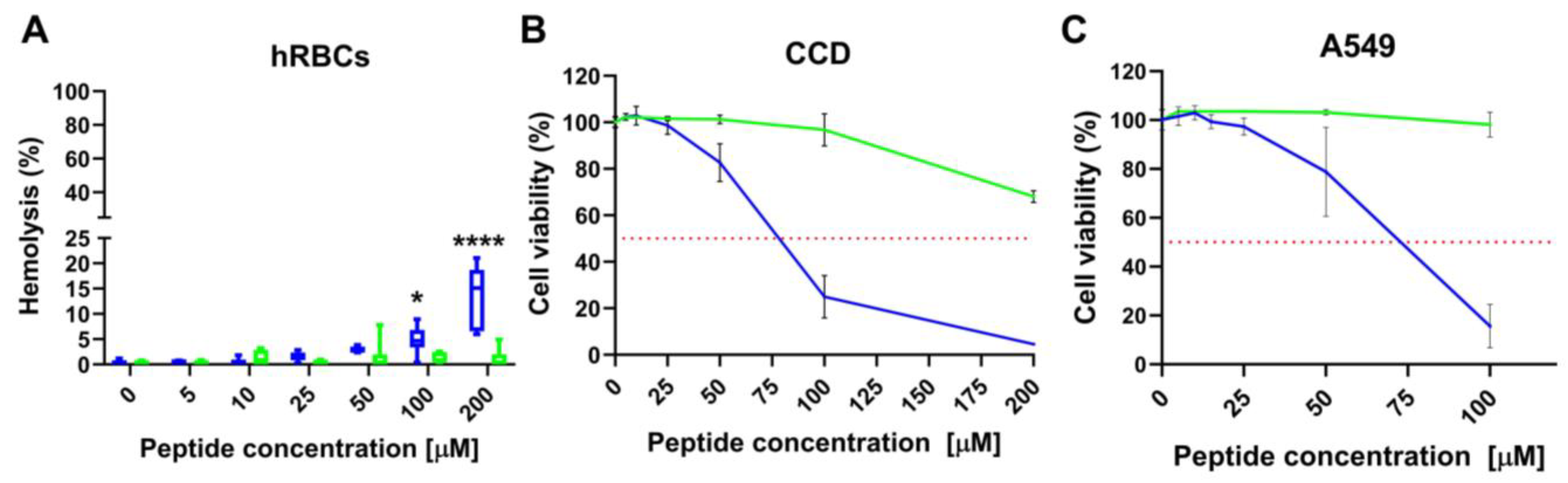
2.8. L- and D-Q53 CecB Hemolytic and Cytotoxic Activity
3. Discussion
4. Materials and Methods
4.1. Microbial Strains and Culture Conditions
4.2. Peptides Synthesis
4.3. Circular Dichroism Analysis
4.4. Enzymatic Digestion of Peptides
4.5. MIC and MBC Tests
4.6. Resistance of L- and D-Q53 CecB to Salts, Serum, and Heat Treatments
4.7. Inhibition of Biofilm Formation
4.8. Biofilm Degradation
4.9. In Vitro Interactions between DNA and CecB Peptides
4.10. P. aeruginosa Growth Curves with sub-MIC Concentrations of L- and D-Q53 CecB Peptides
4.11. Motility Assay
4.12. EPS Determination
4.13. Real-Time qPCR
4.14. Confocal Fluorescence Microscopy
4.15. Hemolytic Assay
4.16. Cytotoxicity Assay
4.17. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- De Oliveira, D.M.P.; Forde, B.M.; Kidd, T.J.; Harris, P.N.A.; Schembri, M.A.; Beatson, S.A.; Paterson, D.L.; Walker, M.J. Antimicrobial Resistance in ESKAPE Pathogens. Clin. Microbiol. Rev. 2020, 33, e00181-19. [Google Scholar] [CrossRef] [PubMed]
- Saiman, L.; Siegel, J. Infection Control in Cystic Fibrosis. Clin. Microbiol. Rev. 2004, 17, 57–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pang, Z.; Raudonis, R.; Glick, B.R.; Lin, T.-J.; Cheng, Z. Antibiotic Resistance in Pseudomonas aeruginosa: Mechanisms and Alternative Therapeutic Strategies. Biotechnol. Adv. 2019, 37, 177–192. [Google Scholar] [CrossRef] [PubMed]
- Cascioferro, S.; Parrino, B.; Carbone, D.; Pecoraro, C.; Diana, P. Novel Strategies in the War against Antibiotic Resistance. Future Med. Chem. 2021, 13, 529–531. [Google Scholar] [CrossRef]
- Rabin, N.; Zheng, Y.; Opoku-Temeng, C.; Du, Y.; Bonsu, E.; Sintim, H.O. Biofilm Formation Mechanisms and Targets for Developing Antibiofilm Agents. Future Med. Chem. 2015, 7, 493–512. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Yoon, S.S. Pseudomonas aeruginosa Biofilm, a Programmed Bacterial Life for Fitness. J. Microbiol. Biotechnol. 2017, 27, 1053–1064. [Google Scholar] [CrossRef] [Green Version]
- Wood, T.K.; Knabel, S.J.; Kwan, B.W. Bacterial Persister Cell Formation and Dormancy. Appl. Environ. Microbiol. 2013, 79, 7116–7121. [Google Scholar] [CrossRef] [Green Version]
- Verderosa, A.D.; Totsika, M.; Fairfull-Smith, K.E. Bacterial Biofilm Eradication Agents: A Current Review. Front. Chem. 2019, 7, 824. [Google Scholar] [CrossRef] [Green Version]
- Pecoraro, C.; Carbone, D.; Deng, D.; Cascioferro, S.M.; Diana, P.; Giovannetti, E. Biofilm Formation as Valuable Target to Fight against Severe Chronic Infections. CMC 2022, 29, 4307–4310. [Google Scholar] [CrossRef]
- Kearns, D.B. A Field Guide to Bacterial Swarming Motility. Nat. Rev. Microbiol. 2010, 8, 634–644. [Google Scholar] [CrossRef] [Green Version]
- Skerker, J.M.; Berg, H.C. Direct Observation of Extension and Retraction of Type IV Pili. Proc. Natl. Acad. Sci. USA 2001, 98, 6901–6904. [Google Scholar] [CrossRef]
- Bradley, D.E. A Function of Pseudomonas aeruginosa PAO Polar Pili: Twitching Motility. Can. J. Microbiol. 1980, 26, 146–154. [Google Scholar] [CrossRef]
- Franklin, M.J.; Nivens, D.E.; Weadge, J.T.; Howell, P.L. Biosynthesis of the Pseudomonas aeruginosa Extracellular Polysaccharides, Alginate, Pel, and Psl. Front. Microbio. 2011, 2, 167. [Google Scholar] [CrossRef] [Green Version]
- Campoccia, D.; Montanaro, L.; Arciola, C.R. Extracellular DNA (EDNA). A Major Ubiquitous Element of the Bacterial Biofilm Architecture. IJMS 2021, 22, 9100. [Google Scholar] [CrossRef]
- Mulcahy, H.; Charron-Mazenod, L.; Lewenza, S. Extracellular DNA Chelates Cations and Induces Antibiotic Resistance in Pseudomonas aeruginosa Biofilms. PLoS Pathog. 2008, 4, e1000213. [Google Scholar] [CrossRef] [Green Version]
- Lewenza, S. Extracellular DNA-Induced Antimicrobial Peptide Resistance Mechanisms in Pseudomonas aeruginosa. Front. Microbio. 2013, 4, 21. [Google Scholar] [CrossRef] [Green Version]
- Billings, N.; Ramirez Millan, M.; Caldara, M.; Rusconi, R.; Tarasova, Y.; Stocker, R.; Ribbeck, K. The Extracellular Matrix Component Psl Provides Fast-Acting Antibiotic Defense in Pseudomonas aeruginosa Biofilms. PLoS Pathog. 2013, 9, e1003526. [Google Scholar] [CrossRef] [Green Version]
- Colvin, K.M.; Gordon, V.D.; Murakami, K.; Borlee, B.R.; Wozniak, D.J.; Wong, G.C.L.; Parsek, M.R. The Pel Polysaccharide Can Serve a Structural and Protective Role in the Biofilm Matrix of Pseudomonas aeruginosa. PLoS Pathog. 2011, 7, e1001264. [Google Scholar] [CrossRef]
- WHO Media Centre. News Release. WHO Publishes List of Bacteria for Which New Antibiotics Are Urgently Needed. Available online: http://www.who.int/mediacentre/news/releases/2017/bacteria-antibiotics-needed/en/ (accessed on 31 March 2017).
- Kumar, P.; Kizhakkedathu, J.; Straus, S. Antimicrobial Peptides: Diversity, Mechanism of Action and Strategies to Improve the Activity and Biocompatibility In Vivo. Biomolecules 2018, 8, 4. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Separovic, F.; O’Brien-Simpson, N.M.; Wade, J.D. Chemically Modified and Conjugated Antimicrobial Peptides against Superbugs. Chem. Soc. Rev. 2021, 50, 4932–4973. [Google Scholar] [CrossRef]
- Hancock, R.E.W.; Alford, M.A.; Haney, E.F. Antibiofilm Activity of Host Defence Peptides: Complexity Provides Opportunities. Nat. Rev. Microbiol. 2021, 19, 786–797. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Gallo, R.L. Antimicrobial Peptides. Curr. Biol. 2016, 26, R14–R19. [Google Scholar] [CrossRef]
- Yu, G.; Baeder, D.Y.; Regoes, R.R.; Rolff, J. Predicting Drug Resistance Evolution: Insights from Antimicrobial Peptides and Antibiotics. Proc. R. Soc. B 2018, 285, 20172687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brady, D.; Grapputo, A.; Romoli, O.; Sandrelli, F. Insect Cecropins, Antimicrobial Peptides with Potential Therapeutic Applications. Int. J. Mol. Sci. 2019, 20, 5862. [Google Scholar] [CrossRef] [Green Version]
- Romoli, O.; Saviane, A.; Bozzato, A.; D’Antona, P.; Tettamanti, G.; Squartini, A.; Cappellozza, S.; Sandrelli, F. Differential Sensitivity to Infections and Antimicrobial Peptide-Mediated Immune Response in Four Silkworm Strains with Different Geographical Origin. Sci. Rep. 2017, 7, 1048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romoli, O.; Mukherjee, S.; Mohid, S.A.; Dutta, A.; Montali, A.; Franzolin, E.; Brady, D.; Zito, F.; Bergantino, E.; Rampazzo, C.; et al. Enhanced Silkworm Cecropin B Antimicrobial Activity against Pseudomonas aeruginosa from Single Amino Acid Variation. ACS Infect. Dis. 2019, 5, 1200–1213. [Google Scholar] [CrossRef]
- Button, B.; Anderson, W.H.; Boucher, R.C. Mucus Hyperconcentration as a Unifying Aspect of the Chronic Bronchitic Phenotype. Ann. Am. Thorac. Soc. 2016, 13 (Suppl. 2), S156–S162. [Google Scholar]
- Joris, L.; Dab, I.; Quinton, P.M. Elemental Composition of Human Airway Surface Fluid in Healthy and Diseased Airways. Am. Rev. Respir. Dis. 1993, 148, 1633–1637. [Google Scholar] [CrossRef]
- Chakraborti, S.; Sarkar, J.; Pramanik, P.K.; Chakraborti, T. Role of Proteases in Lung Disease: A Brief Overview. In Proteases in Human Diseases; Chakraborti, S., Chakraborti, T., Dhalla, N.S., Eds.; Springer Singapore: Singapore, 2017; pp. 333–374. ISBN 978-981-10-3161-8. [Google Scholar]
- Feng, Z.; Xu, B. Inspiration from the Mirror: D-Amino Acid Containing Peptides in Biomedical Approaches. Biomol. Concepts 2016, 7, 179–187. [Google Scholar] [CrossRef]
- Di Grazia, A.; Cappiello, F.; Cohen, H.; Casciaro, B.; Luca, V.; Pini, A.; Di, Y.P.; Shai, Y.; Mangoni, M.L. D-Amino Acids Incorporation in the Frog Skin-Derived Peptide Esculentin-1a(1-21)NH2 Is Beneficial for Its Multiple Functions. Amino Acids 2015, 47, 2505–2519. [Google Scholar] [CrossRef]
- Lucca, A.J.D.; Bland, J.M.; Vigo, C.B.; Jacks, T.J.; Peter, J.; Walsh, T.J. D-Cecropin B: Proteolytic Resistance, Lethality for Pathogenic Fungi and Binding Properties. Med. Mycol. 2000, 38, 301–308. [Google Scholar] [CrossRef] [Green Version]
- Callaway, J.E.; Lai, J.; Haselbeck, B.; Baltaian, M.; Bonnesen, S.P.; Weickmann, J.; Wilcox, G.; Lei, S.P. Modification of the C Terminus of Cecropin Is Essential for Broad-Spectrum Antimicrobial Activity. Antimicrob. Agents Chemother. 1993, 37, 1614–1619. [Google Scholar] [CrossRef] [Green Version]
- Boffoli, D.; Bellato, F.; Avancini, G.; Gurnani, P.; Yilmaz, G.; Romero, M.; Robertson, S.; Moret, F.; Sandrelli, F.; Caliceti, P.; et al. Tobramycin-Loaded Complexes to Prevent and Disrupt Pseudomonas aeruginosa Biofilms. Drug Deliv. Transl. Res. 2022, 12, 1788–1810. [Google Scholar] [CrossRef]
- Casciaro, B.; Lin, Q.; Afonin, S.; Loffredo, M.R.; Turris, V.; Middel, V.; Ulrich, A.S.; Di, Y.P.; Mangoni, M.L. Inhibition of Pseudomonas aeruginosa Biofilm Formation and Expression of Virulence Genes by Selective Epimerization in the Peptide Esculentin-1a(1-21) NH2. FEBS J. 2019, 286, 3874–3891. [Google Scholar] [CrossRef]
- Thöming, J.G.; Häussler, S. Pseudomonas aeruginosa Is More Tolerant Under Biofilm Than Under Planktonic Growth Conditions: A Multi-Isolate Survey. Front. Cell. Infect. Microbiol. 2022, 12, 851784. [Google Scholar] [CrossRef]
- Khaledi, A.; Weimann, A.; Schniederjans, M.; Asgari, E.; Kuo, T.; Oliver, A.; Cabot, G.; Kola, A.; Gastmeier, P.; Hogardt, M.; et al. Predicting Antimicrobial Resistance in Pseudomonas aeruginosa with Machine Learning-enabled Molecular Diagnostics. EMBO Mol. Med. 2020, 12, e10264. [Google Scholar] [CrossRef]
- Dötsch, A.; Schniederjans, M.; Khaledi, A.; Hornischer, K.; Schulz, S.; Bielecka, A.; Eckweiler, D.; Pohl, S.; Häussler, S. The Pseudomonas aeruginosa Transcriptional Landscape Is Shaped by Environmental Heterogeneity and Genetic Variation. mBio 2015, 6, e00749-15. [Google Scholar] [CrossRef] [Green Version]
- Treerat, P.; Widmer, F.; Middleton, P.G.; Iredell, J.; George, A.M. In Vitro Interactions of Tobramycin with Various Nonantibiotics against Pseudomonas aeruginosa and Burkholderia cenocepacia. FEMS Microbiol. Lett. 2008, 285, 40–50. [Google Scholar] [CrossRef] [Green Version]
- Davis, S.D.; Iannetta, A. Antagonistic Effect of Calcium in Serum on the Activity of Tobramycin Against Pseudomonas. Antimicrob. Agents Chemother. 1972, 1, 466–469. [Google Scholar] [CrossRef] [Green Version]
- Wei, G.-X.; Campagna, A.N.; Bobek, L.A. Factors Affecting Antimicrobial Activity of MUC7 12-Mer, a Human Salivary Mucin-Derived Peptide. Ann. Clin. Microbiol. Antimicrob. 2007, 6, 14. [Google Scholar] [CrossRef] [Green Version]
- Mohamed, M.F.; Brezden, A.; Mohammad, H.; Chmielewski, J.; Seleem, M.N. A Short D-Enantiomeric Antimicrobial Peptide with Potent Immunomodulatory and Antibiofilm Activity against Multidrug-Resistant Pseudomonas aeruginosa and Acinetobacter baumannii. Sci. Rep. 2017, 7, 6953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mangoni, M.L.; Maisetta, G.; Di Luca, M.; Gaddi, L.M.H.; Esin, S.; Florio, W.; Brancatisano, F.L.; Barra, D.; Campa, M.; Batoni, G. Comparative Analysis of the Bactericidal Activities of Amphibian Peptide Analogues against Multidrug-Resistant Nosocomial Bacterial Strains. Antimicrob. Agents Chemother. 2008, 52, 85–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de la Fuente-Núñez, C.; Korolik, V.; Bains, M.; Nguyen, U.; Breidenstein, E.B.M.; Horsman, S.; Lewenza, S.; Burrows, L.; Hancock, R.E.W. Inhibition of Bacterial Biofilm Formation and Swarming Motility by a Small Synthetic Cationic Peptide. Antimicrob. Agents Chemother. 2012, 56, 2696–2704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, H.; Xie, Z.; Wei, H.; Zhang, S.; Song, Y.; Wang, M.; Zhang, Y. Antibacterial and Antibiofilm Activity of Temporin-GHc and Temporin-GHd Against Cariogenic Bacteria, Streptococcus mutans. Front. Microbiol. 2019, 10, 2854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Nilsson, M.; Gjermansen, M.; Givskov, M.; Tolker-Nielsen, T. Pyoverdine and PQS Mediated Subpopulation Interactions Involved in Pseudomonas aeruginosa Biofilm Formation. Mol. Microbiol. 2009, 74, 1380–1392. [Google Scholar] [CrossRef] [PubMed]
- Irie, Y.; Starkey, M.; Edwards, A.N.; Wozniak, D.J.; Romeo, T.; Parsek, M.R. Pseudomonas aeruginosa Biofilm Matrix Polysaccharide Psl Is Regulated Transcriptionally by RpoS and Post-Transcriptionally by RsmA. Mol. Microbiol. 2010, 78, 158–172. [Google Scholar] [CrossRef] [Green Version]
- Bouillet, S.; Ba, M.; Houot, L.; Iobbi-Nivol, C.; Bordi, C. Connected Partner-Switches Control the Life Style of Pseudomonas aeruginosa through RpoS Regulation. Sci. Rep. 2019, 9, 6496. [Google Scholar] [CrossRef] [Green Version]
- Leighton, T.L.; Buensuceso, R.N.C.; Howell, P.L.; Burrows, L.L. Biogenesis of Pseudomonas aeruginosa Type IV Pili and Regulation of Their Function: Pseudomonas aeruginosa Type IV Pili. Environ. Microbiol. 2015, 17, 4148–4163. [Google Scholar] [CrossRef]
- Jain, R.; Behrens, A.-J.; Kaever, V.; Kazmierczak, B.I. Type IV Pilus Assembly in Pseudomonas aeruginosa over a Broad Range of Cyclic Di-GMP Concentrations. J. Bacteriol. 2012, 194, 4285–4294. [Google Scholar] [CrossRef] [Green Version]
- Cai, Y.; Hutchin, A.; Craddock, J.; Walsh, M.A.; Webb, J.S.; Tews, I. Differential Impact on Motility and Biofilm Dispersal of Closely Related Phosphodiesterases in Pseudomonas aeruginosa. Sci. Rep. 2020, 10, 6232. [Google Scholar] [CrossRef] [Green Version]
- Colvin, K.M.; Irie, Y.; Tart, C.S.; Urbano, R.; Whitney, J.C.; Ryder, C.; Howell, P.L.; Wozniak, D.J.; Parsek, M.R. The Pel and Psl Polysaccharides Provide Pseudomonas aeruginosa Structural Redundancy within the Biofilm Matrix: Polysaccharides of the P. Aeruginosa Biofilm Matrix. Environ. Microbiol. 2012, 14, 1913–1928. [Google Scholar] [CrossRef] [Green Version]
- Foster, K.A.; Oster, C.G.; Mayer, M.M.; Avery, M.L.; Audus, K.L. Characterization of the A549 Cell Line as a Type II Pulmonary Epithelial Cell Model for Drug Metabolism. Exp. Cell Res. 1998, 243, 359–366. [Google Scholar] [CrossRef]
- Indrayanto, G.; Putra, G.S.; Suhud, F. Validation of In-Vitro Bioassay Methods: Application in Herbal Drug Research. In Profiles of Drug Substances, Excipients and Related Methodology; Elsevier: Amsterdam, The Netherlands, 2021; Volume 46, pp. 273–307. ISBN 978-0-12-824127-1. [Google Scholar]
- Wade, D.; Boman, A.; Wåhlin, B.; Drain, C.M.; Andreu, D.; Boman, H.G.; Merrifield, R.B. All-D Amino Acid-Containing Channel-Forming Antibiotic Peptides. Proc. Natl. Acad. Sci. USA 1990, 87, 4761–4765. [Google Scholar] [CrossRef]
- Bland, J.M.; De Lucca, A.J.; Jacks, T.J.; Vigo, C.B. All-D-Cecropin B: Synthesis, Conformation, Lipopolysaccharide Binding, and Antibacterial Activity. Mol. Cell. Biochem. 2001, 218, 105–111. [Google Scholar] [CrossRef]
- Tang, W.-H.; Wang, C.-F.; Liao, Y.-D. Fetal Bovine Serum Albumin Inhibits Antimicrobial Peptide Activity and Binds Drug Only in Complex with A1-Antitrypsin. Sci. Rep. 2021, 11, 1267. [Google Scholar] [CrossRef]
- Rajesh, K.; Zareena; Hegde, S.; Arun Kumar, M. Assessment of Salivary Calcium, Phosphate, Magnesium, PH, and Flow Rate in Healthy Subjects, Periodontitis, and Dental Caries. Contemp. Clin. Dent. 2015, 6, 461. [Google Scholar] [CrossRef]
- Mardirossian, M.; Pompilio, A.; Degasperi, M.; Runti, G.; Pacor, S.; Di Bonaventura, G.; Scocchi, M. D-BMAP18 Antimicrobial Peptide Is Active In Vitro, Resists to Pulmonary Proteases but Loses Its Activity in a Murine Model of Pseudomonas aeruginosa Lung Infection. Front. Chem. 2017, 5, 40. [Google Scholar] [CrossRef] [Green Version]
- Römling, U.; Balsalobre, C. Biofilm Infections, Their Resilience to Therapy and Innovative Treatment Strategies. J. Intern. Med. 2012, 272, 541–561. [Google Scholar] [CrossRef]
- Thi, M.T.T.; Wibowo, D.; Rehm, B.H.A. Pseudomonas aeruginosa Biofilms. Int. J. Mol. Sci. 2020, 21, 8671. [Google Scholar] [CrossRef]
- Hoffman, L.R.; D’Argenio, D.A.; MacCoss, M.J.; Zhang, Z.; Jones, R.A.; Miller, S.I. Aminoglycoside Antibiotics Induce Bacterial Biofilm Formation. Nature 2005, 436, 1171–1175. [Google Scholar] [CrossRef]
- Tahrioui, A.; Duchesne, R.; Bouffartigues, E.; Rodrigues, S.; Maillot, O.; Tortuel, D.; Hardouin, J.; Taupin, L.; Groleau, M.-C.; Dufour, A.; et al. Extracellular DNA Release, Quorum Sensing, and PrrF1/F2 Small RNAs Are Key Players in Pseudomonas aeruginosa Tobramycin-Enhanced Biofilm Formation. npj Biofilms Microbiomes 2019, 5, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irving, S.E.; Choudhury, N.R.; Corrigan, R.M. The Stringent Response and Physiological Roles of (Pp)PGpp in Bacteria. Nat. Rev. Microbiol. 2021, 19, 256–271. [Google Scholar] [CrossRef] [PubMed]
- Gupta, K.; Liao, J.; Petrova, O.E.; Cherny, K.E.; Sauer, K. Elevated Levels of the Second Messenger C-Di-GMP Contribute to Antimicrobial Resistance of Pseudomonas aeruginosa. Mol. Microbiol. 2014, 92, 488–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walters, M.C.; Roe, F.; Bugnicourt, A.; Franklin, M.J.; Stewart, P.S. Contributions of Antibiotic Penetration, Oxygen Limitation, and Low Metabolic Activity to Tolerance of Pseudomonas aeruginosa Biofilms to Ciprofloxacin and Tobramycin. Antimicrob. Agents Chemother. 2003, 47, 317–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tseng, B.S.; Zhang, W.; Harrison, J.J.; Quach, T.P.; Song, J.L.; Penterman, J.; Singh, P.K.; Chopp, D.L.; Packman, A.I.; Parsek, M.R. The Extracellular Matrix Protects Pseudomonas aeruginosa Biofilms by Limiting the Penetration of Tobramycin: Limited Tobramycin Penetration Protects Biofilms. Environ. Microbiol. 2013, 15, 2865–2878. [Google Scholar] [CrossRef] [Green Version]
- Gorr, S.-U.; Brigman, H.V.; Anderson, J.C.; Hirsch, E.B. The Antimicrobial Peptide DGL13K Is Active against Drug-Resistant Gram-Negative Bacteria and Sub-Inhibitory Concentrations Stimulate Bacterial Growth without Causing Resistance. PLoS ONE 2022, 17, e0273504. [Google Scholar] [CrossRef]
- Biswaro, L.S.; da Costa Sousa, M.G.; Rezende, T.M.B.; Dias, S.C.; Franco, O.L. Antimicrobial Peptides and Nanotechnology, Recent Advances and Challenges. Front. Microbiol. 2018, 9, 855. [Google Scholar] [CrossRef] [Green Version]
- Moreau-Marquis, S.; Stanton, B.A.; O’Toole, G.A. Pseudomonas aeruginosa Biofilm Formation in the Cystic Fibrosis Airway. Pulm. Pharmacol. Ther. 2008, 21, 595–599. [Google Scholar] [CrossRef] [Green Version]
- Fields, G.B.; Noble, R.L. Solid Phase Peptide Synthesis Utilizing 9-Fluorenylmethoxycarbonyl Amino Acids. Int. J. Pept. Protein Res. 2009, 35, 161–214. [Google Scholar] [CrossRef]
- Carpino, L.A.; Imazumi, H.; El-Faham, A.; Ferrer, F.J.; Zhang, C.; Lee, Y.; Foxman, B.M.; Henklein, P.; Hanay, C.; Mügge, C.; et al. The Uronium/Guanidinium Peptide Coupling Reagents: Finally the True Uronium Salts. Angew. Chem. Int. Ed. 2002, 41, 441–445. [Google Scholar] [CrossRef]
- Segev-Zarko, L.; Shai, Y. Methods for Investigating Biofilm Inhibition and Degradation by Antimicrobial Peptides. In Antimicrobial Peptides; Hansen, P.R., Ed.; Methods in Molecular Biology; Springer New York: New York, NY, USA, 2017; Volume 1548, pp. 309–322. ISBN 978-1-4939-6735-3. [Google Scholar]
- Gulube, Z.; Patel, M. Effect of Punica Granatum on the Virulence Factors of Cariogenic Bacteria Streptococcus mutans. Microb. Pathog. 2016, 98, 45–49. [Google Scholar] [CrossRef]
- Edwards, K.J.; Saunders, N.A. Real-Time PCR Used to Measure Stress-Induced Changes in the Expression of the Genes of the Alginate Pathway of Pseudomonas aeruginosa. J. Appl. Microbiol. 2001, 91, 29–37. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | MIC (μM) | MBC (μM) |
|---|---|---|
| D-Q53 CecB | 2.20 | 4.40 (2.20) * |
| L-Q53 CecB | 2.20 | 11 (4.40) * |
| Tobramycin | 2.14 | 4.29 (8.56) * |
| Condition | D-Q53 CecB MIC (μM) | L-Q53 CecB MIC (μM) |
|---|---|---|
| NaCl 100 mM | 2.2 | 2.2 |
| NaCl 200 mM | 4.4 | 2.2 |
| NaCl 300 mM | 8.0 | 8.0 |
| NaCl 400 mM | >8.0 | >8.0 |
| CaCl2 1 mM | 4.4 | 8.0 |
| CaCl2 2 mM | 15 | 15 |
| MgCl2 1 mM | 4.4 | 2.2 |
| MgCl2 0.5 mM | 2.2 | 2.2 |
| FBS 20% | 8.0 | 5.5 |
| FBS 10% | 4.4 | 4.4 |
| 100 °C 5 min | 2.2 | 2.2 |
| 100 °C 10 min | 2.2 | 2.2 |
| 100 °C 30 min | 2.2 | 2.2 |
| P. aeruginosa Strain | Control (CFUs/mL) Mean ± SEM | D-Q53 CecB (CFUs/mL) Mean ± SEM | L-Q53 CecB (CFUs/mL) Mean ± SEM |
|---|---|---|---|
| ATCC 27853 | 3.06 ± 2.07 × 1014 | 1.07 ± 1.06 × 1010 | 7.03 ± 4.10 × 108 |
| ATCC 25668 | 2.67 ± 2.67 × 1015 | 6.70 ± 6.6 × 108 | 2.89 ± 2.38 × 105 |
| Peptide | MHC (μM) | CCD-34 Lu | A549 | ||
|---|---|---|---|---|---|
| IC50 (μM) | SI | IC50 (μM) | SI | ||
| D-Q53 CecB | 100 | 72.59 ± 2.81 | 16.47 | 68.02 ± 3.71 | 15.45 |
| L-Q53 CecB | >200 | >200 | >18.18 | >100 | >9.09 |
| Gene | Primer | Sequence (5′ -> 3′) | Ta (°C) | Ref. |
|---|---|---|---|---|
| lasI | lasI-F | GGCGCGAAGAGTTCGATAAA | 57 | [36] |
| lasI-R | CCATCTCGTCGATGACACTAAC | |||
| rpoS | rpoS-F | CGGCGAGTTGGTCATCATCAAACA | 63 | [36] |
| rpoS-R | ATCGATTGCCCTACCTTGACCTGTC | |||
| fimX | fimX-F | CCTGGCCTATATCCATCTCAAC | 57 | [36] |
| fimX-R | ACTGTTCACGCATCAGTCC | |||
| pelA | pelA-F | CCTTCAGCCATCCGTTCTTCT | 59 | [18] |
| pelA-R | TCGCGTACGAAGTCGACCTT | |||
| pslA | pslA-F | AAGATCAAGAAACGCGTGGAAT | 57 | [18] |
| pslA-R | TGTAGAGGTCGAACCACACCG | |||
| algD | algD-F | GCGACCTGGACCTGGGCT | 58 | [76] |
| algD-R | TCCTCGATCAGCGGGATC | |||
| rplU | rplU-F | CGCAGTGATTGTTACCGGTG | 59 | [36] |
| rplU-R | AGGCCTGAATGCCGGTGATC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Varponi, I.; Ferro, S.; Menilli, L.; Grapputo, A.; Moret, F.; Mastrotto, F.; Marin, O.; Sandrelli, F. Fighting Pseudomonas aeruginosa Infections: Antibacterial and Antibiofilm Activity of D-Q53 CecB, a Synthetic Analog of a Silkworm Natural Cecropin B Variant. Int. J. Mol. Sci. 2023, 24, 12496. https://doi.org/10.3390/ijms241512496
Varponi I, Ferro S, Menilli L, Grapputo A, Moret F, Mastrotto F, Marin O, Sandrelli F. Fighting Pseudomonas aeruginosa Infections: Antibacterial and Antibiofilm Activity of D-Q53 CecB, a Synthetic Analog of a Silkworm Natural Cecropin B Variant. International Journal of Molecular Sciences. 2023; 24(15):12496. https://doi.org/10.3390/ijms241512496
Chicago/Turabian StyleVarponi, Irene, Stefania Ferro, Luca Menilli, Alessandro Grapputo, Francesca Moret, Francesca Mastrotto, Oriano Marin, and Federica Sandrelli. 2023. "Fighting Pseudomonas aeruginosa Infections: Antibacterial and Antibiofilm Activity of D-Q53 CecB, a Synthetic Analog of a Silkworm Natural Cecropin B Variant" International Journal of Molecular Sciences 24, no. 15: 12496. https://doi.org/10.3390/ijms241512496