

Genome Sequencing Provides Novel Insights into Mudflat Burrowing Adaptations in Eel Goby Taenioides sp. (Teleost: Amblyopinae)
Abstract
:1. Introduction
2. Results
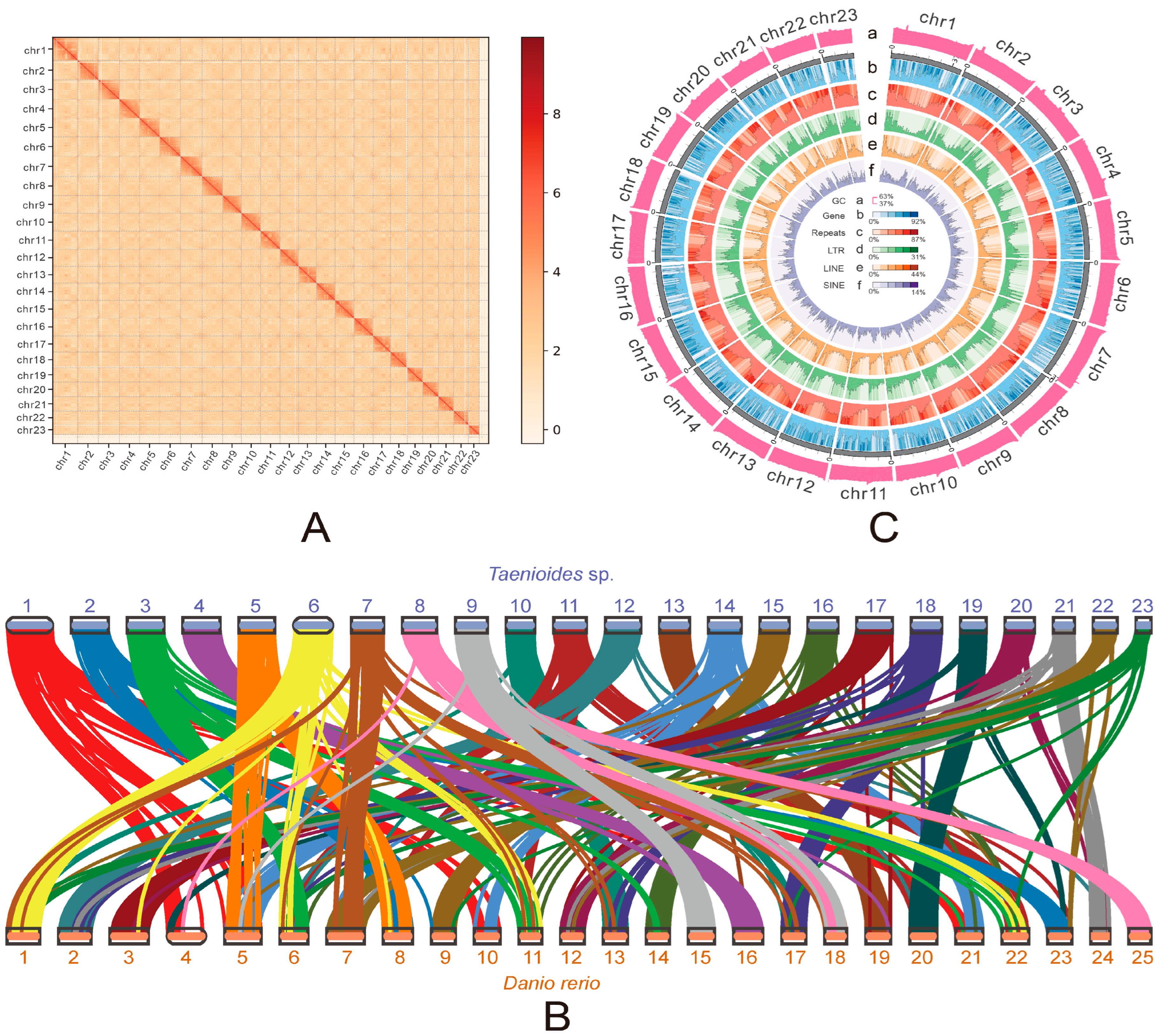
2.1. Assembly and Characterization of Taenioides sp. Genome
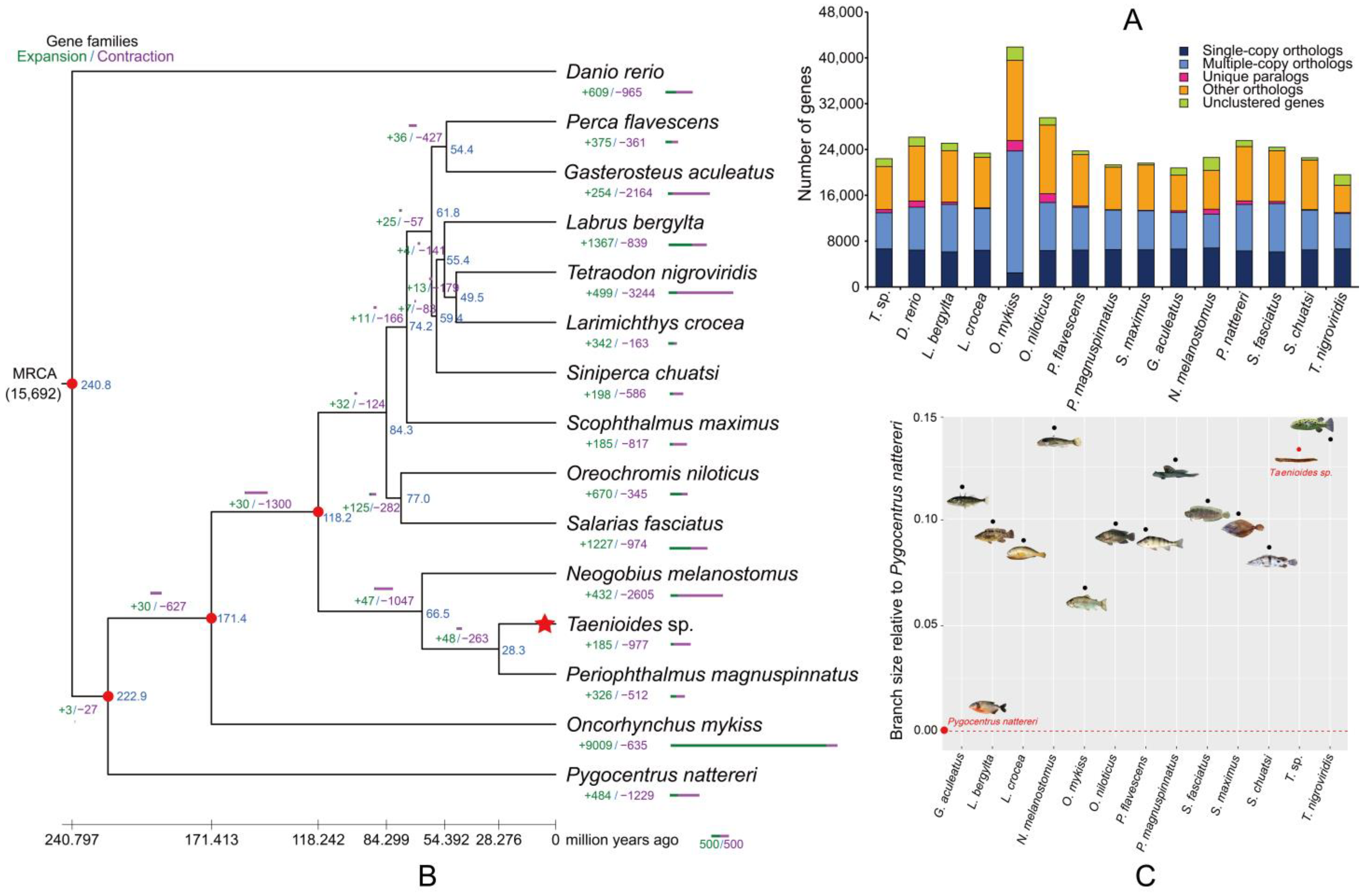
2.2. Phylogenetic Origin and Fast Evolution of Taenioides sp.
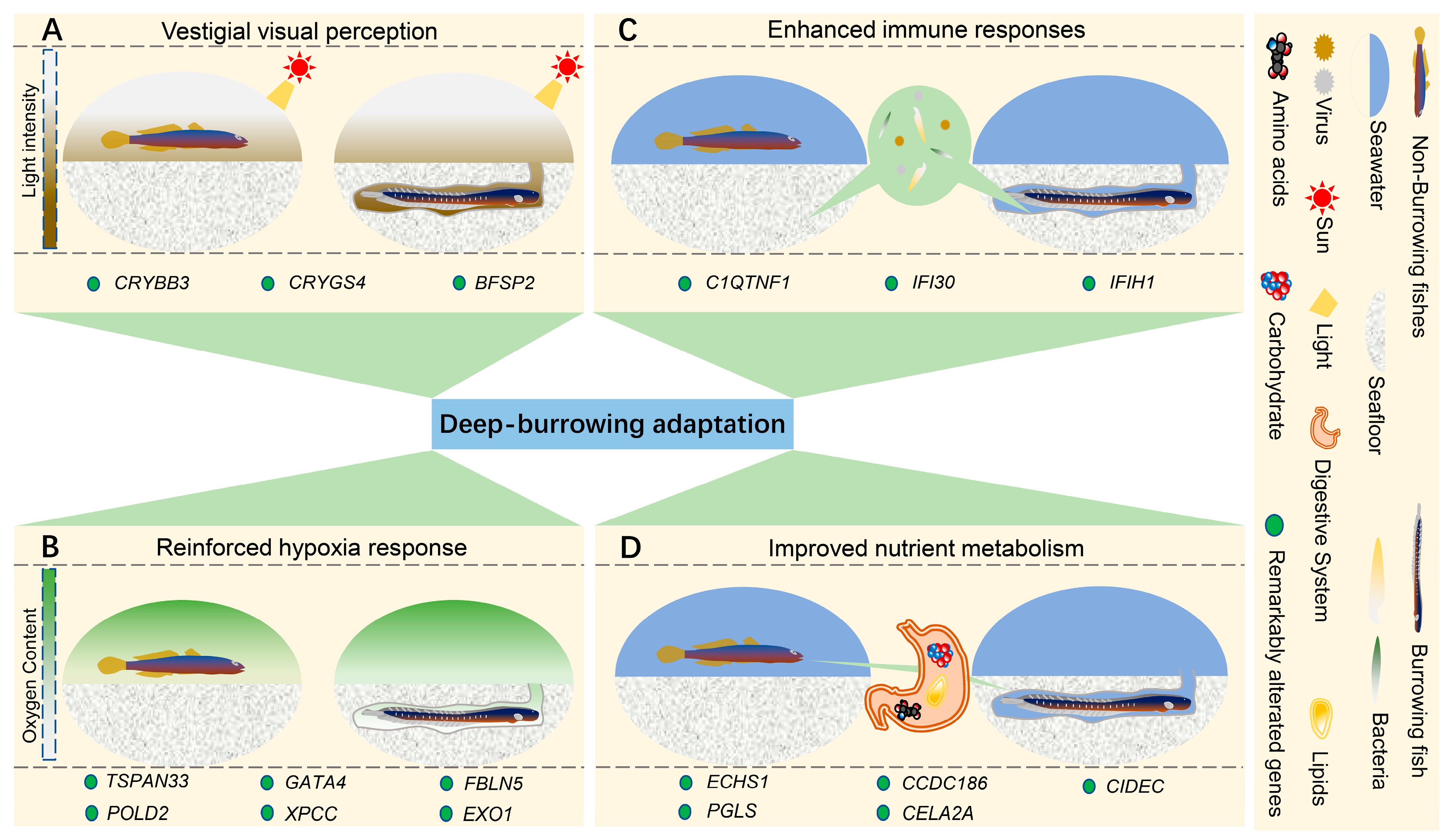
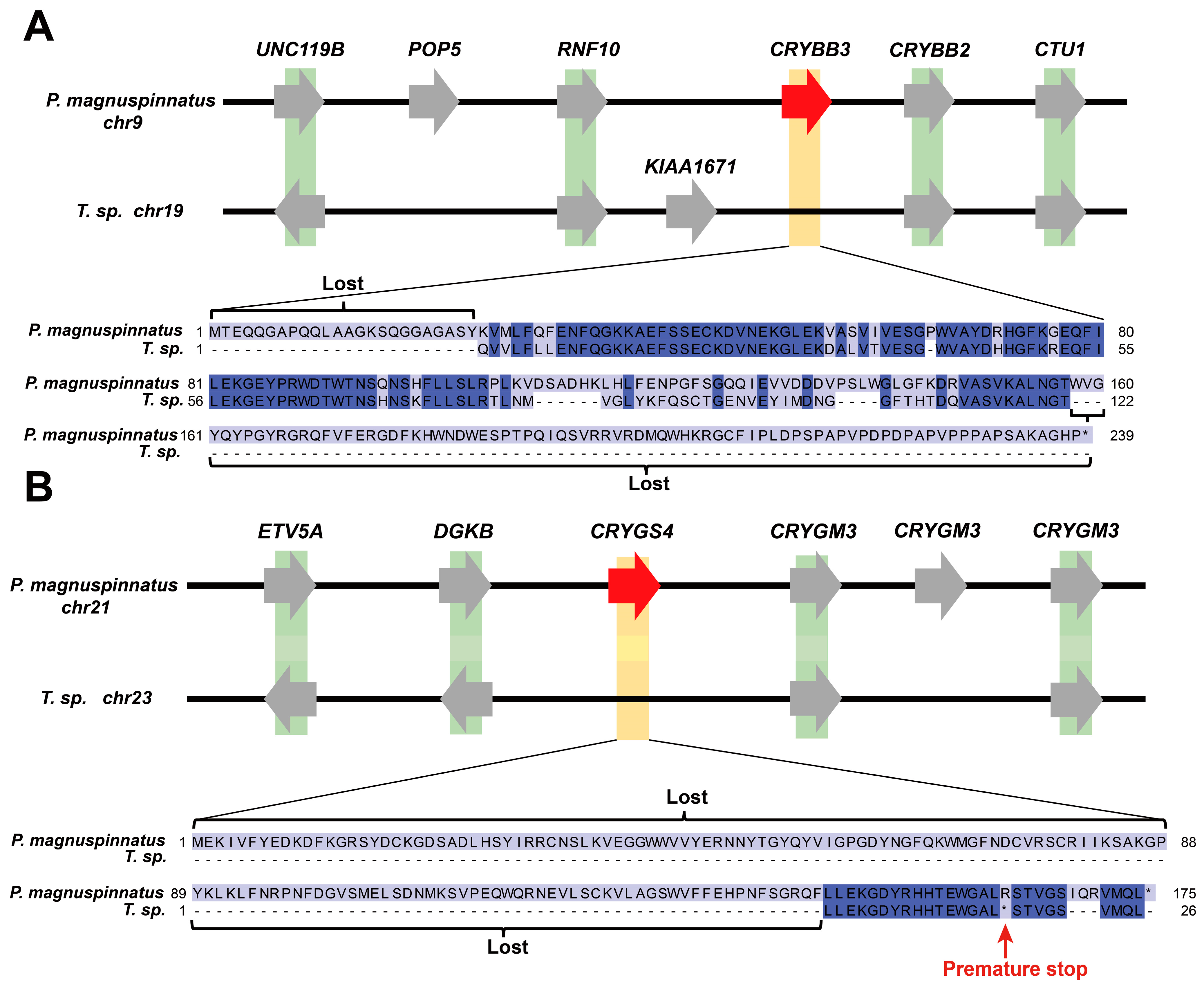
2.3. Genetic Alterations in Taenioides sp. Possibly Correlated wth Deep-Burrowing Adaptations
3. Discussion
3.1. Possible Genetic Origin of Visual Modification Adapted to Burrowing Habitats in Taenioides sp.
3.2. Possible Genetic Origin of Improved Hypoxia Resistance Adapted to Burrowing Habitats in Taenioides sp.
3.3. Genetic Changes Possibly Confer Enhanced Immune Response Adapted to Burrowing Habitats in Taenioides sp.
3.4. Genetic Changes Possibly Confer Altered Nutri-Physiology Adapted to Burrowing Habitats in Taenioides sp.
3.5. Future Directions for Pursuing the Burrowing Adaptation Mechanisms in Taenioides sp.
4. Materials and Methods
4.1. DNA and RNA Extraction
4.2. Library Constructions and Sequencing
4.3. Genome Assemblies and Chromosome Construction
4.4. Genome Annotations
4.5. Phylogenetic Tree Constructions and Divergence Time Evaluation
4.6. Estimation of Relative Evolutionary Rates
4.7. Estimation of Gene Family Expansion and Contraction
4.8. Detection of Positive Selection
4.9. Identification of Gene Losses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, K.; Wang, J.; Zhu, C.L.; Yang, L.D.; Ren, Y.D.; Ruan, J.; Fan, G.Y.; Hu, J.; Xu, W.J.; Bi, X.P.; et al. African lungfifish genome sheds light on the vertebrate water-to-land transition. Cell 2021, 184, 1362–1376. [Google Scholar] [CrossRef] [PubMed]
- Lü, Z.M.; Liu, Y.T.; Zhao, S.J.; Fang, J.Q.; Zhu, K.H.; Liu, J.; Gong, L.; Liu, L.Q.; Liu, B.J. Amblyopinae mitogenomes provide novel insights into the paraphyletic origin of their adaptation to mudflat habitats. Int. J. Mol. Sci. 2023, 24, 4362. [Google Scholar] [CrossRef] [PubMed]
- Meyer, A.; Schloissnig, S.; Franchini, P.; Du, K.; Woltering, J.M.; Irisarri, I.; Wong, W.Y.; Nowoshilow, S.; Kneitz, S.; Kawaguchi, A.; et al. Giant lungfish genome elucidates the conquest of land by vertebrates. Nature 2021, 590, 285–289. [Google Scholar] [CrossRef]
- Long, J.A.; Gordon, M.S. The greatest step in vertebrate history: A paleobiological review of the fish-tetrapod transition. Physiol. Biochem. Zool. 2004, 77, 700–719. [Google Scholar] [CrossRef]
- Martin, K.L.M. Time and tide wait for no fish: Intertidal fishes out of water. Environ. Biol. Fishes 1995, 44, 165–181. [Google Scholar] [CrossRef]
- You, X.X.; Bian, C.; Zan, Q.J.; Xu, X.; Liu, X.; Chen, J.M.; Wang, J.T.; Qiu, Y.; Li, W.J.; Zhang, X.H.; et al. Mudskipper genome provide insights into the terrestrial adaptation of amphibious fishes. Nat. Commun. 2014, 5, 5594. [Google Scholar] [CrossRef]
- Ord, T.J.; Cooke, G.M. Repeated evolution of amphibious behavior in fifish and its implications for the colonization of novel environments. Evolution 2016, 70, 1747–1759. [Google Scholar] [CrossRef]
- Steppan, S.J.; Meyer, A.A.; Barrow, L.N.; Alhajeri, B.H.; Al-Zaidan, A.S.Y.; Gignac, P.M.; Erickson, G.M. Phylogenetics and the evolution of terrestriality in mudskippers (Gobiidae: Oxudercinae). Mol. Phylogenet. Evol. 2022, 169, 107416. [Google Scholar] [CrossRef]
- Murdy, E.O. Systematics of Amblyopinae. In The Biology of Gobies; Panzer, R.E.A., Ed.; CRC Press: Boca Raton, FL, USA, 2011; pp. 107–118. [Google Scholar]
- Maeda, K.; Hanahara, N.; Uehara, M.; Tachihara, K. Larval study revealed diversity and life-history traits of crypto-benthic eel gobies. J. Fish Biol. 2022, 101, 1411–1427. [Google Scholar] [CrossRef]
- Gonzales, T.T.; Katoh, M.; Ishimatsu, A. Intertidal burrows of the air-breathing eel goby, Odontamblyopus lacepedii (Gobiidae: Amblyopinae). Ichthyol. Res. 2008, 55, 303–306. [Google Scholar] [CrossRef]
- Yao, C.H.; Zhao, S.H.; Chen, J.; Zhua, K.H.; Fang, J.Q.; Liu, L.Q.; Lü, Z.M. Molecular and morphological analyses suggest cryptic diversity of eel gobies, genus Taenioides (Gobiidae), in coastal waters of China. J. Ichthyol. 2022, 62, 1025–1033. [Google Scholar] [CrossRef]
- Gonzales, T.T.; Katoh, M.; Ishimatsu, A. Air breathing of aquatic burrow-dwelling eel goby, Odontamblyopus lacepedii (Gobiidae: Amblyopinae). J. Exp. Biol. 2006, 209, 1085–1092. [Google Scholar] [CrossRef]
- Thacker, C.E. Molecular phylogeny of the gobioid fishes (Teleostei: Perciformes: Gobioidei). Mol. Phylogenet. Evol. 2003, 26, 354–368. [Google Scholar] [CrossRef] [PubMed]
- Haq, B.U.; Hardenbol, J.; Vail, P.R. Chronology of fluctuating sea levels since the triassic. Science 1987, 235, 1156–1167. [Google Scholar] [CrossRef] [PubMed]
- Hochmuth, K.; Whittaker, J.M.; Sauermilc, I.; Klocker, A.; Gohl, K.; LaCasce, J.H. Southern Ocean biogenic blooms freezing-in Oligocene colder climates. Nat. Commun. 2022, 13, 6785. [Google Scholar] [CrossRef]
- Kimura, M.; Ohta, T. On the rate of molecular evolution. J. Mol. Evol. 1971, 1, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Heikens, M.J.; Cao, T.M.; Morita, C.; Dehart, S.L.; Tsai, S. Penumbra encodes a novel tetraspanin that is highly expressed in erythroid progenitors and promotes effective erythropoiesis. Blood 2007, 109, 3244–3252. [Google Scholar] [CrossRef]
- Sullivan, K.M.; Bissonnette, R.; Yanagisawa, H.; Hussain, S.N.; Davis, E.C. Fibulin-5 functions as an endogenous angiogenesis inhibitor. Lab. Investig. 2007, 87, 818–827. [Google Scholar] [CrossRef] [PubMed]
- Li, H.X.; Zuo, S.; He, Z.S.; Yang, Y.T.; Pasha, Z.; Wang, Y.G.; Xu, M.F. Paracrine factors released by GATA-4 overexpressed esenchymal stem cells increase angiogenesis and cell survival. Am. J. Physiol.-Heart Circ. Physiol. 2010, 299, 1772–1781. [Google Scholar] [CrossRef]
- Keijzers, G.; Liu, D.; Rasmussen, L.J. Exonuclease 1 and its versatile roles in DNA repair. Crit. Rev. Biochem. Mol. Biol. 2016, 51, 440–451. [Google Scholar] [CrossRef]
- Nicolas, E.; Golemis, E.A.; Arora, S. POLD1: Central mediator of DNA replication and repair, and implication in cancer and other pathologies. Gene 2016, 590, 128–141. [Google Scholar] [CrossRef] [PubMed]
- Zebian, A.; Shaito, A.; Mazurier, F.; Rezvani, H.R.; Zibara, K. XPC beyond nucleotide excision repair and skin cancers. Mutat. Res.-Rev. Mutat. Res. 2019, 782, 108286. [Google Scholar] [CrossRef] [PubMed]
- Zhu, K.C.; Yu, W.B.; Guo, H.Y.; Zhang, N.; Guo, L.; Liu, B.S.; Jiang, S.G.; Zhang, D.C. Genomic structure, expression pattern and polymorphisms of GILT in golden pompano Trachinotus ovatus (Linnaeus 1758). Gene 2018, 665, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Schäffler, A.; Buechler, C. CTRP family: Linking immunity to metabolism. Trends Endocrinol. Metab. 2012, 23, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.X.; Jin, S.T.; Jiang, J.; Han, K.H.; Min, X.P.; Liu, X.R. Predict the relationship between gene and large yellow croaker’s economic traits. Molecules 2017, 22, 1978. [Google Scholar] [CrossRef] [PubMed]
- Kupor, S.R.; Fraenkel, D.G. Glucose metabolism in 6~phosphogluconolactonase mutants of Escherichia coli. J. Biol. Chem. 1972, 247, 1904–1910. [Google Scholar] [CrossRef]
- Zhang, X.Q.; Yang, J.T.; Guo, Y.B. Functional proteomic analysis of nonalcoholic fatty liver disease in rat models: Enoyl–coenzyme a hydratase down-regulation exacerbates hepatic steatosis. Hepatology 2010, 51, 1190–1199. [Google Scholar] [CrossRef]
- Wang, G.X.; Li, R.B.; Yang, Y.; Cai, L.; Ding, S.; Xu, T.; Han, M.; Wu, X.H. Disruption of the Golgi protein Otg1 gene causes defective hormone secretion and aberrant glucose homeostasis in mice. Cell Biosci. 2016, 6, 41. [Google Scholar] [CrossRef]
- Esteghamat, F.; Broughton, J.S.; Smith, E.; Cardone, R.; Tyagi, T.; Guerra, M.; Szabó, A.; Ugwu, N.; Mani, M.V.; Azari, B.; et al. CELA2A mutations predispose to early-onset atherosclerosis and metabolic syndrome and affect plasma insulin and platelet activation. Nat. Genet. 2019, 51, 1233–1243. [Google Scholar] [CrossRef]
- Huang, L.; Liao, Q.C.; Pan, T.L.; Sun, Y.; Aluo, Z.; Xiao, L.G.; Yu, J.S.; Liu, S.Q.; Xiao, Y.; Yang, Y.F.; et al. Small intestine-specific knockout of CIDEC improves obesity and hepatic steatosis by inhibiting synthesis of phosphatidic acid. Int. J. Biol. Sci. 2022, 18, 5740–5752. [Google Scholar] [CrossRef]
- Perng, M.D.; Zhang, Q.J.; Quinlan, R.A. Insights into the beaded filament of the eye lens. Exp. Cell Res. 2007, 313, 2180–2188. [Google Scholar] [CrossRef] [PubMed]
- Limi, S.; Zhao, Y.L.; Guo, P.; Lopez-Jones, M.; Zheng, D.Y.; Singer, R.H.; Skoultchi, A.I.; Cvekl, A. Bidirectional analysis of Cryba4-Crybb1 nascent transcription and nuclear accumulation of Crybb3 mRNAs in lens fibers. Biochem. Mol. Biol. 2019, 60, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Farnsworth, D.R.; Posner, M.; Mille, A.C. Single cell transcriptomics of the developing zebrafish lens and identification of putative controllers of lens development. Exp. Eye Res. 2021, 206, 108535. [Google Scholar] [CrossRef] [PubMed]
- Yildiz, S.H.; Karaosmanoğlu, C.; Duman, R.; Varol, N.; Erdoan, M.Z.; Solak, M.; Duman, R.; Elmas, M. Relationship between expression levels of TDRD7 and CRYBB3 and development of age-related cortico-nuclear cataracts. Egypt. J. Med. Hum. Genet. 2023, 24, 16. [Google Scholar] [CrossRef]
- Sandilands, A.; Prescott, A.R.; Wegener, A. Knockout of the intermediate filament protein CP49 destabilises the lens fibre cell cytoskeleton and decreases lens optical quality, but does not induce cataract. Exp. Eye Res. 2003, 76, 385–391. [Google Scholar] [CrossRef]
- Ma, Z.W.; Chauss, D.; Disatham, J.; Jiao, X.D.; Brennan, L.A.; Menko, A.S.; Kantorow, M.; Hejtmancik, J.F. Patterns of crystallin gene expression in differentiation state specific regions of the embryonic chicken lens. Investig. Ophthalmol. Vis. Sci. 2022, 63, 8. [Google Scholar] [CrossRef]
- Riazuddin, S.A.; Yasmeen Afshan Yao, W.L. Mutations in βB3-Crystallin associated with autosomal recessive cataract in two Pakistani families. Investig. Ophthalmol. Vis. Sci. 2005, 46, 2100–2106. [Google Scholar] [CrossRef]
- Zin, O.A.; Neves, L.M.; Motta, F.L.; Horovitz, D.D.G.; Guida, L.; Gomes, L.H.F.; Cunha, D.P.; Rodrigues, A.P.S.; Zin, A.A.; Sallum, J.M.F.; et al. Novel mutation in CRYBB3 causing pediatric cataract and microphthalmia. Genes 2021, 12, 1069. [Google Scholar] [CrossRef]
- Rechsteiner, D.; Issler, L.; Koller, S.; Lang, E.; Bähr, L.; Feil, S.; Rüegger, C.M.; Kottke, R.; Toelle, S.P.; Zweifel, N.; et al. Genetic analysis in a Swiss cohort of bilateral congenital cataract. JAMA Ophthalmol. 2021, 139, 691–700. [Google Scholar] [CrossRef]
- Saito, S.; Lin, Y.C.; Tsai, M.H.; Lin, C.S.; Murayama, Y.; Sato, R.; Yokoyama, K.K. Emerging roles of hypoxia-inducible factors and reactive oxygen species in cancer and pluripotent stem cells. Kaohsiung J. Med. Sci. 2015, 31, 279–286. [Google Scholar] [CrossRef]
- Morita, R.; Nakane, S.H.; Shimada, A.; Inoue, M.; Iino, H.; Wakamatsu, T.; Fukui, K.; Nakagawa, N.; Masui, R.; Kuramitsu, S. Molecular mechanisms of the Whole DNA Repair System: A Comparison of Bacterial and Eukaryotic Systems. J. Nucleic Acids 2010, 2010, 179594. [Google Scholar] [CrossRef] [PubMed]
- Whitman, W.B.; Coleman, D.C.; Wiebe, W.J. Prokaryotes: The unseen majority. Proc. Natl. Acad. Sci. USA 1998, 95, 6578–6583. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.L.; Liu, X.L.; Meng, Y.M.; Wu, H.X.; Wu, Y.K.; Yang, B.; Wang, L. Autoimmune disease associated IFIH1 single nucleotide polymorphism related with IL-18 serum levels in Chinese systemic lupus erythematosus patients. Sci. Rep. 2018, 8, 9442. [Google Scholar] [CrossRef]
- Vujkovic, M.; Ramdas, S.; Lorenz, K.M.; Guo, X.Q.; Darlay, R.; Cordell, H.J.; He, J.; Gindin, Y.; Chung, C.; Myers, R.P.; et al. A multiancestry genome-wide association study of unexplained chronic ALT elevation as a proxy for nonalcoholic fatty liver disease with histological and radiological validation. Nat. Genet. 2022, 54, 761–771. [Google Scholar] [CrossRef]
- Sharpe, A.J.; McKenzie, M. Mitochondrial fatty acid oxidation disorders associated with short-chain enoyl-CoA hydratase (ECHS1) defificiency. Cells 2018, 7, 46. [Google Scholar] [CrossRef]
- Fang, X.D.; Seim, I.; Huang, Z.Y.; Gerashchenko, M.V.; Xiong, Z.Q.; Turanov, A.A.; Zhu, Y.B.; Lobanov, A.V.; Fan, D.; Yim, S.H.; et al. Adaptations to a subterranean environment and longevity revealed by the analysis of mole rat genomes. Cell Rep. 2014, 8, 1354–1364. [Google Scholar] [CrossRef]
- Aspiras, A.C.; Rohnera, N.; Martineau, B.; Borowsky, R.L.; Tabin, C.J. Melanocortin 4 receptor mutations contribute to the adaptation of cavefish to nutrient-poor conditions. Proc. Natl. Acad. Sci. USA 2015, 112, 9668–9673. [Google Scholar] [CrossRef] [PubMed]
- Riddle, M.R.; Aspiras, A.C.; Gaudenz, K.; Peuß, R.; Sung, J.Y.; Martineau, B.; Peavey, M.; Box, A.C.; Tabin, J.A.; McGaugh, S.; et al. Insulin resistance in cavefish as an adaptation to a nutrient-limited environment. Nature 2020, 555, 647–651. [Google Scholar] [CrossRef]
- Kim, E.B.; Fang, X.D.; Fushan, A.A.; Huang, Z.Y.; Lobanov, A.V.; Han, L.J.; Marino, S.M.; Sun, X.Q.; Turanov, A.A.; Yang, P.C.; et al. Genome sequencing reveals insights into physiology and longevity of the naked mole rat. Nature 2011, 479, 223–227. [Google Scholar] [CrossRef]
- Carlini, D.B.; Fong, D.W. The transcriptomes of cave and surface populations of Gammarus minus (Crustacea: Amphipoda) provide evidence for positive selection on cave downregulated transcripts. PLoS ONE 2017, 12, e0186173. [Google Scholar] [CrossRef]
- Zhao, Q.Y.; Shao, F.; Li, Y.P.; Yi, S.V.; Peng, Z.G. Novel genome sequence of Chinese cavefish (Triplophysa rosa) reveals pervasive relaxation of natural selection in cavefish genomes. Mol. Ecol. 2022, 31, 5831–5845. [Google Scholar] [CrossRef]
- Rétaux, S.; Casane, D. Evolution of eye development in the darkness of caves: Adaptation, drift, or both? EvoDevo 2013, 4, 26. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, 884–890. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.H.; Shi, Y.J.; Yuan, J.Y.; Hu, X.S.; Zhang, H.; Li, N.; Li, Z.Y.; Chen, Y.X.; Mu, D.S.; Fan, W. Estimation of genomic characteristics by analyzing k-mer frequency in de novo genome projects. Quant. Biol. 2013, 35, 62–67. [Google Scholar]
- Ranallo-Benavidez, T.R.; Jaron, K.S.; Schatz, M.C. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat. Commun. 2020, 11, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Concepcion, G.T.; Feng, X.; Zhang, H.W.; Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hififiasm. Nat. Methods 2021, 18, 170–175. [Google Scholar] [CrossRef]
- Durand, N.C.; Shamim, M.S.; Machol, I.; Rao, S.S.P.; Huntley, M.H.; Lander, E.S.; Aiden, E.L. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 2016, 3, 95–98. [Google Scholar] [CrossRef]
- Dudchenko, O.; Batra, S.S.; Omer, A.D.; Nyquist, S.K.; Hoeger, M.; Durand, N.C.; Shamim, M.S.; Machol, I.; Lander, E. S, Aiden, A.P.; et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 2017, 356, 92–95. [Google Scholar] [CrossRef]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Marçais, G.; Delcher, A.L.; Phillippy, A.M.; Coston, R.; Salzberg, S.L.; Zimin, A. MUMmer4: A fast and versatile genome alignment system. PLoS Comput. Biol. 2018, 14, e1005944. [Google Scholar] [CrossRef]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Bedell, J.A.; Korf, I.; Gish, W. MaskerAid: A performance enhancement to RepeatMasker. Bioinformatics 2000, 16, 1040–1041. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Bridges, S.; Magbanua, Z.V.; Peterson, D.G. Empirical comparison of ab initio repeat finding programs. Nucleic Acids Res. 2008, 36, 2284–2294. [Google Scholar] [CrossRef] [PubMed]
- Stanke, M.; Waack, S. Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics 2003, 19, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Burge, C.; Karlin, S. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 1997, 268, 78–94. [Google Scholar] [CrossRef]
- Chang, Z.; Li, G.; Liu, J.; Zhang, Y.; Ashby, C.; Liu, D.L.; Cramer, C.L.; Huang, X.Z. Bridger: A new framework for de novo transcriptome assembly using RNA-seq data. Genome Biol. 2015, 16, 30. [Google Scholar] [CrossRef]
- Kent, W.J. BLAT-The BLAST-like alignment tool. Genome Res. 2002, 12, 656–664. [Google Scholar]
- Haas, B.J.; Delcher, A.L.; Mount, S.M.; Wortman, J.R.; Smith Jr, R.K.; Hannick, L.I.; Maiti, R.; Ronning, C.M.; Rusch, D.B.; Town, C.D.; et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 2003, 31, 5654–5666. [Google Scholar] [CrossRef]
- Mount, D.W. Using the Basic Local Alignment Search Tool (BLAST); Cold Spring Harbor Protocols: Cold Spring Harbor, NY, USA, 2007. [Google Scholar]
- Birney, E.; Clamp, M.; Durbin, R. GeneWise and genomewise. Genome Res. 2004, 14, 988–995. [Google Scholar] [CrossRef]
- Campbell, M.S.; Holt, C.; Moore, B.; Yandelll, M. Genome annotation and curation using MAKER and MAKER-P. Curr. Protoc. Bioinform. 2014, 48, 4111–41139. [Google Scholar] [CrossRef]
- Jones, P.; Binns, D.; Chang, H.Y.; Fraser, M.; Li, W.Z.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genomescale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyotoencyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Boeckmann, B.; Bairoch, A.; Apweiler, R.; Blatter, M.C.; Estreicher, A.; Gasteiger, E.; Martin, M.J.; Michoud, K.; O’Donovan, C.; Phan, I.; et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 2003, 31, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Stoeckert, C.J.; Roos, D.S. OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 2003, 13, 2178–2189. [Google Scholar] [CrossRef]
- Edgar, R.C. Muscle: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Yang, Z.H. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Suleski, M.; Hedges, S.B. TimeTree: A resource for timelines, timetrees, and divergence times. Mol. Biol. Evol. 2017, 34, 1812–1819. [Google Scholar] [CrossRef]
- Takezaki, N.; Rzhetsky, A.; Nei, M. Phylogenetic test of the molecular clock and linearized trees. Mol. Biol. Evol. 1995, 12, 823–833. [Google Scholar]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis Version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- De Bie, T.; Cristianini, N.; Demuth, J.P.; Hahn, M.W. CAFE: A computational tool for the study of gene family evolution. Bioinformatics 2006, 22, 1269–1271. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.Q.; Wang, P.Q.; Li, J.; Zhao, Q.Z.; Ji, C.M.; Zhu, Z.B.; Zhai, Y.F.; Qin, X.D.; Zhou, J.G.; Yu, H.Y.; et al. Whole-genome sequence of synthesized allopolyploids in Cucumis reveals insights into the genome evolution of allopolyploidization. Adv. Sci. 2021, 8, 2004222. [Google Scholar] [CrossRef]
- Tang, H.; Lyons, E.; Pedersen, B.; Schnable, J.C.; Paterson, A.H.; Freeling, M. Screening synteny blocks in pairwise genome comparisons through integer programming. BMC Bioinform. 2011, 12, 102. [Google Scholar] [CrossRef] [PubMed]
- Keilwagen, J.; Wenk, M.; Erickson, J.L.; Schattat, M.H.; Grau, J.; Hartung, F. Using intron position conservation for homology-based gene prediction. Nucleic Acids Res. 2016, 44, e89. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nucleotide Type | Sequencing Strategy | Library Size (bp) | Clean Data (Gb) | Sequencing Depth (x) |
|---|---|---|---|---|
| Genome | PacBioHiFi | 15,000 | 37.05 | 47.93 |
| Genome | MGISEQ-T7 | 300 | 75.59 | 97.79 |
| Genome | Hi-C | 300 | 118.16 | 152.86 |
| RNA | MGISEQ-T7 | 300 | 6.61 | — |
| Term | HiFiasm Contigs | Hi-C Scaffolds | ||
|---|---|---|---|---|
| Size (bp) | Number | Size (bp) | Number | |
| N90 | 2,215,384 | 52 | 27,022,322 | 21 |
| N80 | 6,471,807 | 34 | 29,825,879 | 18 |
| N70 | 9,977,670 | 25 | 31,731,700 | 16 |
| N60 | 16,903,772 | 19 | 33,492,621 | 13 |
| N50 | 19,260,084 | 15 | 34,184,133 | 11 |
| Max length (bp) | 40,497,657 | 43,849,507 | ||
| Total length (bp) | 774,058,921 | 773,284,894 | ||
| Total number (>100 bp) | 287 | 106 | ||
| Total number (>10 kb) | 287 | 106 | ||
| Gene Number | Percentage | |
|---|---|---|
| Complete Buscos | 3522 | 96.76 |
| Complete and single-copy BUSCOs | 3488 | 95.82 |
| Complete and duplicated BUSCOs | 34 | 0.93 |
| Fragment BUSCOs | 21 | 0.58 |
| Missing BUSCOs | 97 | 2.66 |
| Total BUSCOs groups searched | 3640 | 100.00 |
| Database | Annotated Gene Number | Percentage (%) |
|---|---|---|
| Go | 15,263 | 68.14 |
| INTERPRO | 19,975 | 89.18 |
| KEGG | 21,053 | 93.99 |
| SWISSPROT | 19,194 | 85.69 |
| TREMBL | 21,270 | 94.96 |
| All annotated genes | 21,445 | 95.74 |
| All predicted genes | 22,399 | 100.00 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Liu, T.; Wang, Y.; Liu, J.; Liu, B.; Gong, L.; Lü, Z.; Liu, L. Genome Sequencing Provides Novel Insights into Mudflat Burrowing Adaptations in Eel Goby Taenioides sp. (Teleost: Amblyopinae). Int. J. Mol. Sci. 2023, 24, 12892. https://doi.org/10.3390/ijms241612892
Liu Y, Liu T, Wang Y, Liu J, Liu B, Gong L, Lü Z, Liu L. Genome Sequencing Provides Novel Insights into Mudflat Burrowing Adaptations in Eel Goby Taenioides sp. (Teleost: Amblyopinae). International Journal of Molecular Sciences. 2023; 24(16):12892. https://doi.org/10.3390/ijms241612892
Chicago/Turabian StyleLiu, Yantao, Tianwei Liu, Yuzhen Wang, Jing Liu, Bingjian Liu, Li Gong, Zhenming Lü, and Liqin Liu. 2023. "Genome Sequencing Provides Novel Insights into Mudflat Burrowing Adaptations in Eel Goby Taenioides sp. (Teleost: Amblyopinae)" International Journal of Molecular Sciences 24, no. 16: 12892. https://doi.org/10.3390/ijms241612892