
Effects of Tofacitinib on Muscle Remodeling in Experimental Rheumatoid Sarcopenia

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Weight and CRP
2.2. Structural and Histological Changes in Skeletal Muscle
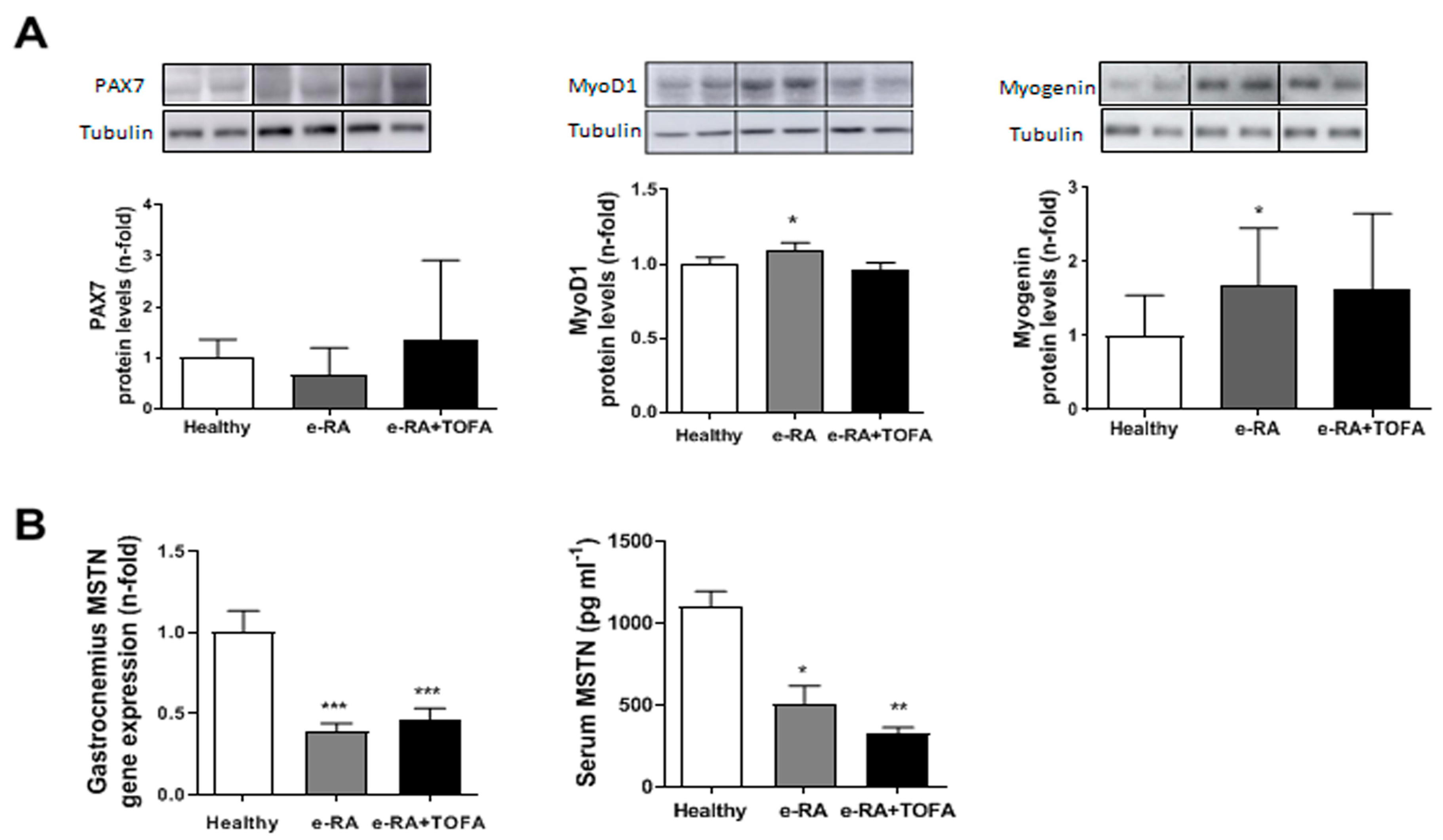
2.3. Myogenic Markers in Skeletal Muscle
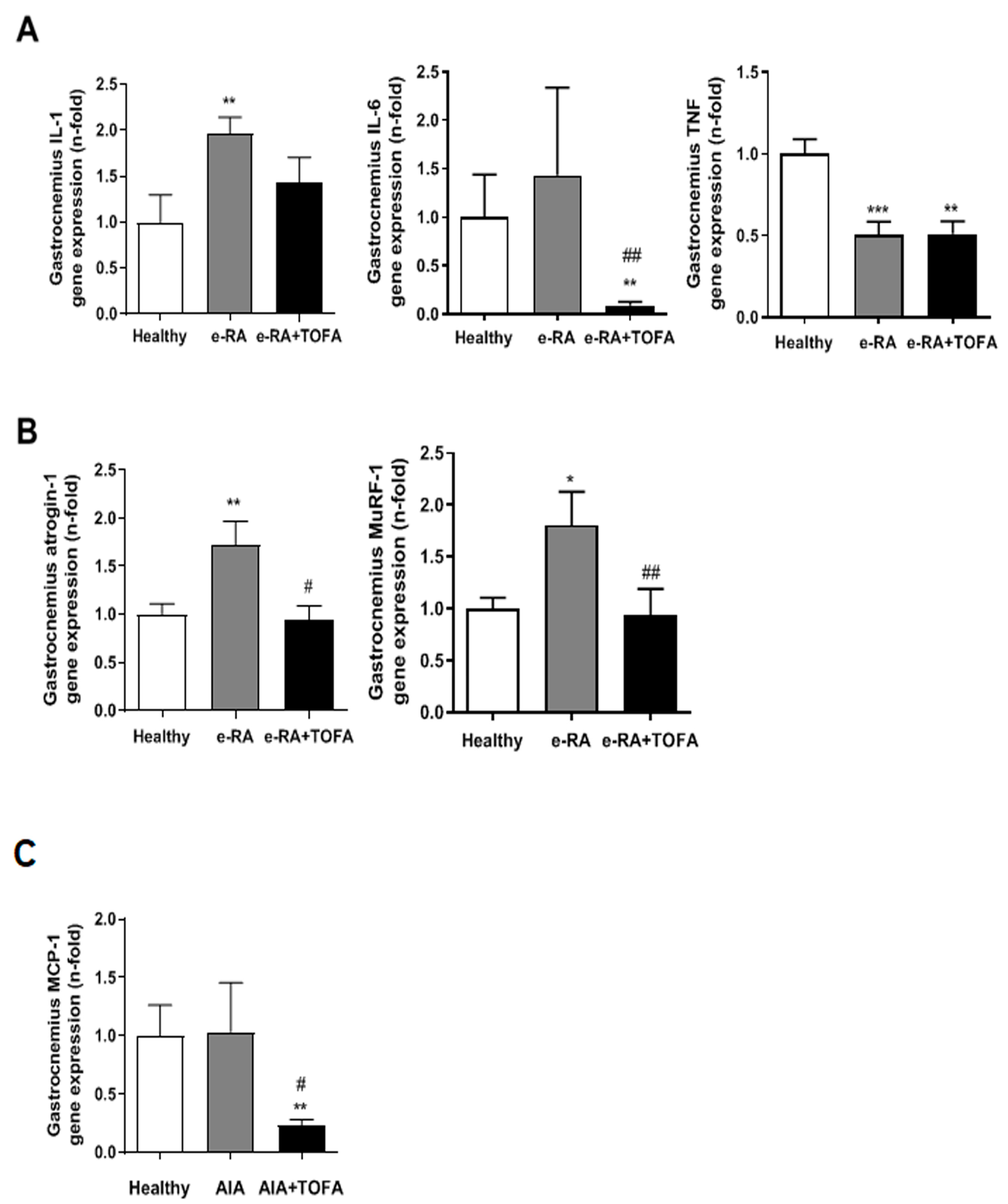
2.4. Atrogenes and Proinflammatory Mediators in Skeletal Muscle
2.5. Creatine Kinase, Creatine, and Pyruvate Activation
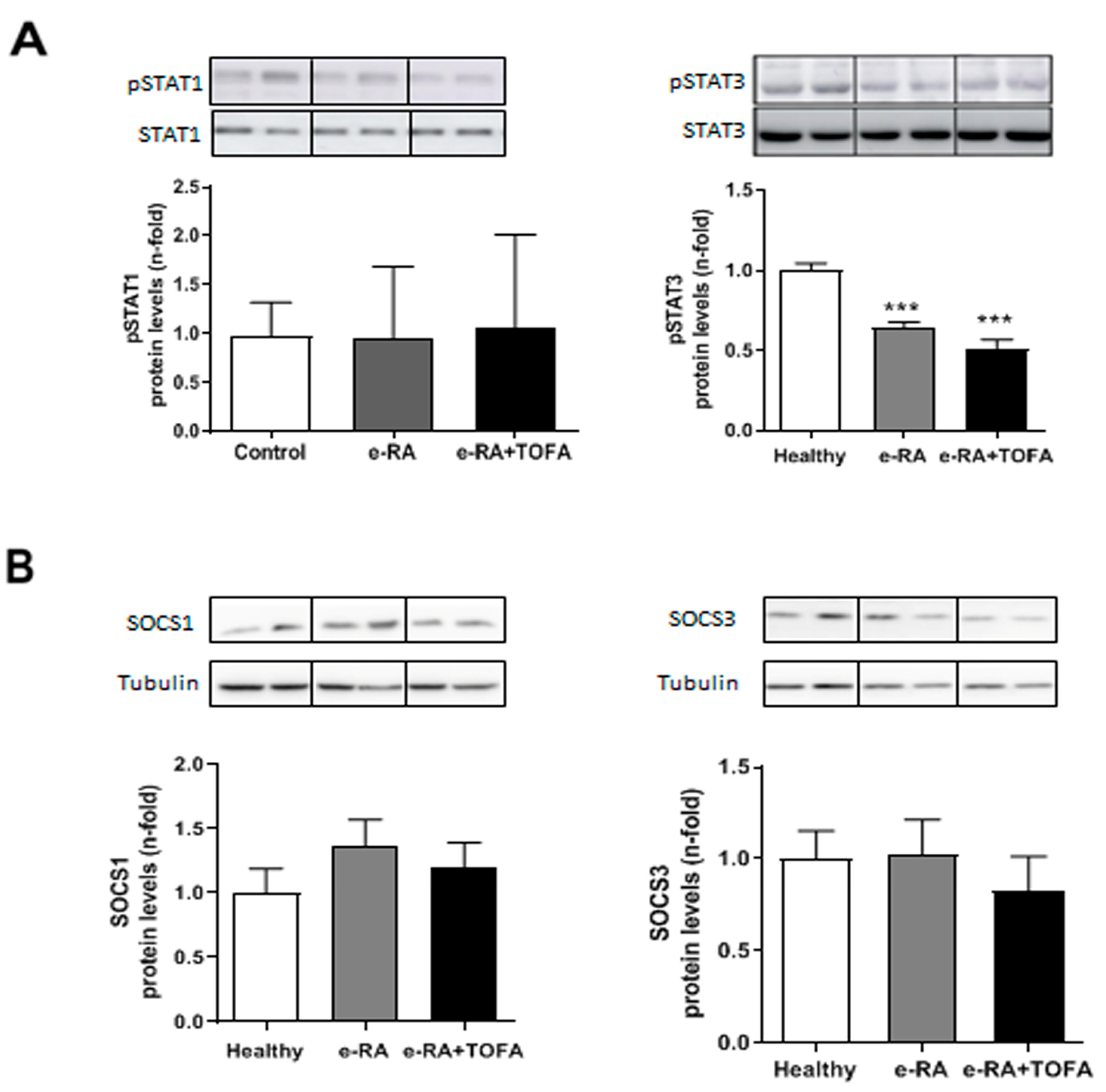
2.6. Levels of pSTAT1, pSTAT3, SOCS1, and SOCS3 in Skeletal Muscle
3. Discussion
4. Materials and Methods
4.1. Experimental Model of Arthritis in Rabbits
4.2. Tissue Collection
4.3. Determination of Serum Biochemical Markers
4.4. Histological Studies
4.5. Fluorescent Labeling of EDL Fibers
4.6. RNA Isolation and RT-PCR
4.7. Western Blot
4.8. Creatine Kinase, Creatine, and Pyruvate Analysis
4.9. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Michaud, K.; Wolfe, F. Comorbidities in Rheumatoid Arthritis. Best. Pract. Res. Clin. Rheumatol. 2007, 21, 885–906. [Google Scholar] [CrossRef] [PubMed]
- Ångström, L.; Hörnberg, K.; Sundström, B.; Södergren, A. Rheumatoid Cachexia in Early Rheumatoid Arthritis: Prevalence and Associated Variables. Scand. J. Rheumatol. 2023, 52, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Morley, J.E.; Thomas, D.R.; Wilson, M.-M.G. Cachexia: Pathophysiology and Clinical Relevance. Am. J. Clin. Nutr. 2006, 83, 735–743. [Google Scholar] [CrossRef] [PubMed]
- Santo, R.C.E.; Fernandes, K.Z.; Lora, P.S.; Filippin, L.I.; Xavier, R.M. Prevalence of Rheumatoid Cachexia in Rheumatoid Arthritis: A Systematic Review and Meta-Analysis: Systematic Review of RA Cachexia Prevalence. J. Cachexia Sarcopenia Muscle 2018, 9, 816–825. [Google Scholar] [CrossRef] [PubMed]
- Schiaffino, S.; Dyar, K.A.; Ciciliot, S.; Blaauw, B.; Sandri, M. Mechanisms Regulating Skeletal Muscle Growth and Atrophy. FEBS J. 2013, 280, 4294–4314. [Google Scholar] [CrossRef] [PubMed]
- Webster, J.M.; Kempen, L.J.A.P.; Hardy, R.S.; Langen, R.C.J. Inflammation and Skeletal Muscle Wasting During Cachexia. Front. Physiol. 2020, 11, 597–675. [Google Scholar] [CrossRef]
- Lemmey, A.B.; Wilkinson, T.J.; Clayton, R.J.; Sheikh, F.; Whale, J.; Jones, H.S.J.; Ahmad, Y.A.; Chitale, S.; Jones, J.G.; Maddison, P.J.; et al. Tight Control of Disease Activity Fails to Improve Body Composition or Physical Function in Rheumatoid Arthritis Patients. Rheumathology 2016, 55, 1736–1745. [Google Scholar] [CrossRef]
- Roubenoff, R. The “Cytokine for Gerontologists” Has Some Company. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2014, 69, 163–164. [Google Scholar] [CrossRef]
- Moresi, V.; Adamo, S.; Berghella, L. The JAK / STAT Pathway in Skeletal Muscle Pathophysiology. Front. Physiol. 2019, 10, 500. [Google Scholar] [CrossRef]
- Kami, K.; Senba, E. In Vivo Activation of STAT3 Signaling in Satellite Cells and Myofibers in Regenerating Rat Skeletal Muscles. J. Histochem. Cytochem. 2002, 50, 1579–1589. [Google Scholar] [CrossRef]
- Spangenburg, E.E.; Booth, F.W. Multiple Signaling Pathways Mediate LIF-Induced Skeletal Muscle Satellite Cell Proliferation. Am. J. Physiol. Cell Physiol. 2002, 283, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Xu, Y.; Li, W.; Wang, G.; Song, Y.; Yang, G.; Han, X.; Du, Z.; Sun, L.; Ma, K. STAT3 Induces Muscle Stem Cell Differentiation by Interaction with MyoD. Cytokine 2009, 46, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Cánoves, P.; Scheele, C.; Pedersen, B.K.; Serrano, A.L. Interleukin-6 Myokine Signaling in Skeletal Muscle: A Double-Edged Sword? FEBS J. 2013, 280, 4131–4148. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, B.K.; Febbraio, M.A. Muscle as an Endocrine Organ: Focus on Muscle-Derived Interleukin-6. Physiol. Rev. 2008, 88, 1379–1406. [Google Scholar] [CrossRef]
- Sun, L.; Ma, K.; Wang, H.; Xiao, F.; Gao, Y.; Zhang, W.; Wang, K.; Gao, X.; Ip, N.; Wu, Z. JAK1-STAT1-STAT3, a Key Pathway Promoting Proliferation and Preventing Premature Differentiation of Myoblasts. J. Cell Biol. 2007, 179, 129–138. [Google Scholar] [CrossRef]
- Little, R.D.; Prieto-Potin, I.; Pérez-Baos, S.; Villalvilla, A.; Gratal, P.; Cicuttini, F.; Largo, R.; Herrero-Beaumont, G. Compensatory Anabolic Signaling in the Sarcopenia of Experimental Chronic Arthritis. Sci. Rep. 2017, 7, 6311. [Google Scholar] [CrossRef]
- Baird, M.F.; Graham, S.M.; Baker, J.S.; Bickerstaff, G.F. Creatine-Kinase- and Exercise-Related Muscle Damage Implications for Muscle Performance and Recovery. J. Nutr. Metab. 2012, 2012, 960363. [Google Scholar] [CrossRef]
- O’Connor, R.S.; Steeds, C.M.; Wiseman, R.W.; Pavlath, G.K. Phosphocreatine as an Energy Source for Actin Cytoskeletal Rearrangements during Myoblast Fusion: Phosphocreatine Regulates Cell Fusion. J. Physiol. 2008, 586, 2841–2853. [Google Scholar] [CrossRef]
- Saks, V. The Phosphocreatine-Creatine Kinase System Helps to Shape Muscle Cells and Keep Them Healthy and Alive. J. Physiol. 2008, 586, 2817–2818. [Google Scholar] [CrossRef]
- Kyriakides, T.; Angelini, C.; Schaefer, J.; Sacconi, S.; Siciliano, G.; Vilchez, J.J.; Hilton-Jones, D. EFNS Guidelines on the Diagnostic Approach to Pauci- or Asymptomatic HyperCKemia. Eur. J. Neurol. 2010, 17, 767–773. [Google Scholar] [CrossRef]
- Moghadam-Kia, S.; Oddis, C.V.; Aggarwal, R. Approach to Asymptomatic Creatine Kinase Elevation. CCJM 2016, 83, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Panaccione, R.; Isaacs, J.D.; Chen, L.A.; Wang, W.; Marren, A.; Kwok, K.; Wang, L.; Chan, G.; Su, C. Characterization of Creatine Kinase Levels in Tofacitinib-Treated Patients with Ulcerative Colitis: Results from Clinical Trials. Dig. Dis. Sci. 2021, 66, 2732–2743. [Google Scholar] [CrossRef] [PubMed]
- Anjara, P.; Jiang, M.; Mundae, M. Symptomatic Elevation Creatine Kinase Following Treatment of Rheumatoid Arthritis with Baricitinib. Clin. Rheumatol. 2020, 39, 613–614. [Google Scholar] [CrossRef] [PubMed]
- Giles, J.T.; Ling, S.M.; Ferrucci, L.; Bartlett, S.J.; Andersen, R.E.; Towns, M.; Muller, D.; Fontaine, K.R.; Bathon, J.M. Abnormal Body Composition Phenotypes in Older Rheumatoid Arthritis Patients: Association with Disease Characteristics and Pharmacotherapies. Arthritis Rheum. 2008, 59, 807–815. [Google Scholar] [CrossRef]
- Isaacs, J.D.; Zuckerman, A.; Krishnaswami, S.; Nduaka, C.; Lan, S.; Hutmacher, M.M.; Boy, M.G.; Kowalski, K.; Menon, S.; Riese, R. Changes in Serum Creatinine in Patients with Active Rheumatoid Arthritis Treated with Tofacitinib: Results from Clinical Trials. Arthritis Res. Ther. 2014, 16, R158. [Google Scholar] [CrossRef]
- Bissonnette, R.; Iversen, L.; Sofen, H.; Griffiths, C.E.M.; Foley, P.; Romiti, R.; Bachinsky, M.; Rottinghaus, S.T.; Tan, H.; Proulx, J.; et al. Tofacitinib Withdrawal and Retreatment in Moderate-to-Severe Chronic Plaque Psoriasis: A Randomized Controlled Trial. Br. J. Dermatol. 2015, 172, 1395–1406. [Google Scholar] [CrossRef]
- Romero, F.; Martínez-Calatrava, M.; Sánchez-Pernaute, O.; Gualillo, O.; Largo, R.; Herrero-Beaumont, G. Pharmacological Modulation by Celecoxib of Cachexia Associated with Experimental Arthritis and Atherosclerosis in Rabbits: PGE2 Inhibition in RA-Associated Cachexia. Br. J. Pharmacol. 2010, 161, 1012–1022. [Google Scholar] [CrossRef]
- Filippin, L.I.; Teixeira, V.N.; Viacava, P.R.; Lora, P.S.; Xavier, L.L.; Xavier, R.M. Temporal Development of Muscle Atrophy in Murine Model of Arthritis Is Related to Disease Severity. J. Cachexia Sarcopenia Muscle 2013, 4, 231–238. [Google Scholar] [CrossRef]
- Castillero, E.; Martín, A.I.; López-Menduiña, M.; Granado, M.; Villanúa, M.Á.; López-Calderón, A. IGF-I System, Atrogenes and Myogenic Regulatory Factors in Arthritis Induced Muscle Wasting. Mol. Cell. Endocrinol. 2009, 309, 8–16. [Google Scholar] [CrossRef]
- Trendelenburg, A.U.; Meyer, A.; Rohner, D.; Boyle, J.; Hatakeyama, S.; Glass, D.J. Myostatin Reduces Akt/TORC1/P70S6K Signaling, Inhibiting Myoblast Differentiation and Myotube Size. Am. J. Physiol. Cell Physiol. 2009, 296, C1258–C1270. [Google Scholar] [CrossRef]
- Wang, D.-T.; Yang, Y.-J.; Huang, R.-H.; Zhang, Z.-H.; Lin, X. Myostatin Activates the Ubiquitin-Proteasome and Autophagy-Lysosome Systems Contributing to Muscle Wasting in Chronic Kidney Disease. Oxidative Med. Cell. Longev. 2015, 2015, 684965. [Google Scholar] [CrossRef] [PubMed]
- Vainshtein, A.; Sandri, M. Signaling Pathways That Control Muscle Mass. Int. J. Mol. Sci. 2020, 21, 4759. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.-M.; Zhao, Y.-P.; Zhao, Y.; Deng, S.-L.; Yu, K. Regulation of Myostatin on the Growth and Development of Skeletal Muscle. Front. Cell Dev. Biol. 2021, 9, 785712. [Google Scholar] [CrossRef] [PubMed]
- Evers-van Gogh, I.; Oteng, A.-B.; Alex, S.; Hamers, N.; Catoire, M.; Stienstra, R.; Kalkhoven, E.; Kersten, S. Muscle-Specific Inflammation Induced by MCP-1 Overexpression Does Not Affect Whole-Body Insulin Sensitivity in Mice. Diabetologia 2016, 59, 624–633. [Google Scholar] [CrossRef] [PubMed]
- Luciano-Mateo, F.; Cabré, N.; Fernández-Arroyo, S.; Baiges-Gaya, G.; Hernández-Aguilera, A.; Rodríguez-Tomàs, E.; Muñoz-Pinedo, C.; Menéndez, J.A.; Camps, J.; Joven, J. Chemokine C–C Motif Ligand 2 Overexpression Drives Tissue-Specific Metabolic Responses in the Liver and Muscle of Mice. Sci. Rep. 2020, 10, 11954. [Google Scholar] [CrossRef]
- Broholm, C.; Laye, M.J.; Brandt, C.; Vadalasetty, R.; Pilegaard, H.; Pedersen, B.K.; Scheele, C. LIF Is a Contraction-Induced Myokine Stimulating Human Myocyte Proliferation. J. Appl. Physiol. 2011, 111, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Serrano, A.L.; Baeza-Raja, B.; Perdiguero, E.; Jardí, M.; Muñoz-Cánoves, P. Interleukin-6 Is an Essential Regulator of Satellite Cell-Mediated Skeletal Muscle Hypertrophy. Cell Metab. 2008, 7, 33–44. [Google Scholar] [CrossRef]
- Guerci, A.; Lahoute, C.; Hébrard, S.; Collard, L.; Graindorge, D.; Favier, M.; Cagnard, N.; Batonnet-Pichon, S.; Précigout, G.; Garcia, L.; et al. Srf-Dependent Paracrine Signals Produced by Myofibers Control Satellite Cell-Mediated Skeletal Muscle Hypertrophy. Cell Metab. 2012, 15, 25–37. [Google Scholar] [CrossRef]
- Nunes, A.M.; Wuebbles, R.D.; Sarathy, A.; Fontelonga, T.M.; Deries, M.; Burkin, D.J.; Thorsteinsdóttir, S. Impaired Fetal Muscle Development and JAK-STAT Activation Mark Disease Onset and Progression in a Mouse Model for Merosin-Deficient Congenital Muscular Dystrophy. Hum. Mol. Genet. 2017, 26, 2018–2033. [Google Scholar] [CrossRef]
- Wang, K.; Wang, C.; Xiao, F.; Wang, H.; Wu, Z. JAK2/STAT2/STAT3 Are Required for Myogenic Differentiation. J. Biol. Chem. 2008, 283, 34029–34036. [Google Scholar] [CrossRef]
- Kataoka, Y.; Matsumura, I.; Ezoe, S.; Nakata, S.; Takigawa, E.; Sato, Y.; Kawasaki, A.; Yokota, T.; Nakajima, K.; Felsani, A.; et al. Reciprocal Inhibition between MyoD and STAT3 in the Regulation of Growth and Differentiation of Myoblasts. J. Biol. Chem. 2003, 278, 44178–44187. [Google Scholar] [CrossRef] [PubMed]
- Wormald, S.; Hilton, D.J. Inhibitors of Cytokine Signal Transduction. J. Biol. Chem. 2004, 279, 821–824. [Google Scholar] [CrossRef] [PubMed]
- Lieskovska, J.; Guo, D.; Derman, E. Growth Impairment in IL-6-Overexpressing Transgenic Mice Is Associated with Induction of SOCS3 MRNA. Growth Horm. IGF Res. 2003, 13, 26–35. [Google Scholar] [CrossRef]
- Bonetto, A.; Aydogdu, T.; Jin, X.; Zhang, Z.; Zhan, R.; Puzis, L.; Koniaris, L.G.; Zimmers, T.A. JAK/STAT3 Pathway Inhibition Blocks Skeletal Muscle Wasting Downstream of IL-6 and in Experimental Cancer Cachexia. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E410–E421. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Baos, S.; Gratal, P.; Barrasa, J.I.; Lamuedra, A.; Sánchez-pernaute, O.; Herrero-beaumont, G.; Largo, R. Inhibition of PSTAT1 by Tofacitinib Accounts for the Early Improvement of Experimental Chronic Synovitis. J. Inflamm. 2019, 16, 2. [Google Scholar] [CrossRef]
- Pérez-Baos, S.; Prieto-Potin, I.; Román-Blas, J.A.; Sánchez-Pernaute, O.; Largo, R.; Herrero-Beaumont, G. Mediators and Patterns of Muscle Loss in Chronic Systemic Inflammation. Front. Physiol. 2018, 9, 409. [Google Scholar] [CrossRef]
- Hein, T.R. The Effect of Disease-Modifying Anti-Rheumatic Drugs on Skeletal Muscle Mass in Rheumatoid Arthritis Patients: A Systematic Review with Meta-Analysis. Arthritis Res. Ther. 2022, 24, 171. [Google Scholar] [CrossRef]
- Bennett, J.L.; Egail, M.; Anderson, A.E.; Dodds, R.; Feeney, C. The Rheumatoid Arthritis and MUScle (RAMUS) Study: Protocol for an Observational Single-Arm Study of Skeletal Muscle in Patients with Rheumatoid Arthritis Receiving Tofacitinib. J. Frailty Sarcopenia Falls 2023, 8, 53–59. [Google Scholar] [CrossRef]
- Stucki, G.; Brühlmann, P.; Stoll, T.; Stucki, S.; Willer, B.; Michel, B. Low Serum Creatine Kinase Activity Is Associated with Muscle Weakness in Patients with Rheumatoid Arthritis. J. Rheumatol. 1996, 23, 603–608. [Google Scholar]
- Yildirim, K.; Erdal, A.; Karatay, S.; Ugur, M.; Senel, K. Serum Creatine Kinase Levels as Inflammatory Marker in Patients with Rheumatoid Arthritis. Pain Clin. 2002, 14, 303–307. [Google Scholar] [CrossRef]
- Lee, Y.; Choi, S.; Ji, D.; Song, G. Serum Creatine Kinase in Patients with Rheumatic Diseases. Clin. Rheumatol. 2000, 19, 296–300. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, A.A.; Strunz, C.M.; Takada, J.Y.; Mansur, A.P. Biochemical Markers of Muscle Damage and High Serum Concentration of Creatine Kinase in Patients on Statin Therapy. Biomark. Med. 2019, 13, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Sanmartí, R.; Collado, A.; Gratacós, J.; Bedini, J.L.; Pañella, D.; Filella, X.; Llena, J.; Muñoz-Gomez, J. Reduced Activity of Serum Creatine Kinase in Rheumatoid Arthritis: A Phenomenon Linked to the Inflammatory Response. Br. J. Rheumatol. 1994, 33, 231–234. [Google Scholar] [CrossRef] [PubMed]
- Pasut, A.; Jones, A.E.; Rudnicki, M.A. Isolation and Culture of Individual Myofibers and Their Satellite Cells from Adult Skeletal Muscle. J. Vis. Exp. 2013, 73, e50074. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bermejo-Álvarez, I.; Pérez-Baos, S.; Gratal, P.; Medina, J.P.; Largo, R.; Herrero-Beaumont, G.; Mediero, A. Effects of Tofacitinib on Muscle Remodeling in Experimental Rheumatoid Sarcopenia. Int. J. Mol. Sci. 2023, 24, 13181. https://doi.org/10.3390/ijms241713181
Bermejo-Álvarez I, Pérez-Baos S, Gratal P, Medina JP, Largo R, Herrero-Beaumont G, Mediero A. Effects of Tofacitinib on Muscle Remodeling in Experimental Rheumatoid Sarcopenia. International Journal of Molecular Sciences. 2023; 24(17):13181. https://doi.org/10.3390/ijms241713181
Chicago/Turabian StyleBermejo-Álvarez, Ismael, Sandra Pérez-Baos, Paula Gratal, Juan Pablo Medina, Raquel Largo, Gabriel Herrero-Beaumont, and Aránzazu Mediero. 2023. "Effects of Tofacitinib on Muscle Remodeling in Experimental Rheumatoid Sarcopenia" International Journal of Molecular Sciences 24, no. 17: 13181. https://doi.org/10.3390/ijms241713181