
The Ins and Outs of Clusterin: Its Role in Cancer, Eye Diseases and Wound Healing

 and
and 
Abstract
:1. Introduction
1.1. Background
1.2. CLU Expression
1.3. Functions
2. Gene and Protein Structure
2.1. CLU Gene
2.2. CLU Protein and Biosynthesis
2.3. CLU Protein Isoforms
2.3.1. CLU Secretory Isoform (sCLU)
2.3.2. CLU Nuclear Isoform (nCLU)

3. Clusterin Gene Regulation
3.1. Epigenetic Regulation
3.1.1. Modification of Histones
3.1.2. DNA Methylation
3.1.3. MicroRNA
3.2. Stress-Inducible Genes
3.3. Transcription Factors
3.3.1. AP-1
3.3.2. MYC and Ha-RAS
3.3.3. NF-κB
3.3.4. IGFR Pathway
3.3.5. YB-1 and HIF-1a Signaling
3.3.6. PAX6
3.3.7. Nuclear Factor E2-Related Factor 2 (Nrf2)
3.3.8. Hormonal Regulation of CLU Gene Expression
4. Clusterin in Tissue Remodeling
4.1. Differentiation
4.2. Migration and Invasion
4.3. Proliferation
4.4. Apoptosis vs. Cytoprotection
5. Clusterin and Cancer
5.1. Clusterin Isoforms’ Function in Cancer Progression
5.2. Tumorigenesis
5.3. Epithelial to Mesenchymal Transition and Metastasis
5.4. CLU Oncogenic Biomarker
5.5. Chemoresistance and Chemosensitivity with Clusterin
5.6. Clinical Trials of CLU Inhibitors
6. Clusterin in the Eye: Relationship with Eye Disorders
6.1. The Anterior Segment of the Eye
6.1.1. Dry Eye Disease (DED)
6.1.2. Corneal Dystrophies
6.1.3. Stem Cell Culture and Transplantation on the Ocular Surface
6.1.4. Pseudoexfoliative Glaucoma
6.2. The Posterior Segment of the Eye
6.2.1. Age-Related Macular Degeneration (AMD)
6.2.2. Retinal Diseases
6.2.3. Uveal Melanoma (UM)
7. Clusterin and Wound Healing: The Tissue-Engineered Human Cornea as a Model
8. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Blaschuk, O.; Burdzy, K.; Fritz, I.B. Purification and characterization of a cell-aggregating factor (clusterin), the major glycoprotein in ram rete testis fluid. J. Biol. Chem. 1983, 258, 7714–7720. [Google Scholar] [CrossRef] [PubMed]
- Sylvester, S.R.; Skinner, M.K.; Griswold, M.D. A Sulfated Glycoprotein Synthesized by Sertoli Cells and by Epididymal Cells is a Component of the Sperm Membrane. Biol. Reprod. 1984, 31, 1087–1101. [Google Scholar] [CrossRef] [PubMed]
- Léger, J.G.; Montpetit, M.L.; Tenniswood, M.P. Characterization and cloning of androgen-repressed mRNAs from rat ventral prostate. Biochem. Biophys. Res. Commun. 1987, 147, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Herault, Y.; Chatelain, G.; Brun, G.; Michel, D. V-src-induced-transcription of the avian clusterin gene. Nucleic Acids Res. 1992, 20, 6377–6383. [Google Scholar] [CrossRef]
- Palmer, D.J.; Christie, D.L. The primary structure of glycoprotein III from bovine adrenal medullary chromaffin granules. Sequence similarity with human serum protein-40,40 and rat Sertoli cell glycoprotein. J. Biol. Chem. 1990, 265, 6617–6623. [Google Scholar] [CrossRef]
- Murphy, B.F.; Kirszbaum, L.; Walker, I.D.; d’Apice, A.J. SP-40,40, a newly identified normal human serum protein found in the SC5b-9 complex of complement and in the immune deposits in glomerulonephritis. J. Clin. Investig. 1988, 81, 1858–1864. [Google Scholar] [CrossRef]
- De Silva, H.V.; Stuart, W.D.; Duvic, C.R.; Wetterau, J.R.; Ray, M.J.; Ferguson, D.G.; Albers, H.W.; Smith, W.R.; Harmony, J.A. A 70-kDa apolipoprotein designated ApoJ is a marker for subclasses of human plasma high density lipoproteins. J. Biol. Chem. 1990, 265, 13240–13247. [Google Scholar] [CrossRef]
- Jenne, D.E.; Tschopp, J. Molecular structure and functional characterization of a human complement cytolysis inhibitor found in blood and seminal plasma: Identity to sulfated glycoprotein 2, a constituent of rat testis fluid. Proc. Natl. Acad. Sci. USA 1989, 86, 7123–7127. [Google Scholar] [CrossRef]
- May, P.C.; Lampert-Etchells, M.; Johnson, S.A.; Poirier, J.; Masters, J.N.; Finch, C.E. Dynamics of gene expression for a hippocampal glycoprotein elevated in Alzheimer’s disease and in response to experimental lesions in rat. Neuron 1990, 5, 831–839. [Google Scholar] [CrossRef]
- Danik, M.; Chabot, J.G.; Mercier, C.; Benabid, A.L.; Chauvin, C.; Quirion, R.; Suh, M. Human gliomas and epileptic foci express high levels of a mRNA related to rat testicular sulfated glycoprotein 2, a purported marker of cell death. Proc. Natl. Acad. Sci. USA 1991, 88, 8577–8581. [Google Scholar] [CrossRef]
- Gene: CLU. Available online: http://useast.ensembl.org/Homo_sapiens/Gene/Summary?db=core;g=ENSG00000120885;r=8:27596917-27614700 (accessed on 2 February 2022).
- Wong, P.; Taillefer, D.; Lakins, J.; Pineault, J.; Chader, G.; Tenniswood, M. Molecular characterization of human TRPM-2/clusterin, a gene associated with sperm maturation, apoptosis and neurodegeneration. Eur. J. Biochem. 1994, 221, 917–925. [Google Scholar] [CrossRef] [PubMed]
- Aronow, B.J.; Lund, S.D.; Brown, T.L.; Harmony, J.A.; Witte, D.P. Apolipoprotein J expression at fluid-tissue interfaces: Potential role in barrier cytoprotection. Proc. Natl. Acad. Sci. USA 1993, 90, 725–729. [Google Scholar] [CrossRef] [PubMed]
- De Silva, H.V.; Harmony, J.A.K.; Stuart, W.D.; Gil, C.M.; Robbins, J. Apolipoprotein J: Structure and tissue distribution. Biochemistry 1990, 29, 5380–5389. [Google Scholar] [CrossRef] [PubMed]
- Ahuja, H.S.; Tenniswood, M.; Lockshin, R.; Zakeri, Z.F. Expression of clusterin in cell differentiation and cell death. Biochem. Cell Biol. 1994, 72, 523–530. [Google Scholar] [CrossRef] [PubMed]
- French, L.E.; Chonn, A.; Ducrest, D.; Baumann, B.; Belin, D.; Wohlwend, A.; Kiss, J.Z.; Sappino, A.-P.; Tschopp, J.; Schifferli, J.A. Murine clusterin: Molecular cloning and mRNA localization of a gene associated with epithelial differentiation processes during embryogenesis. J. Cell Biol. 1993, 122, 1119–1130. [Google Scholar] [CrossRef]
- Tenniswood, M.P.; Guenette, R.S.; Lakins, J.; Mooibroek, M.; Wong, P.; Welsh, J.E. Active cell death in hormone-dependent tissues. Cancer Metastasis Rev. 1992, 11, 197–220. [Google Scholar] [CrossRef]
- Wong, P.; Pineault, J.; Lakins, J.; Taillefer, D.; Léger, J.; Wang, C.; Tenniswood, M. Genomic organization and expression of the rat TRPM-2 (clusterin) gene, a gene implicated in apoptosis. J. Biol. Chem. 1993, 268, 5021–5031. [Google Scholar] [CrossRef]
- Silkensen, J.R.; Skubitz, K.M.; Skubitz, A.P.; Chmielewski, D.H.; Manivel, J.C.; Dvergsten, J.A.; Rosenberg, M.E. Clusterin promotes the aggregation and adhesion of renal porcine epithelial cells. J. Clin. Investig. 1995, 96, 2646–2653. [Google Scholar] [CrossRef]
- Tung, P.S.; Burdzy, K.; Wong, K.; Fritz, I.B. Competition between cell-substratum interactions and cell-cell interactions. J. Cell. Physiol. 1992, 152, 410–421. [Google Scholar] [CrossRef]
- Zhang, H.; Kim, J.K.; Edwards, C.A.; Xu, Z.; Taichman, R.; Wang, C.-Y. Clusterin inhibits apoptosis by interacting with activated Bax. Nat. Cell Biol. 2005, 7, 909–915. [Google Scholar] [CrossRef]
- Yang, C.-R.; Leskov, K.; Hosley-Eberlein, K.; Criswell, T.; Pink, J.J.; Kinsella, T.J.; Boothman, D.A. Nuclear clusterin/XIP8, an x-ray-induced Ku70-binding protein that signals cell death. Proc. Natl. Acad. Sci. USA 2000, 97, 5907–5912. [Google Scholar] [CrossRef] [PubMed]
- Leskov, K.S.; Klokov, D.Y.; Li, J.; Kinsella, T.J.; Boothman, D.A. Synthesis and functional analyses of nuclear clusterin, a cell death protein. J. Biol. Chem. 2003, 278, 11590–11600. [Google Scholar] [CrossRef] [PubMed]
- Xiu, P.; Dong, X.; Dong, X.; Xu, Z.; Zhu, H.; Liu, F.; Wei, Z.; Zhai, B.; Kanwar, J.R.; Jiang, H.; et al. Secretory clusterin contributes to oxaliplatin resistance by activating Akt pathway in hepatocellular carcinoma. Cancer Sci. 2013, 104, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Redondo, M.; Téllez, T.; Roldan, M.J.; Serrano, A.; García-Aranda, M.; Gleave, M.E.; Hortas, M.L.; Morell, M. Anticlusterin treatment of breast cancer cells increases the sensitivities of chemotherapy and tamoxifen and counteracts the inhibitory action of dexamethasone on chemotherapy-induced cytotoxicity. Breast Cancer Res. BCR 2007, 9, R86. [Google Scholar] [CrossRef] [PubMed]
- Miyake, H.; Nelson, C.; Rennie, P.S.; Gleave, M.E. Acquisition of Chemoresistant Phenotype by Overexpression of the Antiapoptotic Gene Testosterone-repressed Prostate Message-2 in Prostate Cancer Xenograft Models. Cancer Res. 2000, 60, 2547–2554. [Google Scholar]
- Poon, S.; Easterbrook-Smith, S.B.; Rybchyn, M.S.; Carver, J.A.; Wilson, M.R. Clusterin Is an ATP−Independent Chaperone with Very Broad Substrate Specificity that Stabilizes Stressed Proteins in a Folding-Competent State. Biochemistry 2000, 39, 15953–15960. [Google Scholar] [CrossRef]
- Humphreys, D.T.; Carver, J.A.; Easterbrook-Smith, S.B.; Wilson, M.R. Clusterin Has Chaperone-like Activity Similar to That of Small Heat Shock Proteins. J. Biol. Chem. 1999, 274, 6875–6881. [Google Scholar] [CrossRef]
- Wilson, M.R.; Easterbrook-Smith, S.B. Clusterin is a secreted mammalian chaperone. Trends Biochem. Sci. 2000, 25, 95–98. [Google Scholar] [CrossRef]
- Bailey, R.W.; Dunker, A.K.; Brown, C.J.; Garner, E.C.; Griswold, M.D. Clusterin, a Binding Protein with a Molten Globule-like Region †. Biochemistry 2001, 40, 11828–11840. [Google Scholar] [CrossRef]
- Poon, S.; Rybchyn, M.S.; Easterbrook-Smith, S.B.; Carver, J.A.; Pankhurst, G.J.; Wilson, M.R. Mildly Acidic pH Activates the Extracellular Molecular Chaperone Clusterin. J. Biol. Chem. 2002, 277, 39532–39540. [Google Scholar] [CrossRef]
- Yerbury, J.J.; Poon, S.; Meehan, S.; Thompson, B.; Kumita, J.R.; Dobson, C.M.; Wilson, M.R. The extracellular chaperone clusterin influences amyloid formation and toxicity by interacting with prefibrillar structures. FASEB J. 2007, 21, 2312–2322. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Yoo, J.C.; Han, J.Y.; Hwang, E.M.; Kim, Y.S.; Jeong, E.Y.; Sun, C.-H.; Yi, G.-S.; Roh, G.S.; Kim, H.J.; et al. Human nuclear clusterin mediates apoptosis by interacting with Bcl-XL through C-terminal coiled coil domain. J. Cell. Physiol. 2012, 227, 1157–1167. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.-R.; Yeh, S.; Leskov, K.; Odegaard, E.; Hsu, H.-L.; Chang, C.; Kinsella, T.J.; Chen, D.J.; Boothman, D.A. Isolation of Ku70-binding proteins (KUBs). Nucleic Acids Res. 1999, 27, 2165–2174. [Google Scholar] [CrossRef] [PubMed]
- Sawada, M.; Hayes, P.; Matsuyama, S. Cytoprotective membrane-permeable peptides designed from the Bax-binding domain of Ku70. Nat. Cell Biol. 2003, 5, 352–357. [Google Scholar] [CrossRef]
- Shim, Y.-J.; Kang, B.-H.; Jeon, H.-S.; Park, I.-S.; Lee, K.-U.; Lee, I.-K.; Park, G.-H.; Lee, K.-M.; Schedin, P.; Min, B.-H. Clusterin induces matrix metalloproteinase-9 expression via ERK1/2 and PI3K/Akt/NF-κB pathways in monocytes/macrophages. J. Leukoc. Biol. 2011, 90, 761–769. [Google Scholar] [CrossRef]
- Ammar, H.; Closset, J.L. Clusterin Activates Survival through the Phosphatidylinositol 3-Kinase/Akt Pathway. J. Biol. Chem. 2008, 283, 12851–12861. [Google Scholar] [CrossRef]
- Leskov, K.S.; Araki, S.; Lavik, J.-P.; Gomez, J.A.; Gama, V.; Gonos, E.S.; Trougakos, I.P.; Matsuyama, S.; Boothman, D.A. CRM1 Protein-mediated Regulation of Nuclear Clusterin (nCLU), an Ionizing Radiation-stimulated, Bax-dependent Pro-death Factor. J. Biol. Chem. 2011, 286, 40083–40090. [Google Scholar] [CrossRef]
- Rizzi, F.; Coletta, M.; Bettuzzi, S. Chapter 2: Clusterin (CLU): From One Gene and Two Transcripts to Many Proteins. In Advances in Cancer Research; Academic Press: Cambridge, MA, USA, 2009; Volume 104, pp. 9–23. [Google Scholar]
- Jenne, D.E.; Tschopp, J. Clusterin: The intriguing guises of a widely expressed glycoprotein. Trends Biochem. Sci. 1992, 17, 154–159. [Google Scholar] [CrossRef]
- Jones, S.E.; Jomary, C. Clusterin. Int. J. Biochem. Cell Biol. 2002, 34, 427–431. [Google Scholar] [CrossRef]
- NCBI CLU Transcripts. Available online: https://www.ncbi.nlm.nih.gov/datasets/tables/genes/?table_type=transcripts&key=6b8b96426de89efc26c6d71d4eef3526 (accessed on 14 April 2023).
- Prochnow, H.; Gollan, R.; Rohne, P.; Hassemer, M.; Koch-Brandt, C.; Baiersdörfer, M. Non-Secreted Clusterin Isoforms Are Translated in Rare Amounts from Distinct Human mRNA Variants and Do Not Affect Bax-Mediated Apoptosis or the NF-κB Signaling Pathway. PLoS ONE 2013, 8, e75303. [Google Scholar] [CrossRef]
- Andersen, C.L.; Schepeler, T.; Thorsen, K.; Birkenkamp-Demtröder, K.; Mansilla, F.; Aaltonen, L.A.; Laurberg, S.; Ørntoft, T.F. Clusterin Expression in Normal Mucosa and Colorectal Cancer. Mol. Cell. Proteom. 2007, 6, 1039–1048. [Google Scholar] [CrossRef] [PubMed]
- Bonacini, M.; Coletta, M.; Ramazzina, I.; Naponelli, V.; Modernelli, A.; Davalli, P.; Bettuzzi, S.; Rizzi, F. Distinct promoters, subjected to epigenetic regulation, drive the expression of two clusterin mRNAs in prostate cancer cells. Biochim. Biophys. Acta (BBA) Gene Regul. Mech. 2015, 1849, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Park, S.Y.; Shin, E.; Lee, S.H.; Kim, Y.S.; Lee, D.H.; Roh, G.S.; Kim, H.J.; Kang, S.S.; Cho, G.J.; et al. Hypoxia Inducible Factor-1α Directly Regulates Nuclear Clusterin Transcription by Interacting with Hypoxia Response Elements in the Clusterin Promoter. Mol. Cells 2014, 37, 178–186. [Google Scholar] [CrossRef] [PubMed]
- Reddy, K.B.; Jin, G.; Karode, M.C.; Harmony, J.A.K.; Howe, P.H. Transforming Growth Factor β (TGFβ)-Induced Nuclear Localization of Apolipoprotein J/Clusterin in Epithelial Cells. Biochemistry 1996, 35, 6157–6163. [Google Scholar] [CrossRef] [PubMed]
- Burkey, B.F.; deSilva, H.V.; Harmony, J.A. Intracellular processing of apolipoprotein J precursor to the mature heterodimer. J. Lipid Res. 1991, 32, 1039–1048. [Google Scholar] [CrossRef]
- Viard, I.; Wehrli, P.; Jornot, L.; Bullani, R.; Vechietti, J.-L.; French, L.E.; Schifferli, J.A.; Tschopp, J. Clusterin Gene Expression Mediates Resistance to Apoptotic Cell Death Induced by Heat Shock and Oxidative Stress. J. Investig. Dermatol. 1999, 112, 290–296. [Google Scholar] [CrossRef]
- Zhang, B.; Zhang, K.; Liu, Z.; Hao, F.; Wang, M.; Li, X.; Yin, Z.; Liang, H. Secreted Clusterin Gene Silencing Enhances Chemosensitivity of A549 Cells to Cisplatin Through AKT and ERK1/2 Pathways in Vitro. Cell. Physiol. Biochem. 2014, 33, 1162–1175. [Google Scholar] [CrossRef]
- Kapron, J.T.; Hilliard, G.M.; Lakins, J.N.; Tenniswood, M.P.R.; West, K.A.; Carr, S.A.; Crabb, J.W. Identification and characterization of glycosylation sites in human serum clusterin. Protein Sci. 2008, 6, 2120–2133. [Google Scholar] [CrossRef]
- Choi-Miura, N.H.; Takahashi, Y.; Nakano, Y.; Tobe, T.; Tomita, M. Identification of the Disulfide Bonds in Human Plasma Protein SP-40,40 (Apolipoprotein-J). J. Biochem. 1992, 112, 557–561. [Google Scholar] [CrossRef]
- Tenniswood, M.; Wang, Z.; Lakins, J.; Morrissey, C.; O’sullivan, J.; Tang, H. Clusterin in the Male Reproductive Tract. J. Androl. 1998, 19, 508–516. [Google Scholar]
- Nizard, P.; Tetley, S.; Dréan, Y.L.; Watrin, T.; Goff, P.L.; Wilson, M.R.; Michel, D. Stress-Induced Retrotranslocation of Clusterin/ApoJ into the Cytosol. Traffic 2007, 8, 554–565. [Google Scholar] [CrossRef] [PubMed]
- Shannan, B.; Seifert, M.; Leskov, K.; Willis, J.; Boothman, D.; Tilgen, W.; Reichrath, J. Challenge and promise: Roles for clusterin in pathogenesis, progression and therapy of cancer. Cell Death Differ. 2006, 13, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Kounnas, M.Z.; Loukinova, E.B.; Stefansson, S.; Harmony, J.A.K.; Brewer, B.H.; Strickland, D.K.; Argraves, W.S. Identification of Glycoprotein 330 as an Endocytic Receptor for Apolipoprotein J/Clusterin. J. Biol. Chem. 1995, 270, 13070–13075. [Google Scholar] [CrossRef]
- NCBI CLU Clusterin [Homo Sapiens (Human)]. Available online: https://www.ncbi.nlm.nih.gov/gene/1191 (accessed on 29 June 2023).
- Schier, A.C.; Taatjes, D.J. Structure and mechanism of the RNA polymerase II transcription machinery. Genes. Dev. 2020, 34, 465–488. [Google Scholar] [CrossRef]
- Pufall, M.A.; Kaplan, C.D. Mechanisms of eukaryotic transcription. Genome Biol. 2013, 14, 311. [Google Scholar] [CrossRef] [PubMed]
- Lambert, S.A.; Jolma, A.; Campitelli, L.F.; Das, P.K.; Yin, Y.; Albu, M.; Chen, X.; Taipale, J.; Hughes, T.R.; Weirauch, M.T. The Human Transcription Factors. Cell 2018, 175, 598–599. [Google Scholar] [CrossRef]
- Zhang, L.; Lu, Q.; Chang, C. Epigenetics in Health and Disease; Springer: Singapore, 2020; pp. 3–55. [Google Scholar]
- NCBI CLU SNPs. Available online: https://www.ncbi.nlm.nih.gov/snp/?term=CLU (accessed on 12 June 2023).
- Padhy, B.; Kapuganti, R.S.; Hayat, B.; Mohanty, P.P.; Alone, D.P. Wide-spread enhancer effect of SNP rs2279590 on regulating epoxide hydrolase-2 and protein tyrosine kinase 2-beta gene expression. Gene 2023, 854, 147096. [Google Scholar] [CrossRef]
- Balcar, V.J.; Zeman, T.; Janout, V.; Janoutova, J.; Lochman, J.; Sery, O. Single Nucleotide Polymorphism rs11136000 of CLU Gene (Clusterin, ApoJ) and the Risk of Late-Onset Alzheimer’s Disease in a Central European Population. Neurochem. Res. 2021, 46, 411–422. [Google Scholar] [CrossRef]
- Burdon, K.P.; Sharma, S.; Hewitt, A.W.; McMellon, A.E.; Wang, J.J.; Mackey, D.A.; Mitchell, P.; Craig, J.E. Genetic analysis of the clusterin gene in pseudoexfoliation syndrome. Mol. Vis. 2008, 14, 1727–1736. [Google Scholar]
- Demirdogen, B.C.; Demirkaya-Budak, S. Influence of clusterin genetic variants on IOP elevation in pseudoexfoliation syndrome and pseudoexfoliative glaucoma in Turkish population. BMC Ophthalmol. 2023, 23, 117. [Google Scholar] [CrossRef]
- Elhawy, E.; Kamthan, G.; Dong, C.Q.; Danias, J. Pseudoexfoliation syndrome, a systemic disorder with ocular manifestations. Hum. Genom. 2012, 6, 22. [Google Scholar] [CrossRef] [PubMed]
- Kuot, A.; Hewitt, A.W.; Griggs, K.; Klebe, S.; Mills, R.; Jhanji, V.; Craig, J.E.; Sharma, S.; Burdon, K.P. Association of TCF4 and CLU polymorphisms with Fuchs’ endothelial dystrophy and implication of CLU and TGFBI proteins in the disease process. Eur. J. Hum. Genet. 2012, 20, 632–638. [Google Scholar] [CrossRef]
- Padhy, B.; Hayat, B.; Nanda, G.G.; Mohanty, P.P.; Alone, D.P. Pseudoexfoliation and Alzheimer’s associated CLU risk variant, rs2279590, lies within an enhancer element and regulates CLU, EPHX2 and PTK2B gene expression. Hum. Mol. Genet. 2017, 26, 4519–4529. [Google Scholar] [CrossRef] [PubMed]
- Padhy, B.; Nanda, G.G.; Chowdhury, M.; Padhi, D.; Rao, A.; Alone, D.P. Role of an extracellular chaperone, Clusterin in the pathogenesis of Pseudoexfoliation Syndrome and Pseudoexfoliation Glaucoma. Exp. Eye Res. 2014, 127, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Rothbart, S.B.; Strahl, B.D. Interpreting the language of histone and DNA modifications. Biochim. Biophys. Acta 2014, 1839, 627–643. [Google Scholar] [CrossRef]
- Roth, S.Y.; Denu, J.M.; Allis, C.D. Histone acetyltransferases. Annu. Rev. Biochem. 2001, 70, 81–120. [Google Scholar] [CrossRef]
- Herman, J.G.; Baylin, S.B. Gene silencing in cancer in association with promoter hypermethylation. N. Engl. J. Med. 2003, 349, 2042–2054. [Google Scholar] [CrossRef]
- Litt, M.D.; Simpson, M.; Gaszner, M.; Allis, C.D.; Felsenfeld, G. Correlation between histone lysine methylation and developmental changes at the chicken beta-globin locus. Science 2001, 293, 2453–2455. [Google Scholar] [CrossRef]
- Liao, F.-T.; Lee, Y.-J.; Ko, J.-L.; Tsai, C.-C.; Tseng, C.-J.; Sheu, G.-T. Hepatitis delta virus epigenetically enhances clusterin expression via histone acetylation in human hepatocellular carcinoma cells. J. Gen. Virol. 2009, 90, 1124–1134. [Google Scholar] [CrossRef]
- Deb, M.; Sengupta, D.; Rath, S.K.; Kar, S.; Parbin, S.; Shilpi, A.; Pradhan, N.; Bhutia, S.K.; Roy, S.; Patra, S.K. Clusterin gene is predominantly regulated by histone modifications in human colon cancer and ectopic expression of the nuclear isoform induces cell death. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2015, 1852, 1630–1645. [Google Scholar] [CrossRef]
- Rauhala, H.E.; Porkka, K.P.; Saramäki, O.R.; Tammela, T.L.J.; Visakorpi, T. Clusterin is epigenetically regulated in prostate cancer. Int. J. Cancer 2008, 123, 1601–1609. [Google Scholar] [CrossRef]
- Hellebrekers, D.M.E.I.; Melotte, V.; Viré, E.; Langenkamp, E.; Molema, G.; Fuks, F.; Herman, J.G.; Criekinge, W.V.; Griffioen, A.W.; van Engeland, M. Identification of Epigenetically Silenced Genes in Tumor Endothelial Cells. Cancer Res. 2007, 67, 4138–4148. [Google Scholar] [CrossRef] [PubMed]
- Suuronen, T.; Nuutinen, T.; Ryhänen, T.; Kaarniranta, K.; Salminen, A. Epigenetic regulation of clusterin/apolipoprotein J expression in retinal pigment epithelial cells. Biochem. Biophys. Res. Commun. 2007, 357, 397–401. [Google Scholar] [CrossRef] [PubMed]
- Corvetta, D.; Chayka, O.; Gherardi, S.; D’Acunto, C.W.; Cantilena, S.; Valli, E.; Piotrowska, I.; Perini, G.; Sala, A. Physical interaction between MYCN oncogene and polycomb repressive complex 2 (PRC2) in neuroblastoma: Functional and therapeutic implications. J. Biol. Chem. 2013, 288, 8332–8341. [Google Scholar] [CrossRef] [PubMed]
- Lund, P.; Weißhaupt, K.; Mikeska, T.; Jammas, D.; Chen, X.; Kuban, R.-J.; Ungethüm, U.; Krapfenbauer, U.; Herzel, H.-P.; Schäfer, R.; et al. Oncogenic HRAS suppresses clusterin expression through promoter hypermethylation. Oncogene 2006, 25, 4890–4903. [Google Scholar] [CrossRef]
- Serrano, A.; Redondo, M.; Tellez, T.; Castro-Vega, I.; Roldan, M.J.; Mendez, R.; Rueda, A.; Jimenez, E. Regulation of Clusterin Expression in Human Cancer via DNA Methylation. Tumor Biol. 2009, 30, 286–291. [Google Scholar] [CrossRef]
- Yang, G.; Zhang, H.; Liu, Y.; Zhou, J.; He, W.; Quick, C.M.; Xie, D.; Smoller, B.R.; Fan, C.-Y. Epigenetic and immunohistochemical characterization of the Clusterin gene in ovarian tumors. Arch. Gynecol. Obstet. 2013, 287, 989–995. [Google Scholar] [CrossRef]
- Rosemblit, N.; Chen, C.-L.C. Regulators for the rat clusterin gene: DNA methylation and cis-acting regulatory elements. J. Mol. Endocrinol. 1994, 13, 69–76. [Google Scholar] [CrossRef]
- Mydlarz, W.; Uemura, M.; Ahn, S.; Hennessey, P.; Chang, S.; Demokan, S.; Sun, W.; Shao, C.; Bishop, J.; Krosting, J.; et al. Clusterin is a Gene Specific Target of MicroRNA-21 in Head and Neck Squamous Cell Carcinoma. Clin. Cancer Res. 2014, 20, 868–877. [Google Scholar] [CrossRef]
- Chayka, O.; Corvetta, D.; Dews, M.; Caccamo, A.E.; Piotrowska, I.; Santilli, G.; Gibson, S.; Sebire, N.J.; Himoudi, N.; Hogarty, M.D.; et al. Clusterin, a Haploinsufficient Tumor Suppressor Gene in Neuroblastomas. JNCI J. Natl. Cancer Inst. 2009, 101, 663–677. [Google Scholar] [CrossRef]
- Chen, X.; Jiang, Y.; Huang, Z.; Li, D.; Chen, X.; Cao, M.; Meng, Q.; Pang, H.; Sun, L.; Zhao, Y.; et al. miRNA-378 reverses chemoresistance to cisplatin in lung adenocarcinoma cells by targeting secreted clusterin. Sci. Rep. 2016, 6, 19455. [Google Scholar] [CrossRef] [PubMed]
- Martindale, J.L.; Holbrook, N.J. Cellular response to oxidative stress: Signaling for suicide and survival. J. Cell Physiol. 2002, 192, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Michel, D.; Chatelain, G.; North, S.; Brun, G. Stress-induced transcription of the clusterin/apoJ gene. Biochem. J. 1997, 328 Pt 1, 45–50. [Google Scholar] [CrossRef]
- Criswell, T.; Klokov, D.; Beman, M.; Lavik, J.P.; Boothman, D.A. Repression of IR-Inducible Clusterin Expression by the p53 Tumor Suppressor Protein. Cancer Biol. Ther. 2003, 2, 372–380. [Google Scholar] [CrossRef] [PubMed]
- Jin, G.; Howe, P.H. Regulation of Clusterin Gene Expression by Transforming Growth Factor β. J. Biol. Chem. 1997, 272, 26620–26626. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Kumano, M.; Beraldi, E.; Fazli, L.; Du, C.; Moore, S.; Sorensen, P.; Zoubeidi, A.; Gleave, M.E. Clusterin facilitates stress-induced lipidation of LC3 and autophagosome biogenesis to enhance cancer cell survival. Nat. Commun. 2014, 5, 5775. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.; Lee, D.-H.; Lee, I.-K.; Park, Y.M.; Jo, I. Far-infrared radiation inhibits proliferation, migration, and angiogenesis of human umbilical vein endothelial cells by suppressing secretory clusterin levels. Cancer Lett. 2014, 346, 74–83. [Google Scholar] [CrossRef]
- Criswell, T.; Beman, M.; Araki, S.; Leskov, K.; Cataldo, E.; Mayo, L.D.; Boothman, D.A. Delayed Activation of Insulin-like Growth Factor-1 Receptor/Src/MAPK/Egr-1 Signaling Regulates Clusterin Expression, a Pro-survival Factor. J. Biol. Chem. 2005, 280, 14212–14221. [Google Scholar] [CrossRef]
- Dumont, P.; Chainiaux, F.; Remacle, J.; Koch-Brandt, C.; Gonos, E.S.; Toussaint, O. Overexpression of apolipoprotein J in human fibroblasts protects against cytotoxicity and premature senescence induced by ethanol and tert-butylhydroperoxide. Cell Stress Chaperones 2002, 7, 23–35. [Google Scholar] [CrossRef]
- Miyake, H.; Hara, I.; Gleave, M.E.; Eto, H. Protection of androgen-dependent human prostate cancer cells from oxidative stress-induced DNA damage by overexpression of clusterin and its modulation by androgen. Prostate 2004, 61, 318–323. [Google Scholar] [CrossRef]
- Carnevali, S.; Luppi, F.; D’Arca, D.; Caporali, A.; Ruggieri, M.P.; Vettori, M.V.; Caglieri, A.; Astancolle, S.; Panico, F.; Davalli, P.; et al. Clusterin Decreases Oxidative Stress in Lung Fibroblasts Exposed to Cigarette Smoke. Am. J. Respir. Crit. Care Med. 2006, 174, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Bailey, R.W.; Aronow, B.; Harmony, J.A.K.; Griswold, M.D. Heat Shock-Initiated Apoptosis Is Accelerated and Removal of Damaged Cells Is Delayed in the Testis of Clusterin/ApoJ Knock-Out Mice. Biol. Reprod. 2002, 66, 1042–1053. [Google Scholar] [CrossRef]
- Chung, J.; Kwak, C.; Jin, R.J.; Lee, C.-H.; Lee, K.H.; Lee, S.E. Enhanced chemosensitivity of bladder cancer cells to cisplatin by suppression of clusterin in vitro. Cancer Lett. 2004, 203, 155–161. [Google Scholar] [CrossRef]
- Han, B.H.; DeMattos, R.B.; Dugan, L.L.; Kim-Han, J.S.; Brendza, R.P.; Fryer, J.D.; Kierson, M.; Cirrito, J.; Quick, K.; Harmony, J.A.; et al. Clusterin contributes to caspase-3-independent brain injury following neonatal hypoxia-ischemia. Nat. Med. 2001, 7, 338–343. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.E.; Meerabux, J.M.A.; Yeats, D.A.; Neal, M.J. Analysis of differentially expressed genes in retinitis pigmentosa retinas Altered expression of clusterin mRNA. FEBS Lett. 1992, 300, 279–282. [Google Scholar] [CrossRef] [PubMed]
- Koltai, T. Clusterin: A key player in cancer chemoresistance and its inhibition. OncoTargets Ther. 2014, 7, 447–456. [Google Scholar] [CrossRef]
- Rosenberg, M.E.; Girton, R.; Finkel, D.; Chmielewski, D.; Barrie, A., III; Witte, D.P.; Zhu, G.; Bissler, J.J.; Harmony, J.A.K.; Aronow, B.J. Apolipoprotein J/Clusterin Prevents a Progressive Glomerulopathy of Aging. Mol. Cell. Biol. 2002, 22, 1893–1902. [Google Scholar] [CrossRef]
- Gelissen, I.C.; Hochgrebe, T.; Wilson, M.R.; Easterbrook-Smith, S.B.; Jessup, W.; Dean, R.T.; Brown, A.J. Apolipoprotein J (clusterin) induces cholesterol export from macrophage-foam cells: A potential anti-atherogenic function? Biochem. J. 1998, 331 Pt 1, 231–237. [Google Scholar] [CrossRef]
- Vakeva, A.; Laurila, P.; Meri, S. Co-deposition of clusterin with the complement membrane attack complex in myocardial infarction. Immunology 1993, 80, 177–182. [Google Scholar]
- Trougakos, I.P.; Poulakou, M.; Stathatos, M.; Chalikia, A.; Melidonis, A.; Gonos, E.S. Serum levels of the senescence biomarker clusterin/apolipoprotein J increase significantly in diabetes type II and during development of coronary heart disease or at myocardial infarction. Exp. Gerontol. 2002, 37, 1175–1187. [Google Scholar] [CrossRef]
- Oda, T.; Wals, P.; Osterburg, H.H.; Johnson, S.A.; Pasinetti, G.M.; Morgan, T.E.; Rozovsky, I.; Stine, W.B.; Snyder, S.W.; Holzman, T.F.; et al. Clusterin (apoJ) alters the aggregation of amyloid beta-peptide (Aβ1-42) and forms slowly sedimenting A beta complexes that cause oxidative stress. Exp. Neurol. 1995, 136, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Boggs, L.N.; Fuson, K.S.; Baez, M.; Churgay, L.; McClure, D.; Becker, G.; May, P.C. Clusterin (Apo J) protects against in vitro amyloid-β (1-40) neurotoxicity. J. Neurochem. 1996, 67, 1324–1327. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, J.; Oyasu, R.; Lang, S.; Sintich, S.; Rademaker, A.; Lee, C.; Kozlowski, J.M.; Sensibar, J.A. Intracellular levels of SGP-2 (Clusterin) correlate with tumor grade in prostate cancer. Clin. Cancer Res. 1997, 3, 1707–1711. [Google Scholar] [PubMed]
- Redondo, M.; Villar, E.; Torres-Munoz, J.; Tellez, T.; Morell, M.; Petito, C.K. Overexpression of clusterin in human breast carcinoma. Am. J. Pathol. 2000, 157, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Xie, D.; Sham, J.S.; Zeng, W.F.; Che, L.H.; Zhang, M.; Wu, H.X.; Lin, H.L.; Wen, J.M.; Lau, S.H.; Hu, L.; et al. Oncogenic role of clusterin overexpression in multistage colorectal tumorigenesis and progression. World J. Gastroenterol. 2005, 11, 3285–3289. [Google Scholar] [CrossRef]
- Michel, D.; Gillet, G.; Volovitch, M.; Pessac, B.; Calothy, G.; Brun, G. Expression of a novel gene encoding a 51.5 kD precursor protein is induced by different retroviral oncogenes in quail neuroretinal cells. Oncogene Res. 1989, 4, 127–136. [Google Scholar]
- Gutacker, C.; Klock, G.; Diel, P.; Koch-Brandt, C. Nerve growth factor and epidermal growth factor stimulate clusterin gene expression in PC12 cells. Biochem. J. 1999, 339 Pt 3, 759–766. [Google Scholar] [CrossRef]
- Jin, G.; Howe, P.H. Transforming growth factor β regulates clusterin gene expression via modulation of transcription factor c-Fos. Eur. J. Biochem. 1999, 263, 534–542. [Google Scholar] [CrossRef]
- Gross, C.; Le-Bel, G.; Desjardins, P.; Benhassine, M.; Germain, L.; Guerin, S.L. Contribution of the Transcription Factors Sp1/Sp3 and AP-1 to Clusterin Gene Expression during Corneal Wound Healing of Tissue-Engineered Human Corneas. Int. J. Mol. Sci. 2021, 22, 12426. [Google Scholar] [CrossRef]
- Klock, G.; Storch, S.; Rickert, J.; Gutacker, C.; Koch-Brandt, C. Differential regulation of the clusterin gene by Ha-ras and c-myc oncogenes and during apoptosis. J. Cell. Physiol. 1998, 177, 593–605. [Google Scholar] [CrossRef]
- Thomas-Tikhonenko, A.; Viard-Leveugle, I.; Dews, M.; Wehrli, P.; Sevignani, C.; Yu, D.; Ricci, S.; el-Deiry, W.; Aronow, B.; Kaya, G.; et al. Myc-Transformed Epithelial Cells Down-Regulate Clusterin, Which Inhibits Their Growth in Vitro and Carcinogenesis in Vivo. Cancer Res. 2004, 64, 3126–3136. [Google Scholar] [CrossRef]
- Shiota, M.; Zardan, A.; Takeuchi, A.; Kumano, M.; Beraldi, E.; Naito, S.; Zoubeidi, A.; Gleave, M.E. Clusterin Mediates TGF-β–Induced Epithelial–Mesenchymal Transition and Metastasis via Twist1 in Prostate Cancer Cells. Cancer Res. 2012, 72, 5261–5272. [Google Scholar] [CrossRef]
- Takeuchi, A.; Shiota, M.; Beraldi, E.; Thaper, D.; Takahara, K.; Ibuki, N.; Pollak, M.; Cox, M.E.; Naito, S.; Gleave, M.E.; et al. Insulin-like growth factor-I induces CLU expression through Twist1 to promote prostate cancer growth. Mol. Cell. Endocrinol. 2014, 384, 117–125. [Google Scholar] [CrossRef]
- Cervellera, M.; Raschella, G.; Santilli, G.; Tanno, B.; Ventura, A.; Mancini, C.; Sevignani, C.; Calabretta, B.; Sala, A. Direct Transactivation of the Anti-apoptotic Gene Apolipoprotein J (Clusterin) by B-MYB. J. Biol. Chem. 2000, 275, 21055–21060. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Massa, P.E.; Hanidu, A.; Peet, G.W.; Aro, P.; Savitt, A.; Mische, S.; Li, J.; Marcu, K.B. IKKα, IKKβ and NEMO/IKKγ are each required for the NF-κB mediated inflammatory response program. J. Biol. Chem. 2002, 277, 45129–45140. [Google Scholar] [CrossRef] [PubMed]
- Saura, J.; Petegnief, V.; Wu, X.; Liang, Y.; Paul, S.M. Microglial apolipoprotein E and astroglial apolipoprotein J expression in vitro: Opposite effects of lipopolysaccharide. J. Neurochem. 2003, 85, 1455–1467. [Google Scholar] [CrossRef] [PubMed]
- Devauchelle, V.; Essabbani, A.; Pinieux, G.D.; Germain, S.; Tourneur, L.; Mistou, S.; Margottin-Goguet, F.; Anract, P.; Migaud, H.; Nen, D.L.; et al. Characterization and Functional Consequences of Underexpression of Clusterin in Rheumatoid Arthritis. J. Immunol. 2006, 177, 6471–6479. [Google Scholar] [CrossRef]
- Santilli, G.; Aronow, B.J.; Sala, A. Essential Requirement of Apolipoprotein J (Clusterin) Signaling for IκB Expression and Regulation of NF-κB Activity. J. Biol. Chem. 2003, 278, 38214–38219. [Google Scholar] [CrossRef] [PubMed]
- Shiota, M.; Zoubeidi, A.; Kumano, M.; Beraldi, E.; Naito, S.; Nelson, C.C.; Sorensen, P.H.B.; Gleave, M.E. Clusterin Is a Critical Downstream Mediator of Stress-Induced YB-1 Transactivation in Prostate Cancer. Mol. Cancer Res. 2011, 9, 1755–1766. [Google Scholar] [CrossRef]
- Kitazawa, K.; Hikichi, T.; Nakamura, T.; Sotozono, C.; Kinoshita, S.; Masui, S. PAX6 regulates human corneal epithelium cell identity. Exp. Eye Res. 2017, 154, 30–38. [Google Scholar] [CrossRef]
- Liu, S.; Tang, S.; Yang, G.; Li, Q. Lysine Demethylase 1B Promotes Tear Secretion Disorder in Sjogren’s Syndrome by Regulating the PAX6/CLU Axis. J. Mol. Neurosci. 2023, 73, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Li, S.; Tang, S.; Ye, S.; Liang, N.; Liang, Y.; Xiao, F. Clusterin protects against Cr(VI)-induced oxidative stress-associated hepatotoxicity by mediating the Akt-Keap1-Nrf2 signaling pathway. Environ. Sci. Pollut. Res. Int. 2022, 29, 52289–52301. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.; Kim, G.H.; Oh, G.-S.; Yoon, J.; Kim, H.W.; Kim, M.-S.; Kim, S.-W. SREBP-1c regulates glucose-stimulated hepatic clusterin expression. Biochem. Biophys. Res. Commun. 2011, 408, 720–725. [Google Scholar] [CrossRef] [PubMed]
- Oh, G.-S.; Kim, G.; Yoon, J.; Kim, G.H.; Kim, S.-W. The E-box-like sterol regulatory element mediates the insulin-stimulated expression of hepatic clusterin. Biochem. Biophys. Res. Commun. 2015, 465, 501–506. [Google Scholar] [CrossRef]
- Reichardt, S.D.; Amouret, A.; Muzzi, C.; Vettorazzi, S.; Tuckermann, J.P.; Luhder, F.; Reichardt, H.M. The Role of Glucocorticoids in Inflammatory Diseases. Cells 2021, 10, 2921. [Google Scholar] [CrossRef]
- Montpetit, M.L.; Lawless, K.R.; Tenniswood, M. Androgen-repressed messages in the rat ventral prostate. Prostate 1986, 8, 25–36. [Google Scholar] [CrossRef]
- Sensibar, J.A.; Sutkowski, D.M.; Raffo, A.; Buttyan, R.; Griswold, M.D.; Sylvester, S.R.; Kozlowski, J.M.; Lee, C. Prevention of Cell Death Induced by Tumor Necrosis Factor α in LNCaP Cells by Overexpression of Sulfated Glycoprotein-2 (Clusterin). Cancer Res. 1995, 55, 2431–2437. [Google Scholar]
- Cochrane, D.R.; Wang, Z.; Muramaki, M.; Gleave, M.E.; Nelson, C.C. Differential Regulation of Clusterin and Its Isoforms by Androgens in Prostate Cells. J. Biol. Chem. 2007, 282, 2278–2287. [Google Scholar] [CrossRef]
- Wünsche, W.; Tenniswood, M.P.; Schneider, M.R.; Vollmer, G. Estrogenic regulation of clusterin mRNA in normal and malignant endometrial tissue. Int. J. Cancer 1998, 76, 684–688. [Google Scholar] [CrossRef]
- Won, Y.S.; Lee, S.J.; Yeo, S.G.; Park, D.C. Effects of Female Sex Hormones on Clusterin Expression and Paclitaxel Resistance in Endometrial Cancer Cell Lines. Int. J. Med. Sci. 2012, 9, 86–92. [Google Scholar] [CrossRef]
- Lin, K.H.; Lee, H.Y.; Shih, C.H.; Yen, C.C.; Chen, S.L.; Yang, R.C.; Wang, C.S. Plasma protein regulation by thyroid hormone. J. Endocrinol. 2003, 179, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Fritz, I.B.; Burdzy, K.; Sétchell, B.; Blaschuk, O. Ram Rete Testis Fluid Contains a Protein (Clusterin) Which Influences Cell-Cell Interactions in Vitro. Biol. Reprod. 1983, 28, 1173–1188. [Google Scholar] [CrossRef]
- Thomas-Salgar, S.; Millis, A.J. Clusterin expression in differentiating smooth muscle cells. J. Biol. Chem. 1994, 269, 17879–17885. [Google Scholar] [CrossRef]
- Miwa, Y.; Takahashi-Yanaga, F.; Morimoto, S.; Sasaguri, T. Involvement of clusterin in 15-deoxy-Δ12,14-prostaglandin J2-induced vascular smooth muscle cell differentiation. Biochem. Biophys. Res. Commun. 2004, 319, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Millis, A.J.T.; Luciani, M.; McCue, H.M.; Rosenberg, M.E.; Moulson, C.L. Clusterin regulates vascular smooth muscle cell nodule formation and migration. J. Cell. Physiol. 2001, 186, 210–219. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.M.; Kim, S.Y.; Lee, S.; Shin, Y.J.; Min, B.H.; Bendayan, M.; Park, I.S. Clusterin induces differentiation of pancreatic duct cells into insulin-secreting cells. Diabetologia 2006, 49, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Min, B.-H.; Kim, B.-M.; Lee, S.-H.; Kang, S.-W.; Bendayan, M.; Park, I.-S. Clusterin Expression in the Early Process of Pancreas Regeneration in the Pancreatectomized Rat. J. Histochem. Cytochem. 2003, 51, 1355–1365. [Google Scholar] [CrossRef] [PubMed]
- Sivamurthy, N.; Stone, D.H.; LoGerfo, F.W.; Quist, W.C. Apolipoprotein J inhibits the migration, adhesion, and proliferation of vascular smooth muscle cells. J. Vasc. Surg. 2001, 34, 716–723. [Google Scholar] [CrossRef]
- Han-Jong, K.; Eun-Kyung, Y.; Joon-Young, K.; Young-Keun, C.; Hyo-Jeong, L.; Jeong-Kook, K.; Ho, J.N.; Ki-Up, L.; In-Sun, P.; Bon-Hong, M.; et al. Protective Role of Clusterin/Apolipoprotein J Against Neointimal Hyperplasia via Antiproliferative Effect on Vascular Smooth Muscle Cells and Cytoprotective Effect on Endothelial Cells. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1558–1564. [Google Scholar]
- Jeong, S.; Ledee, D.R.; Gordon, G.M.; Itakura, T.; Patel, N.; Martin, A.; Fini, M.E. Interaction of Clusterin and Matrix Metalloproteinase-9 and Its Implication for Epithelial Homeostasis and Inflammation. Am. J. Pathol. 2012, 180, 2028–2039. [Google Scholar] [CrossRef]
- Li, J.; Jia, L.; Zhao, P.; Jiang, Y.; Zhong, S.; Chen, D. Stable Knockdown of Clusterin by Vector-Based RNA Interference in a Human Breast Cancer Cell Line Inhibits Tumour Cell Invasion and Metastasis. J. Int. Med. Res. 2012, 40, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Couture, C.; Desjardins, P.; Zaniolo, K.; Germain, L.; Guerin, S.L. Enhanced wound healing of tissue-engineered human corneas through altered phosphorylation of the CREB and AKT signal transduction pathways. Acta Biomater. 2018, 73, 312–325. [Google Scholar] [CrossRef] [PubMed]
- Zeng, S.; Pan, Y.; Liu, F.; Yin, J.; Jiang, M.; Long, Y.; Zhao, X.; Lash, G.E.; Yang, H. Role of clusterin in the regulation of trophoblast development and preeclampsia. Biochem. Biophys. Res. Commun. 2021, 583, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Dong, B.; Song, Y.; Li, Y.; Kou, L.; Qin, Q. Apolipoprotein J Attenuates Vascular Restenosis by Promoting Autophagy and Inhibiting the Proliferation and Migration of Vascular Smooth Muscle Cells. J. Cardiovasc. Transl. Res. 2022, 15, 1086–1099. [Google Scholar] [CrossRef]
- Liu, X.; Meng, L.; Li, J.; Meng, J.; Teng, X.; Gu, H.; Hu, S.; Wei, Y. Secretory clusterin is upregulated in rats with pulmonary arterial hypertension induced by systemic-to-pulmonary shunts and exerts important roles in pulmonary artery smooth muscle cells. Acta Physiol. 2015, 213, 505–518. [Google Scholar] [CrossRef]
- Kang, B.-H.; Shim, Y.-J.; Tae, Y.-K.; Song, J.-A.; Choi, B.-K.; Park, I.-S.; Min, B.-H. Clusterin stimulates the chemotactic migration of macrophages through a pertussis toxin sensitive G-protein-coupled receptor and Gβγ-dependent pathways. Biochem. Biophys. Res. Commun. 2014, 445, 645–650. [Google Scholar] [CrossRef]
- Shim, Y.-J.; Kang, B.-H.; Choi, B.-K.; Park, I.-S.; Min, B.-H. Clusterin induces the secretion of TNF-α and the chemotactic migration of macrophages. Biochem. Biophys. Res. Commun. 2012, 422, 200–205. [Google Scholar] [CrossRef]
- Nguan, C.Y.C.; Guan, Q.; Gleave, M.E.; Du, C. Promotion of cell proliferation by clusterin in the renal tissue repair phase after ischemia-reperfusion injury. Am. J. Physiol. Ren. Physiol. 2014, 306, F724–F733. [Google Scholar] [CrossRef]
- Shin, Y.-J.; Kang, S.-W.; Jeong, S.-Y.; Shim, Y.-J.; Kim, Y.-H.; Kim, B.-M.; Kee, S.-H.; Park, J.-J.; Park, I.-S.; Min, B.-H. Clusterin enhances proliferation of primary astrocytes through extracellular signal-regulated kinase activation. NeuroReport 2006, 17, 1871–1875. [Google Scholar] [CrossRef]
- Kim, B.-M.; Han, Y.-M.; Shin, Y.-J.; Min, B.-H.; Park, I.-S. Clusterin expression during regeneration of pancreatic islet cells in streptozotocin-induced diabetic rats. Diabetologia 2001, 44, 2192–2202. [Google Scholar] [CrossRef]
- Shirasawa, T.; Miyata, M.; Eto, H.; Hamada, N.; Akasaki, Y.; Miyauchi, T.; Furusho, Y.; Orihara, K.; Hamasaki, S.; Aronow, B.J.; et al. Deficiency of clusterin inhibits neointimal hyperplasia after vascular injury. J. Atheroscler. Thromb. 2009, 16, 772–781. [Google Scholar] [CrossRef]
- Bettuzzi, S.; Astancolle, S.; Guidetti, G.; Moretti, M.; Tiozzo, R.; Corti, A. Clusterin (SGP-2) gene expression is cell cycle dependent in normal human dermal fibroblasts. FEBS Lett. 1999, 448, 297–300. [Google Scholar] [CrossRef] [PubMed]
- Bettuzzi, S.; Scorcioni, F.; Astancolle, S.; Davalli, P.; Scaltriti, M.; Corti, A. Clusterin (SGP-2) transient overexpression decreases proliferation rate of SV40-immortalized human prostate epithelial cells by slowing down cell cycle progression. Oncogene 2002, 21, 4328–4334. [Google Scholar] [CrossRef] [PubMed]
- Peix, L.; Evans, I.C.; Pearce, D.R.; Simpson, J.K.; Maher, T.M.; McAnulty, R.J. Diverse functions of clusterin promote and protect against the development of pulmonary fibrosis. Sci. Rep. 2018, 8, 1906. [Google Scholar] [CrossRef] [PubMed]
- Trougakos, I.P.; Lourda, M.; Antonelou, M.H.; Kletsas, D.; Gorgoulis, V.G.; Papassideri, I.S.; Zou, Y.; Margaritis, L.H.; Boothman, D.A.; Gonos, E.S. Intracellular Clusterin Inhibits Mitochondrial Apoptosis by Suppressing p53-Activating Stress Signals and Stabilizing the Cytosolic Ku70-Bax Protein Complex. Clin. Cancer Res. 2009, 15, 48–59. [Google Scholar] [CrossRef]
- Essabbani, A.; Garcia, L.; Zonetti, M.J.; Fisco, T.; Pucci, S.; Chiocchia, G. Exon-Skipping Strategy by Ratio Modulation between Cytoprotective versus Pro-Apoptotic Clusterin Forms Increased Sensitivity of LNCaP to Cell Death. PLoS ONE 2013, 8, e54920. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, X.; Zhao, H.; Liang, B.; Du, Q. Clusterin confers resistance to TNF-alpha-induced apoptosis in breast cancer cells through NF-kappaB activation and Bcl-2 overexpression. J. Chemother. 2012, 24, 348–357. [Google Scholar] [CrossRef]
- Kim, J.H.; Kim, J.H.; Yu, Y.S.; Min, B.-H.; Kim, K.-W. The role of clusterin in retinal development and free radical damage. Br. J. Ophthalmol. 2007, 91, 1541–1546. [Google Scholar] [CrossRef]
- Kim, J.H.; Kim, J.H.; Jun, H.O.; Yu, Y.S.; Min, B.H.; Park, K.H.; Kim, K.-W. Protective Effect of Clusterin from Oxidative Stress-Induced Apoptosis in Human Retinal Pigment Epithelial Cells. Investig. Ophthalmol. Vis. Sci. 2010, 51, 561–566. [Google Scholar] [CrossRef]
- Bursch, W.; Gleeson, T.; Kleine, L.; Tenniswood, M. Expression of clusterin (testosterone-repressed prostate message-2) mRNA during growth and regeneration of rat liver. Arch. Toxicol. 1995, 69, 253–258. [Google Scholar] [CrossRef]
- Loison, F.; Debure, L.; Nizard, P.; le Goff, P.; Michel, D.; le Drean, Y. Up-regulation of the clusterin gene after proteotoxic stress: Implication of HSF1-HSF2 heterocomplexes. Biochem. J. 2006, 395, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Pucci, S.; Bonanno, E.; Pichiorri, F.; Angeloni, C.; Spagnoli, L.G. Modulation of different clusterin isoforms in human colon tumorigenesis. Oncogene 2004, 23, 2298–2304. [Google Scholar] [CrossRef] [PubMed]
- Scaltriti, M.; Bettuzzi, S.; Sharrard, R.M.; Caporali, A.; Caccamo, A.E.; Maitland, N.J. Clusterin overexpression in both malignant and nonmalignant prostate epithelial cells induces cell cycle arrest and apoptosis. Br. J. Cancer 2004, 91, 1842–1850. [Google Scholar] [CrossRef] [PubMed]
- Akakura, K.; Bruchovsky, N.; Rennie, P.S.; Coldman, A.J.; Goldenberg, S.L.; Tenniswood, M.; Fox, K. Effects of intermittent androgen suppression on the stem cell composition and the expression of the TRPM-2 (clusterin) gene in the Shionogi carcinoma. J. Steroid Biochem. Mol. Biol. 1996, 59, 501–511. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Li, X.; Gu, W.; Li, X.; Zhao, J.; Wu, J.; Cai, J.; Feng, X.; Tao, T. Cytoplasmic Clusterin Suppresses Lung Cancer Metastasis by Inhibiting the ROCK1-ERK Axis. Cancers 2022, 14, 2463. [Google Scholar] [CrossRef]
- Nicholson, G.; Lawrence, A.; Ala, F.A.; Bird, G.W. Semi-quantitative assay of D antigen site density by flow cytometric analysis. Transfus. Med. 1991, 1, 87–90. [Google Scholar] [CrossRef]
- Ma, X.; Bai, Y. IGF-1 activates the P13K/AKT signaling pathway via upregulation of secretory clusterin. Mol. Med. Rep. 2012, 6, 1433–1437. [Google Scholar] [CrossRef]
- Wang, C.; Jiang, K.; Gao, D.; Kang, X.; Sun, C.; Zhang, Q.; Li, Y.; Sun, L.; Zhang, S.; Guo, K.; et al. Clusterin protects hepatocellular carcinoma cells from endoplasmic reticulum stress induced apoptosis through GRP78. PLoS ONE 2013, 8, e55981. [Google Scholar] [CrossRef]
- Blume, A.J.; Foster, C.J. Mouse neuroblastoma cell adenylate cyclase: Regulation by 2-chloroadenosine, prostaglandin E1 and the cations Mg2+, Ca2+ and Mn2+. J. Neurochem. 1976, 26, 305–311. [Google Scholar] [CrossRef]
- Flanagan, L.; Whyte, L.; Chatterjee, N.; Tenniswood, M. Effects of clusterin over-expression on metastatic progression and therapy in breast cancer. BMC Cancer 2010, 10, 107. [Google Scholar] [CrossRef]
- Heimann, W.G.; Karns, R.E.; Snyder, H.L. Xeromammography at a community hospital. A year’s experience. Conn. Med. 1975, 39, 465–467. [Google Scholar] [PubMed]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Guidelines and definitions for research on epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2020, 21, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Wang, W.; Liu, Z.; Sun, X.; Liao, Z.; Chen, F.; Jiang, G.; Huo, G. Blocking ERK signaling pathway lowers MMP-9 expression to alleviate brain edema after traumatic brain injury in rats. Nan Fang Yi Ke Da Xue Xue Bao 2020, 40, 1018–1022. [Google Scholar] [PubMed]
- Wang, X.; Luo, L.; Dong, D.; Yu, Q.; Zhao, K. Clusterin plays an important role in clear renal cell cancer metastasis. Urol. Int. 2014, 92, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Lai, Y.; Wang, Q.; Liu, X.; He, W.; Zhang, H.; Fan, C.; Yang, G. Overexpression of clusterin promotes angiogenesis via the vascular endothelial growth factor in primary ovarian cancer. Mol. Med. Rep. 2013, 7, 1726–1732. [Google Scholar] [CrossRef]
- Li, Y.; Lu, J.; Zhou, S.; Wang, W.; Tan, G.; Zhang, Z.; Dong, Z.; Kang, T.; Tang, F. Clusterin induced by N,N′-Dinitrosopiperazine is involved in nasopharyngeal carcinoma metastasis. Oncotarget 2016, 7, 5548–5563. [Google Scholar] [CrossRef]
- Tian, Y.; Wang, C.; Chen, S.; Liu, J.; Fu, Y.; Luo, Y. Extracellular Hsp90alpha and clusterin synergistically promote breast cancer epithelial-to-mesenchymal transition and metastasis via LRP1. J. Cell Sci. 2019, 132, jcs228213. [Google Scholar] [CrossRef]
- Wang, C.; Jiang, K.; Kang, X.; Gao, D.; Sun, C.; Li, Y.; Sun, L.; Zhang, S.; Liu, X.; Wu, W.; et al. Tumor-derived secretory clusterin induces epithelial-mesenchymal transition and facilitates hepatocellular carcinoma metastasis. Int. J. Biochem. Cell Biol. 2012, 44, 2308–2320. [Google Scholar] [CrossRef]
- Radziwon-Balicka, A.; Santos-Martinez, M.J.; Corbalan, J.J.; O’Sullivan, S.; Treumann, A.; Gilmer, J.F.; Radomski, M.W.; Medina, C. Mechanisms of platelet-stimulated colon cancer invasion: Role of clusterin and thrombospondin 1 in regulation of the P38MAPK-MMP-9 pathway. Carcinogenesis 2014, 35, 324–332. [Google Scholar] [CrossRef]
- Yang, P.; Yang, Z.; Dong, Y.; Yang, L.; Peng, S.; Yuan, L.; Hu, X.; Chen, S.; Tang, H.; Yang, X.; et al. Clusterin is a biomarker of breast cancer prognosis and correlated with immune microenvironment. Transl. Cancer Res. 2023, 12, 31–45. [Google Scholar] [CrossRef]
- Yang, S.; Tang, W.; Azizian, A.; Gaedcke, J.; Strobel, P.; Wang, L.; Cawley, H.; Ohara, Y.; Valenzuela, P.; Zhang, L.; et al. Dysregulation of HNF1B/Clusterin axis enhances disease progression in a highly aggressive subset of pancreatic cancer patients. Carcinogenesis 2022, 43, 1198–1210. [Google Scholar] [CrossRef] [PubMed]
- Nafee, A.M.; Pasha, H.F.; Abd El Aal, S.M.; Mostafa, N.A. Clinical significance of serum clusterin as a biomarker for evaluating diagnosis and metastasis potential of viral-related hepatocellular carcinoma. Clin. Biochem. 2012, 45, 1070–1074. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Yao, M.; Sai, W.; Qian, Q.; Pan, L.; Qiu, L.; Huang, J.; Wu, W.; Yao, D. Diagnostic and prognostic significance of secretory clusterin expression in patients with hepatocellular carcinoma. Tumour Biol. 2016, 37, 999–1008. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Halberg, R.B.; Ehrhardt, W.M.; Torrealba, J.; Dove, W.F. Clusterin as a biomarker in murine and human intestinal neoplasia. Proc. Natl. Acad. Sci. USA 2003, 100, 9530–9535. [Google Scholar] [CrossRef]
- Gao, G.; Luan, X. Diagnostic performance of clusterin in hepatocellular carcinoma: A meta-analysis. Int. J. Biol. Markers 2022, 37, 404–411. [Google Scholar] [CrossRef] [PubMed]
- Beheshti Namdar, A.; Kabiri, M.; Mosanan Mozaffari, H.; Aminifar, E.; Mehrad-Majd, H. Circulating Clusterin Levels and Cancer Risk: A Systematic Review and Meta-Analysis. Cancer Control 2022, 29, 10732748211038437. [Google Scholar] [CrossRef]
- Mansoori, B.; Mohammadi, A.; Davudian, S.; Shirjang, S.; Baradaran, B. The Different Mechanisms of Cancer Drug Resistance: A Brief Review. Adv. Pharm. Bull. 2017, 7, 339–348. [Google Scholar] [CrossRef]
- Whitesell, L.; Lindquist, S.L. HSP90 and the chaperoning of cancer. Nat. Rev. Cancer 2005, 5, 761–772. [Google Scholar] [CrossRef]
- Zoubeidi, A.; Chi, K.; Gleave, M. Targeting the cytoprotective chaperone, clusterin, for treatment of advanced cancer. Clin. Cancer Res. 2010, 16, 1088–1093. [Google Scholar] [CrossRef]
- Tang, M.; Li, J.; Liu, B.; Song, N.; Wang, Z.; Yin, C. Clusterin expression and human testicular seminoma. Med. Hypotheses 2013, 81, 635–637. [Google Scholar] [CrossRef]
- Mu, L.; Yang, F.; Guo, D.; Li, P.; Zhang, M. Overexpression of secretory clusterin (sCLU) induces chemotherapy resistance in human gastric cancer cells by targeting miR-195-5p. Bioengineered 2020, 11, 472–483. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zou, F.; Zhong, J.; Yue, L.; Wang, F.; Wei, H.; Yang, G.; Jin, T.; Dong, X.; Li, J.; et al. Secretory Clusterin Mediates Oxaliplatin Resistance via the Gadd45a/PI3K/Akt Signaling Pathway in Hepatocellular Carcinoma. J. Cancer 2018, 9, 1403–1413. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Sai, W.; Yao, M.; Gu, H.; Yao, Y.; Qian, Q.; Yao, D. Silencing clusterin gene transcription on effects of multidrug resistance reversing of human hepatoma HepG2/ADM cells. Tumour Biol. 2015, 36, 3995–4003. [Google Scholar] [CrossRef]
- Xu, M.; Chen, X.; Han, Y.; Ma, C.; Ma, L.; Li, S. Clusterin silencing sensitizes pancreatic cancer MIA-PaCa-2 cells to gmcitabine via regulation of NF-kB/Bcl-2 signaling. Int. J. Clin. Exp. Med. 2015, 8, 12476–12486. [Google Scholar] [PubMed]
- Praharaj, P.P.; Patra, S.; Panigrahi, D.P.; Patra, S.K.; Bhutia, S.K. Clusterin as modulator of carcinogenesis: A potential avenue for targeted cancer therapy. Biochim. Biophys. Acta Rev. Cancer 2021, 1875, 188500. [Google Scholar] [CrossRef] [PubMed]
- Chi, K.N.; Zoubeidi, A.; Gleave, M.E. Custirsen (OGX-011): A second-generation antisense inhibitor of clusterin for the treatment of cancer. Expert. Opin. Investig. Drugs 2008, 17, 1955–1962. [Google Scholar] [CrossRef]
- Gleave, M.; Jansen, B. Clusterin and IGFBPs as antisense targets in prostate cancer. Ann. N. Y. Acad. Sci. 2003, 1002, 95–104. [Google Scholar] [CrossRef]
- Kususda, Y.; Miyake, H.; Gleave, M.E.; Fujisawa, M. Clusterin inhibition using OGX-011 synergistically enhances antitumour activity of sorafenib in a human renal cell carcinoma model. Br. J. Cancer 2012, 106, 1945–1952. [Google Scholar] [CrossRef]
- Mustafi, S.; Sant, D.W.; Liu, Z.J.; Wang, G. Ascorbate induces apoptosis in melanoma cells by suppressing Clusterin expression. Sci. Rep. 2017, 7, 3671. [Google Scholar] [CrossRef]
- Lamoureux, F.; Baud’huin, M.; Ory, B.; Guiho, R.; Zoubeidi, A.; Gleave, M.; Heymann, D.; Redini, F. Clusterin inhibition using OGX-011 synergistically enhances zoledronic acid activity in osteosarcoma. Oncotarget 2014, 5, 7805–7819. [Google Scholar] [CrossRef]
- Lamoureux, F.; Thomas, C.; Yin, M.J.; Kuruma, H.; Beraldi, E.; Fazli, L.; Zoubeidi, A.; Gleave, M.E. Clusterin inhibition using OGX-011 synergistically enhances Hsp90 inhibitor activity by suppressing the heat shock response in castrate-resistant prostate cancer. Cancer Res. 2011, 71, 5838–5849. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekar, T.; Yang, J.C.; Gao, A.C.; Evans, C.P. Mechanisms of resistance in castration-resistant prostate cancer (CRPC). Transl. Androl. Urol. 2015, 4, 365–380. [Google Scholar] [PubMed]
- Matsumoto, H.; Yamamoto, Y.; Shiota, M.; Kuruma, H.; Beraldi, E.; Matsuyama, H.; Zoubeidi, A.; Gleave, M. Cotargeting Androgen Receptor and Clusterin Delays Castrate-Resistant Prostate Cancer Progression by Inhibiting Adaptive Stress Response and AR Stability. Cancer Res. 2013, 73, 5206–5217. [Google Scholar] [CrossRef]
- Laskin, J.J.; Nicholas, G.; Lee, C.; Gitlitz, B.; Vincent, M.; Cormier, Y.; Stephenson, J.; Ung, Y.; Sanborn, R.; Pressnail, B.; et al. Phase I/II trial of custirsen (OGX-011), an inhibitor of clusterin, in combination with a gemcitabine and platinum regimen in patients with previously untreated advanced non-small cell lung cancer. J. Thorac. Oncol. 2012, 7, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Chia, S.; Dent, S.; Ellard, S.; Ellis, P.M.; Vandenberg, T.; Gelmon, K.; Powers, J.; Walsh, W.; Seymour, L.; Eisenhauer, E.A. Phase II trial of OGX-011 in combination with docetaxel in metastatic breast cancer. Clin. Cancer Res. 2009, 15, 708–713. [Google Scholar] [CrossRef] [PubMed]
- Saad, F.; Hotte, S.; North, S.; Eigl, B.; Chi, K.; Czaykowski, P.; Wood, L.; Pollak, M.; Berry, S.; Lattouf, J.B.; et al. Randomized phase II trial of Custirsen (OGX-011) in combination with docetaxel or mitoxantrone as second-line therapy in patients with metastatic castrate-resistant prostate cancer progressing after first-line docetaxel: CUOG trial P-06c. Clin. Cancer Res. 2011, 17, 5765–5773. [Google Scholar] [CrossRef]
- Chi, K.N.; Hotte, S.J.; Yu, E.Y.; Tu, D.; Eigl, B.J.; Tannock, I.; Saad, F.; North, S.; Powers, J.; Gleave, M.E.; et al. Randomized phase II study of docetaxel and prednisone with or without OGX-011 in patients with metastatic castration-resistant prostate cancer. J. Clin. Oncol. 2010, 28, 4247–4254. [Google Scholar] [CrossRef]
- Beer, T.M.; Hotte, S.J.; Saad, F.; Alekseev, B.; Matveev, V.; Flechon, A.; Gravis, G.; Joly, F.; Chi, K.N.; Malik, Z.; et al. Custirsen (OGX-011) combined with cabazitaxel and prednisone versus cabazitaxel and prednisone alone in patients with metastatic castration-resistant prostate cancer previously treated with docetaxel (AFFINITY): A randomised, open-label, international, phase 3 trial. Lancet Oncol. 2017, 18, 1532–1542. [Google Scholar] [PubMed]
- Jomary, C.; Neal, M.J.; Jones, S.E. Comparison of clusterin gene expression in normal and dystrophic human retinas. Mol. Brain Res. 1993, 20, 279–284. [Google Scholar] [CrossRef]
- Gwon, J.S.; Kim, I.B.; Lee, M.Y.; Oh, S.J.; Chun, M.H. Expression of clusterin in Muller cells of the rat retina after pressure-induced ischemia. Glia 2004, 47, 35–45. [Google Scholar] [CrossRef]
- Wong, P.; Pfeffer, B.A.; Bernstein, S.L.; Chambers, M.L.; Chader, G.J.; Zakeri, Z.F.; Wu, Y.Q.; Wilson, M.R.; Becerra, S.P. Clusterin protein diversity in the primate eye. Mol. Vis. 2000, 6, 184–191. [Google Scholar]
- Wong, P.; Ulyanova, T.; Organisciak, D.T.; Bennett, S.; Lakins, J.; Arnold, J.M.; Kutty, R.K.; Tenniswood, M.; vanVeen, T.; Darrow, R.M.; et al. Expression of multiple forms of clusterin during light-induced retinal degeneration. Curr. Eye Res. 2001, 23, 157–165. [Google Scholar] [CrossRef]
- Zenkel, M.; Kruse, F.E.; Junemann, A.G.; Naumann, G.O.; Schlotzer-Schrehardt, U. Clusterin deficiency in eyes with pseudoexfoliation syndrome may be implicated in the aggregation and deposition of pseudoexfoliative material. Investig. Ophthalmol. Vis. Sci. 2006, 47, 1982–1990. [Google Scholar] [CrossRef]
- Kim, J.H.; Yu, Y.S.; Kim, J.H.; Kim, K.-W.; Min, B.-H. The Role of Clusterin in In Vitro Ischemia of Human Retinal Endothelial Cells. Curr. Eye Res. 2007, 32, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Reeder, D.J.; Stuart, W.D.; Witte, D.P.; Brown, T.L.; Harmony, J.A.K. Local synthesis of apolipoprotein J in the eye. Exp. Eye Res. 1995, 60, 495–504. [Google Scholar] [CrossRef] [PubMed]
- National Institutes of Health. NEI Bank. Available online: https://neibank.nei.nih.gov/index.shtml (accessed on 1 August 2023).
- Lemp, M.A.; Baudouin, C.; Baum, J.; Dogru, M.; Foulks, G.N.; Kinoshita, S.; Laibson, P.; McCulley, J.P.; Murube, J.; Pflugfelder, S.C.; et al. The definition and classification of dry eye disease: Report of the Definition and Classification Subcommittee of the International Dry Eye WorkShop (2007). Ocul. Surf. 2007, 5, 75–92. [Google Scholar]
- Hogasen, K.; Mollnes, T.E.; Harboe, M.; Gotze, O.; Hammer, H.B.; Oppermann, M. Terminal complement pathway activation and low lysis inhibitors in rheumatoid arthritis synovial fluid. J. Rheumatol. 1995, 22, 24–28. [Google Scholar] [PubMed]
- Lemp, M.A. The mucin-deficient dry eye. Int. Ophthalmol. Clin. 1973, 13, 185–189. [Google Scholar] [CrossRef]
- Newkirk, M.M.; Apostolakos, P.; Neville, C.; Fortin, P.R. Systemic lupus erythematosus, a disease associated with low levels of clusterin/apoJ, an antiinflammatory protein. J. Rheumatol. 1999, 26, 597–603. [Google Scholar]
- Okada, N.; Kawakita, T.; Mishima, K.; Saito, I.; Miyashita, H.; Yoshida, S.; Shimmura, S.; Tsubota, K. Clusterin Promotes Corneal Epithelial Cell Growth through Upregulation of Hepatocyte Growth Factor by Mesenchymal Cells In Vitro. Investig. Ophthalmol. Vis. Sci. 2011, 52, 2905–2910. [Google Scholar] [CrossRef]
- Karring, H.; Runager, K.; Thogersen, I.B.; Klintworth, G.K.; Hojrup, P.; Enghild, J.J. Composition and proteolytic processing of corneal deposits associated with mutations in the TGFBI gene. Exp. Eye Res. 2012, 96, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Nishida, K.; Quantock, A.J.; Dota, A.; Choi-Miura, N.-H.; Kinoshita, S. Apolipoproteins J and E co-localise with amyloid in gelatinous drop-like and lattice type I corneal dystrophies. Br. J. Ophthalmol. 1999, 83, 1178–1182. [Google Scholar] [CrossRef]
- Gain, P.; Jullienne, R.; He, Z.; Aldossary, M.; Acquart, S.; Cognasse, F.; Thuret, G. Global Survey of Corneal Transplantation and Eye Banking. JAMA Ophthalmol. 2016, 134, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Jurkunas, U.V.; Bitar, M.S.; Rawe, I.; Harris, D.L.; Colby, K.; Joyce, N.C. Increased Clusterin Expression in Fuchs’ Endothelial Dystrophy. Investig. Ophthalmol. Vis. Sci. 2008, 49, 2946–2955. [Google Scholar] [CrossRef] [PubMed]
- Fini, M.E.; Jeong, S.; Wilson, M.R. Therapeutic Potential of the Molecular Chaperone and Matrix Metalloproteinase Inhibitor Clusterin for Dry Eye. Int. J. Mol. Sci. 2020, 22, 116. [Google Scholar] [CrossRef]
- Yu, V.; Bhattacharya, D.; Webster, A.; Bauskar, A.; Flowers, C.; Heur, M.; Chintala, S.K.; Itakura, T.; Wilson, M.R.; Barr, J.T.; et al. Clusterin from human clinical tear samples: Positive correlation between tear concentration and Schirmer strip test results. Ocul. Surf. 2018, 16, 478–486. [Google Scholar] [CrossRef] [PubMed]
- Bauskar, A.; Mack, W.J.; Mauris, J.; Argueso, P.; Heur, M.; Nagel, B.A.; Kolar, G.R.; Gleave, M.E.; Nakamura, T.; Kinoshita, S.; et al. Clusterin Seals the Ocular Surface Barrier in Mouse Dry Eye. PLoS ONE 2015, 10, e0138958. [Google Scholar] [CrossRef] [PubMed]
- Rinsky, B.; Beykin, G.; Grunin, M.; Amer, R.; Khateb, S.; Tiosano, L.; Almeida, D.; Hagbi-Levi, S.; Elbaz-Hayoun, S.; Chowers, I. Analysis of the Aqueous Humor Proteome in Patients With Age-Related Macular Degeneration. Investig. Ophthalmol. Vis. Sci. 2021, 62, 18. [Google Scholar] [CrossRef] [PubMed]
- Zenkel, M.; Poschl, E.; von der Mark, K.; Hofmann-Rummelt, C.; Naumann, G.O.; Kruse, F.E.; Schlotzer-Schrehardt, U. Differential gene expression in pseudoexfoliation syndrome. Investig. Ophthalmol. Vis. Sci. 2005, 46, 3742–3752. [Google Scholar] [CrossRef]
- Fan, B.J.; Pasquale, L.R.; Kang, J.H.; Levkovitch-Verbin, H.; Haines, J.L.; Wiggs, J.L. Association of clusterin (CLU) variants and exfoliation syndrome: An analysis in two Caucasian studies and a meta-analysis. Exp. Eye Res. 2015, 139, 115–122. [Google Scholar] [CrossRef]
- Jung, G.S.; Kim, M.K.; Jung, Y.A.; Kim, H.S.; Park, I.S.; Min, B.H.; Lee, K.U.; Kim, J.G.; Park, K.G.; Lee, I.K. Clusterin attenuates the development of renal fibrosis. J. Am. Soc. Nephrol. 2012, 23, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Crabb, J.W.; Miyagi, M.; Gu, X.; Shadrach, K.; West, K.A.; Sakaguchi, H.; Kamei, M.; Hasan, A.; Yan, L.; Rayborn, M.E.; et al. Drusen proteome analysis: An approach to the etiology of age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2002, 99, 14682–14687. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.V.; Leitner, W.P.; Staples, M.K.; Anderson, D.H. Complement activation and inflammatory processes in Drusen formation and age related macular degeneration. Exp. Eye Res. 2001, 73, 887–896. [Google Scholar] [CrossRef] [PubMed]
- Mullins, R.F.; Russell, S.R.; Anderson, D.H.; Hageman, G.S. Drusen associated with aging and age-related macular degeneration contain proteins common to extracellular deposits associated with atherosclerosis, elastosis, amyloidosis, and dense deposit disease. FASEB J. 2000, 14, 835–846. [Google Scholar] [CrossRef]
- Jackson, J.K.; Gleave, M.E.; Gleave, J.; Burt, H.M. The inhibition of angiogenesis by antisense oligonucleotides to clusterin. Angiogenesis 2005, 8, 229–238. [Google Scholar] [CrossRef]
- Zhang, C.; Nie, J.; Feng, L.; Luo, W.; Yao, J.; Wang, F.; Wang, H. The emerging roles of clusterin on reduction of both blood retina barrier breakdown and neural retina damage in diabetic retinopathy. Discov. Med. 2016, 21, 227–237. [Google Scholar]
- Vargas, A.; Kim, H.S.; Baral, E.; Yu, W.Q.; Craft, C.M.; Lee, E.J. Protective effect of clusterin on rod photoreceptor in rat model of retinitis pigmentosa. PLoS ONE 2017, 12, e0182389. [Google Scholar] [CrossRef]
- Vargas, A.; Yamamoto, K.L.; Craft, C.M.; Lee, E.J. Clusterin enhances cell survival by suppressing neuronal nitric-oxide synthase expression in the rhodopsin S334ter-line3 retinitis pigmentosa model. Brain Res. 2021, 1768, 147575. [Google Scholar] [CrossRef]
- Zhou, M.; Shen, D.; Head, J.E.; Chew, E.Y.; Chevez-Barrios, P.; Green, W.R.; Chan, C.C. Ocular clusterin expression in von Hippel-Lindau disease. Mol. Vis. 2007, 13, 2129–2136. [Google Scholar]
- Asbell, P.; Messmer, E.; Chan, C.; Johnson, G.; Sloesen, B.; Cook, N. Defining the needs and preferences of patients with dry eye disease. BMJ Open Ophthalmol. 2019, 4, e000315. [Google Scholar] [CrossRef]
- Bakkar, M.M.; Shihadeh, W.A.; Haddad, M.F.; Khader, Y.S. Epidemiology of symptoms of dry eye disease (DED) in Jordan: A cross-sectional non-clinical population-based study. Cont. Lens Anterior Eye 2016, 39, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Hashemi, H.; Khabazkhoob, M.; Kheirkhah, A.; Emamian, M.H.; Mehravaran, S.; Shariati, M.; Fotouhi, A. Prevalence of dry eye syndrome in an adult population. Clin. Exp. Ophthalmol. 2014, 42, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.J.; Lee, J.; Saw, S.M.; Gazzard, G.; Koh, D.; Widjaja, D.; Tan, D.T. Prevalence and risk factors associated with dry eye symptoms: A population based study in Indonesia. Br. J. Ophthalmol. 2002, 86, 1347–1351. [Google Scholar] [CrossRef]
- Moss, S.E.; Klein, R.; Klein, B.E. Prevalence of and risk factors for dry eye syndrome. Arch. Ophthalmol. 2000, 118, 1264–1268. [Google Scholar] [CrossRef]
- Onwubiko, S.N.; Eze, B.I.; Udeh, N.N.; Arinze, O.C.; Onwasigwe, E.N.; Umeh, R.E. Dry eye disease: Prevalence, distribution and determinants in a hospital-based population. Cont. Lens Anterior Eye 2014, 37, 157–161. [Google Scholar] [CrossRef]
- Sendecka, M.; Baryluk, A.; Polz-Dacewicz, M. Prevalence and risk factors of dry eye syndrome. Przegl Epidemiol. 2004, 58, 227–233. [Google Scholar]
- Shanti, Y.; Shehada, R.; Bakkar, M.M.; Qaddumi, J. Prevalence and associated risk factors of dry eye disease in 16 northern West bank towns in Palestine: A cross-sectional study. BMC Ophthalmol. 2020, 20, 26. [Google Scholar] [CrossRef]
- Uchino, M.; Nishiwaki, Y.; Michikawa, T.; Shirakawa, K.; Kuwahara, E.; Yamada, M.; Dogru, M.; Schaumberg, D.A.; Kawakita, T.; Takebayashi, T.; et al. Prevalence and risk factors of dry eye disease in Japan: Koumi study. Ophthalmology 2011, 118, 2361–2367. [Google Scholar] [CrossRef]
- Vehof, J.; Kozareva, D.; Hysi, P.G.; Hammond, C.J. Prevalence and risk factors of dry eye disease in a British female cohort. Br. J. Ophthalmol. 2014, 98, 1712–1717. [Google Scholar] [CrossRef]
- Craig, J.P.; Nelson, J.D.; Azar, D.T.; Belmonte, C.; Bron, A.J.; Chauhan, S.K.; de Paiva, C.S.; Gomes, J.A.P.; Hammitt, K.M.; Jones, L.; et al. TFOS DEWS II Report Executive Summary. Ocul. Surf. 2017, 15, 802–812. [Google Scholar] [CrossRef]
- Gipson, I.K. The ocular surface: The challenge to enable and protect vision: The Friedenwald lecture. Investig. Ophthalmol. Vis. Sci. 2007, 48, 4391–4398. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Koh, J.H.; Kwok, S.K.; Park, S.H. The EULAR Sjogren’s Syndrome Patient-Reported Index is an independent determinant of health-related utility values of Korean patients with primary Sjogren’s syndrome. Clin. Exp. Rheumatol. 2016, 34, 663–667. [Google Scholar] [PubMed]
- Saboo, U.S.; Amparo, F.; Abud, T.B.; Schaumberg, D.A.; Dana, R. Vision-Related Quality of Life in Patients with Ocular Graft-versus-Host Disease. Ophthalmology 2015, 122, 1669–1674. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.C.; Chai, X.; Inamoto, Y.; Pidala, J.; Martin, P.J.; Flowers, M.E.; Shen, T.T.; Lee, S.J.; Jagasia, M. Impact of Ocular Chronic Graft-versus-Host Disease on Quality of Life. Biol. Blood Marrow Transpl. 2015, 21, 1687–1691. [Google Scholar] [CrossRef] [PubMed]
- Bordron, A.; Devauchelle-Pensec, V.; Le Dantec, C.; Capdeville, A.; Brooks, W.H.; Renaudineau, Y. Epigenetics in Primary Sjogren’s Syndrome. Adv. Exp. Med. Biol. 2020, 1253, 285–308. [Google Scholar]
- Chotikavanich, S.; de Paiva, C.S.; Li de, Q.; Chen, J.J.; Bian, F.; Farley, W.J.; Pflugfelder, S.C. Production and activity of matrix metalloproteinase-9 on the ocular surface increase in dysfunctional tear syndrome. Investig. Ophthalmol. Vis. Sci. 2009, 50, 3203–3209. [Google Scholar] [CrossRef]
- Luo, L.; Li, D.Q.; Doshi, A.; Farley, W.; Corrales, R.M.; Pflugfelder, S.C. Experimental dry eye stimulates production of inflammatory cytokines and MMP-9 and activates MAPK signaling pathways on the ocular surface. Investig. Ophthalmol. Vis. Sci. 2004, 45, 4293–4301. [Google Scholar] [CrossRef]
- Pflugfelder, S.C.; Solomon, A.; Dursun, D.; Li, D.Q. Dry eye and delayed tear clearance: “A call to arms”. Adv. Exp. Med. Biol. 2002, 506 Pt B, 739–743. [Google Scholar]
- Solomon, A.; Dursun, D.; Liu, Z.; Xie, Y.; Macri, A.; Pflugfelder, S.C. Pro- and anti-inflammatory forms of interleukin-1 in the tear fluid and conjunctiva of patients with dry-eye disease. Investig. Ophthalmol. Vis. Sci. 2001, 42, 2283–2292. [Google Scholar]
- Matsuda, A.; Itoh, Y.; Koshikawa, N.; Akizawa, T.; Yana, I.; Seiki, M. Clusterin, an abundant serum factor, is a possible negative regulator of MT6-MMP/MMP-25 produced by neutrophils. J. Biol. Chem. 2003, 278, 36350–36357. [Google Scholar] [CrossRef]
- Chintala, S.K.; Pan, J.; Satapathy, S.; Condruti, R.; Hao, Z.; Liu, P.W.; O’Conner, C.F.; Barr, J.T.; Wilson, M.R.; Jeong, S.; et al. Recombinant Human Clusterin Seals Damage to the Ocular Surface Barrier in a Mouse Model of Ophthalmic Preservative-Induced Epitheliopathy. Int. J. Mol. Sci. 2023, 24, 981. [Google Scholar] [CrossRef]
- Cunin, P.; Beauvillain, C.; Miot, C.; Augusto, J.F.; Preisser, L.; Blanchard, S.; Pignon, P.; Scotet, M.; Garo, E.; Fremaux, I.; et al. Clusterin facilitates apoptotic cell clearance and prevents apoptotic cell-induced autoimmune responses. Cell Death Dis. 2016, 7, e2215. [Google Scholar] [CrossRef]
- Chiang, C.C.; Lin, J.M.; Chen, W.L.; Tsai, Y.Y. Allogeneic serum eye drops for the treatment of severe dry eye in patients with chronic graft-versus-host disease. Cornea 2007, 26, 861–863. [Google Scholar] [CrossRef] [PubMed]
- Dogru, M.; Tsubota, K. Pharmacotherapy of dry eye. Expert. Opin. Pharmacother. 2011, 12, 325–334. [Google Scholar] [CrossRef]
- Na, K.S.; Kim, M.S. Allogeneic serum eye drops for the treatment of dry eye patients with chronic graft-versus-host disease. J. Ocul. Pharmacol. Ther. 2012, 28, 479–483. [Google Scholar] [CrossRef]
- Ogawa, Y.; Okamoto, S.; Mori, T.; Yamada, M.; Mashima, Y.; Watanabe, R.; Kuwana, M.; Tsubota, K.; Ikeda, Y.; Oguchi, Y. Autologous serum eye drops for the treatment of severe dry eye in patients with chronic graft-versus-host disease. Bone Marrow Transpl. 2003, 31, 579–583. [Google Scholar] [CrossRef] [PubMed]
- Pan, Q.; Angelina, A.; Zambrano, A.; Marrone, M.; Stark, W.J.; Heflin, T.; Tang, L.; Akpek, E.K. Autologous serum eye drops for dry eye. Cochrane Database Syst. Rev. 2013, 8, CD009327. [Google Scholar] [PubMed]
- Tsubota, K.; Goto, E.; Fujita, H.; Ono, M.; Inoue, H.; Saito, I.; Shimmura, S. Treatment of dry eye by autologous serum application in Sjogren’s syndrome. Br. J. Ophthalmol. 1999, 83, 390–395. [Google Scholar] [CrossRef] [PubMed]
- Wilson, S.E.; Bourne, W.M. Fuchs’ dystrophy. Cornea 1988, 7, 2–18. [Google Scholar] [CrossRef]
- Dota, A.; Nishida, K.; Quantock, A.J.; Kinoshita, S. Clusterin in human corneal endothelium and aqueous humor. Exp. Eye Res. 1999, 69, 705–708. [Google Scholar] [CrossRef]
- Imhof, A.; Charnay, Y.; Vallet, P.G.; Aronow, B.; Kovari, E.; French, L.E.; Bouras, C.; Giannakopoulos, P. Sustained astrocytic clusterin expression improves remodeling after brain ischemia. Neurobiol. Dis. 2006, 22, 274–283. [Google Scholar] [CrossRef] [PubMed]
- An, E.; Lu, X.; Flippin, J.; Devaney, J.M.; Halligan, B.; Hoffman, E.P.; Strunnikova, N.; Csaky, K.; Hathout, Y. Secreted proteome profiling in human RPE cell cultures derived from donors with age related macular degeneration and age matched healthy donors. J. Proteome Res. 2006, 5, 2599–2610. [Google Scholar] [CrossRef] [PubMed]
- Lidstrom, A.M.; Bogdanovic, N.; Hesse, C.; Volkman, I.; Davidsson, P.; Blennow, K. Clusterin (apolipoprotein J) protein levels are increased in hippocampus and in frontal cortex in Alzheimer’s disease. Exp. Neurol. 1998, 154, 511–521. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, E.; Soto, C.; Governale, S.; Frangione, B.; Ghiso, J. Apolipoprotein J and Alzheimer’s amyloid beta solubility. Biochem. J. 1996, 316 Pt 2 Pt 2, 671–679. [Google Scholar] [CrossRef]
- Buddi, R.; Lin, B.; Atilano, S.R.; Zorapapel, N.C.; Kenney, M.C.; Brown, D.J. Evidence of oxidative stress in human corneal diseases. J. Histochem. Cytochem. 2002, 50, 341–351. [Google Scholar] [CrossRef]
- Wang, Z.; Handa, J.T.; Green, W.R.; Stark, W.J.; Weinberg, R.S.; Jun, A.S. Advanced glycation end products and receptors in Fuchs’ dystrophy corneas undergoing Descemet’s stripping with endothelial keratoplasty. Ophthalmology 2007, 114, 1453–1460. [Google Scholar] [CrossRef]
- Dua, H.S.; Azuara-Blanco, A. Limbal stem cells of the corneal epithelium. Surv. Ophthalmol. 2000, 44, 415–425. [Google Scholar] [CrossRef]
- Sejpal, K.; Bakhtiari, P.; Deng, S.X. Presentation, diagnosis and management of limbal stem cell deficiency. Middle East. Afr. J. Ophthalmol. 2013, 20, 5–10. [Google Scholar]
- Tseng, S.C.; Kruse, F.E.; Merritt, J.; Li, D.Q. Comparison between serum-free and fibroblast-cocultured single-cell clonal culture systems: Evidence showing that epithelial anti-apoptotic activity is present in 3T3 fibroblast-conditioned media. Curr. Eye Res. 1996, 15, 973–984. [Google Scholar] [CrossRef]
- Mishima, K.; Inoue, H.; Nishiyama, T.; Mabuchi, Y.; Amano, Y.; Ide, F.; Matsui, M.; Yamada, H.; Yamamoto, G.; Tanaka, J.; et al. Transplantation of side population cells restores the function of damaged exocrine glands through clusterin. Stem Cells 2012, 30, 1925–1937. [Google Scholar] [CrossRef]
- Ritch, R.; Schlotzer-Schrehardt, U. Exfoliation syndrome. Surv. Ophthalmol. 2001, 45, 265–315. [Google Scholar] [CrossRef] [PubMed]
- Hardenborg, E.; Botling-Taube, A.; Hanrieder, J.; Andersson, M.; Alm, A.; Bergquist, J. Protein content in aqueous humor from patients with pseudoexfoliation (PEX) investigated by capillary LC MALDI-TOF/TOF MS. Proteom. Clin. Appl. 2009, 3, 299–306. [Google Scholar] [CrossRef]
- Sharma, S.; Chataway, T.; Burdon, K.P.; Jonavicius, L.; Klebe, S.; Hewitt, A.W.; Mills, R.A.; Craig, J.E. Identification of LOXL1 protein and Apolipoprotein E as components of surgically isolated pseudoexfoliation material by direct mass spectrometry. Exp. Eye Res. 2009, 89, 479–485. [Google Scholar] [CrossRef] [PubMed]
- Doudevski, I.; Rostagno, A.; Cowman, M.; Liebmann, J.; Ritch, R.; Ghiso, J. Clusterin and complement activation in exfoliation glaucoma. Investig. Ophthalmol. Vis. Sci. 2014, 55, 2491–2499. [Google Scholar] [CrossRef]
- Thomas, C.J.; Mirza, R.G.; Gill, M.K. Age-Related Macular Degeneration. Med. Clin. N. Am. 2021, 105, 473–491. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.L.; Su, X.; Li, X.; Cheung, C.M.; Klein, R.; Cheng, C.Y.; Wong, T.Y. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: A systematic review and meta-analysis. Lancet Glob Health 2014, 2, e106–e116. [Google Scholar] [CrossRef] [PubMed]
- Gheorghe, A.; Mahdi, L.; Musat, O. Age-Related Macular Degeneration. Rom. J. Ophthalmol. 2015, 59, 74–77. [Google Scholar]
- Anderson, D.H.; Mullins, R.F.; Hageman, G.S.; Johnson, L.V. A role for local inflammation in the formation of drusen in the aging eye. Am. J. Ophthalmol. 2002, 134, 411–431. [Google Scholar] [CrossRef]
- Edwards, A.O.; Ritter, R., 3rd; Abel, K.J.; Manning, A.; Panhuysen, C.; Farrer, L.A. Complement factor H polymorphism and age-related macular degeneration. Science 2005, 308, 421–424. [Google Scholar] [CrossRef]
- Fagerness, J.A.; Maller, J.B.; Neale, B.M.; Reynolds, R.C.; Daly, M.J.; Seddon, J.M. Variation near complement factor I is associated with risk of advanced AMD. Eur. J. Hum. Genet. 2009, 17, 100–104. [Google Scholar] [CrossRef]
- Zhan, X.; Larson, D.E.; Wang, C.; Koboldt, D.C.; Sergeev, Y.V.; Fulton, R.S.; Fulton, L.L.; Fronick, C.C.; Branham, K.E.; Bragg-Gresham, J.; et al. Identification of a rare coding variant in complement 3 associated with age-related macular degeneration. Nat. Genet. 2013, 45, 1375–1379. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Dong, N.; Yang, M.; Wang, J.; Feng, X.; Wang, Y. Complement Inhibitors in Age-Related Macular Degeneration: A Potential Therapeutic Option. J. Immunol. Res. 2021, 2021, 9945725. [Google Scholar] [CrossRef] [PubMed]
- Cho, N.H.; Shaw, J.E.; Karuranga, S.; Huang, Y.; da Rocha Fernandes, J.D.; Ohlrogge, A.W.; Malanda, B. IDF Diabetes Atlas: Global estimates of diabetes prevalence for 2017 and projections for 2045. Diabetes Res Clin Pract. 2018, 138, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Morrish, N.J.; Wang, S.L.; Stevens, L.K.; Fuller, J.H.; Keen, H. Mortality and causes of death in the WHO Multinational Study of Vascular Disease in Diabetes. Diabetologia 2001, 44 (Suppl. 2), S14–S21. [Google Scholar] [CrossRef] [PubMed]
- Klein, R.; Klein, B.E.; Moss, S.E.; Davis, M.D.; DeMets, D.L. The Wisconsin Epidemiologic Study of Diabetic Retinopathy. X. Four-year incidence and progression of diabetic retinopathy when age at diagnosis is 30 years or more. Arch. Ophthalmol. 1989, 107, 244–249. [Google Scholar] [CrossRef]
- GBD 2019 Blindness and Vision Impairment Collaborators; on behalf of the Vision Loss Expert Group of the Global Burden of Disease Study. Causes of blindness and vision impairment in 2020 and trends over 30 years, and prevalence of avoidable blindness in relation to VISION 2020: The Right to Sight: An analysis for the Global Burden of Disease Study. Lancet Glob Health 2021, 9, e144–e160. [Google Scholar]
- Modjtahedi, B.S.; Wu, J.; Luong, T.Q.; Gandhi, N.K.; Fong, D.S.; Chen, W. Severity of Diabetic Retinopathy and the Risk of Future Cerebrovascular Disease, Cardiovascular Disease, and All-Cause Mortality. Ophthalmology 2021, 128, 1169–1179. [Google Scholar] [CrossRef]
- Cheung, N.; Mitchell, P.; Wong, T.Y. Diabetic retinopathy. Lancet 2010, 376, 124–136. [Google Scholar] [CrossRef]
- Campochiaro, P.A. Ocular neovascularization. J. Mol. Med. 2013, 91, 311–321. [Google Scholar] [CrossRef]
- Hamel, C. Retinitis pigmentosa. Orphanet J. Rare Dis. 2006, 1, 40. [Google Scholar] [CrossRef]
- Karsdorp, N.; Elderson, A.; Wittebol-Post, D.; Hene, R.J.; Vos, J.; Feldberg, M.A.; van Gils, A.P.; Jansen-Schillhorn van Veen, J.M.; Vroom, T.M.; Hoppener, J.W.; et al. Von Hippel-Lindau disease: New strategies in early detection and treatment. Am. J. Med. 1994, 97, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Maher, E.R.; Webster, A.R.; Moore, A.T. Clinical features and molecular genetics of Von Hippel-Lindau disease. Ophthalmic Genet. 1995, 16, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Latif, F.; Tory, K.; Gnarra, J.; Yao, M.; Duh, F.M.; Orcutt, M.L.; Stackhouse, T.; Kuzmin, I.; Modi, W.; Geil, L.; et al. Identification of the von Hippel-Lindau disease tumor suppressor gene. Science 1993, 260, 1317–1320. [Google Scholar] [CrossRef] [PubMed]
- Seizinger, B.R.; Rouleau, G.A.; Ozelius, L.J.; Lane, A.H.; Farmer, G.E.; Lamiell, J.M.; Haines, J.; Yuen, J.W.; Collins, D.; Majoor-Krakauer, D.; et al. Von Hippel-Lindau disease maps to the region of chromosome 3 associated with renal cell carcinoma. Nature 1988, 332, 268–269. [Google Scholar] [CrossRef] [PubMed]
- Kaelin, W.G., Jr. The von Hippel-Lindau protein, HIF hydroxylation, and oxygen sensing. Biochem. Biophys. Res. Commun. 2005, 338, 627–638. [Google Scholar] [CrossRef]
- Kondo, K.; Klco, J.; Nakamura, E.; Lechpammer, M.; Kaelin, W.G., Jr. Inhibition of HIF is necessary for tumor suppression by the von Hippel-Lindau protein. Cancer Cell 2002, 1, 237–246. [Google Scholar] [CrossRef]
- Raval, R.R.; Lau, K.W.; Tran, M.G.; Sowter, H.M.; Mandriota, S.J.; Li, J.L.; Pugh, C.W.; Maxwell, P.H.; Harris, A.L.; Ratcliffe, P.J. Contrasting properties of hypoxia-inducible factor 1 (HIF-1) and HIF-2 in von Hippel-Lindau-associated renal cell carcinoma. Mol. Cell Biol. 2005, 25, 5675–5686. [Google Scholar] [CrossRef]
- Nakamura, E.; Abreu-e-Lima, P.; Awakura, Y.; Inoue, T.; Kamoto, T.; Ogawa, O.; Kotani, H.; Manabe, T.; Zhang, G.J.; Kondo, K.; et al. Clusterin is a secreted marker for a hypoxia-inducible factor-independent function of the von Hippel-Lindau tumor suppressor protein. Am. J. Pathol. 2006, 168, 574–584. [Google Scholar] [CrossRef]
- Landreville, S.; Agapova, O.A.; Harbour, J.W. Emerging insights into the molecular pathogenesis of uveal melanoma. Future Oncol. 2008, 4, 629–636. [Google Scholar] [CrossRef]
- Ortega, M.A.; Fraile-Martinez, O.; Garcia-Honduvilla, N.; Coca, S.; Alvarez-Mon, M.; Bujan, J.; Teus, M.A. Update on uveal melanoma: Translational research from biology to clinical practice (Review). Int. J. Oncol. 2020, 57, 1262–1279. [Google Scholar] [CrossRef]
- Pessuti, C.L.; Costa, D.F.; Ribeiro, K.S.; Abdouh, M.; Tsering, T.; Nascimento, H.; Commodaro, A.G.; Marcos, A.A.A.; Torrecilhas, A.C.; Belfort, R.N.; et al. Characterization of extracellular vesicles isolated from different liquid biopsies of uveal melanoma patients. J. Circ. Biomark. 2022, 11, 36–47. [Google Scholar] [CrossRef]
- Zhao, W.; Wang, X.; Jiang, Y.; Jia, X.; Guo, Y. miR-217-5p Inhibits Invasion and Metastasis of Prostate Cancer by Targeting Clusterin. Mamm. Genome 2021, 32, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Yao, M.; Wu, M.; Yang, J.; Yao, D.; Wang, L. Secretory clusterin promotes hepatocellular carcinoma progression by facilitating cancer stem cell properties via AKT/GSK-3beta/beta-catenin axis. J. Transl. Med. 2020, 18, 81. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Cao, W.; Su, Q.; Liu, Z.; Zhang, L. Clusterin silencing inhibits proliferation and reduces invasion in human laryngeal squamous carcinoma cells. World J. Surg. Oncol. 2014, 12, 124. [Google Scholar] [CrossRef]
- Lu, J.; Luo, J.H.; Pang, J.; Cao, J.Z.; Wu, R.H.; Tong, Z.T.; Chen, W.; Xie, D. Lentivirus-mediated RNA interference of clusterin enhances the chemosensitivity of EJ bladder cancer cells to epirubicin in vitro. Mol. Med. Rep. 2012, 6, 1133–1139. [Google Scholar] [CrossRef] [PubMed]
- Habiel, D.M.; Camelo, A.; Espindola, M.; Burwell, T.; Hanna, R.; Miranda, E.; Carruthers, A.; Bell, M.; Coelho, A.L.; Liu, H.; et al. Divergent roles for Clusterin in Lung Injury and Repair. Sci. Rep. 2017, 7, 15444. [Google Scholar] [CrossRef]
- Guo, J.; Guan, Q.; Liu, X.; Wang, H.; Gleave, M.E.; Nguan, C.Y.; Du, C. Relationship of clusterin with renal inflammation and fibrosis after the recovery phase of ischemia-reperfusion injury. BMC Nephrol. 2016, 17, 133. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, H.S.; Malda, J.; Richardson, J.B.; Roberts, S. Increased Production of Clusterin in Biopsies of Repair Tissue following Autologous Chondrocyte Implantation. Cartilage 2013, 4, 227–238. [Google Scholar] [CrossRef]
- Deming, Y.; Xia, J.; Cai, Y.; Lord, J.; Holmans, P.; Bertelsen, S.; Holtzman, D.; Morris, J.C.; Bales, K.; Pickering, E.H.; et al. A potential endophenotype for Alzheimer’s disease: Cerebrospinal fluid clusterin. Neurobiol. Aging 2016, 37, 208.e1–208.e9. [Google Scholar] [CrossRef]
- Chen, D.; Wang, Y.; Zhang, K.; Jiao, X.; Yan, B.; Liang, J. Antisense oligonucleotide against clusterin regulates human hepatocellular carcinoma invasion through transcriptional regulation of matrix metalloproteinase-2 and E-cadherin. Int. J. Mol. Sci. 2012, 13, 10594–10607. [Google Scholar] [CrossRef]
- Ljubimov, A.V.; Saghizadeh, M. Progress in corneal wound healing. Prog. Retin. Eye Res. 2015, 49, 17–45. [Google Scholar] [CrossRef] [PubMed]
- Nishida, T.; Tanaka, T. Extracellular matrix and growth factors in corneal wound healing. Curr. Opin. Ophthalmol. 1996, 7, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Al-Hazmi, N.; Thomas, G.J.; Speight, P.M.; Whawell, S.A. The 120 kDa cell-binding fragment of fibronectin up-regulates migration of alphavbeta6-expressing cells by increasing matrix metalloproteinase-2 and -9 secretion. Eur. J. Oral. Sci. 2007, 115, 454–458. [Google Scholar] [CrossRef] [PubMed]



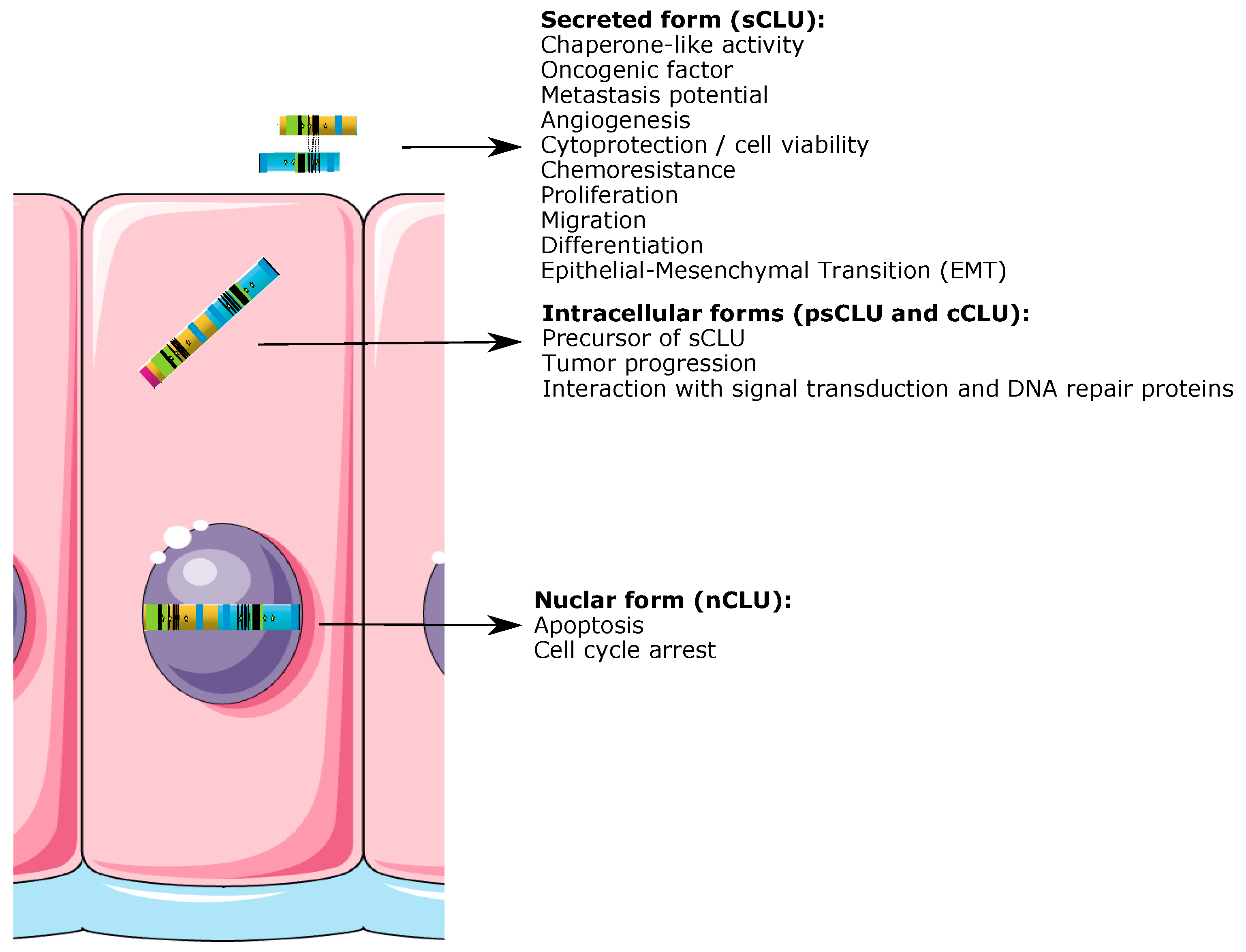


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure | CLU Expression | Pathology | Function |
|---|---|---|---|
| Cornea | Yes | LSCD |
|
| Dystrophies (lattice corneal dystrophy type 1, gelatinous drop-like dystrophy, non-amyloid deposits in granular corneal dystrophy type 2 and Fuch’s dystrophy) |
| ||
| DED | |||
| Anterior chamber (aquous humor) | Yes | AMD |
|
| Iris | Yes | PXG |
|
| Posterior chamber | Yes | ||
| Ciliary body | Yes | PXG |
|
| Vitreous body | Yes | UM |
|
| Lens | Yes | PXG |
|
| Retina | Yes | AMD |
|
| DM |
| ||
| RP |
| ||
| Hemangioblastoma |
| ||
| Choroïd | Yes | ||
| Sclera | Yes | ||
| Optic nerve | Yes | Hemangioblastoma |
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gross, C.; Guérin, L.-P.; Socol, B.G.; Germain, L.; Guérin, S.L. The Ins and Outs of Clusterin: Its Role in Cancer, Eye Diseases and Wound Healing. Int. J. Mol. Sci. 2023, 24, 13182. https://doi.org/10.3390/ijms241713182
Gross C, Guérin L-P, Socol BG, Germain L, Guérin SL. The Ins and Outs of Clusterin: Its Role in Cancer, Eye Diseases and Wound Healing. International Journal of Molecular Sciences. 2023; 24(17):13182. https://doi.org/10.3390/ijms241713182
Chicago/Turabian StyleGross, Christelle, Louis-Philippe Guérin, Bianca G. Socol, Lucie Germain, and Sylvain L. Guérin. 2023. "The Ins and Outs of Clusterin: Its Role in Cancer, Eye Diseases and Wound Healing" International Journal of Molecular Sciences 24, no. 17: 13182. https://doi.org/10.3390/ijms241713182