
Genome-Scale Characterization of Mycobacterium abscessus Complex Isolates from Portugal
 , , , and
, , , and
Abstract
:1. Introduction
2. Results
2.1. Portuguese Dataset Characterization
2.2. Antimicrobial Susceptibility Profiling
2.3. MABC Phylogenetic Analysis

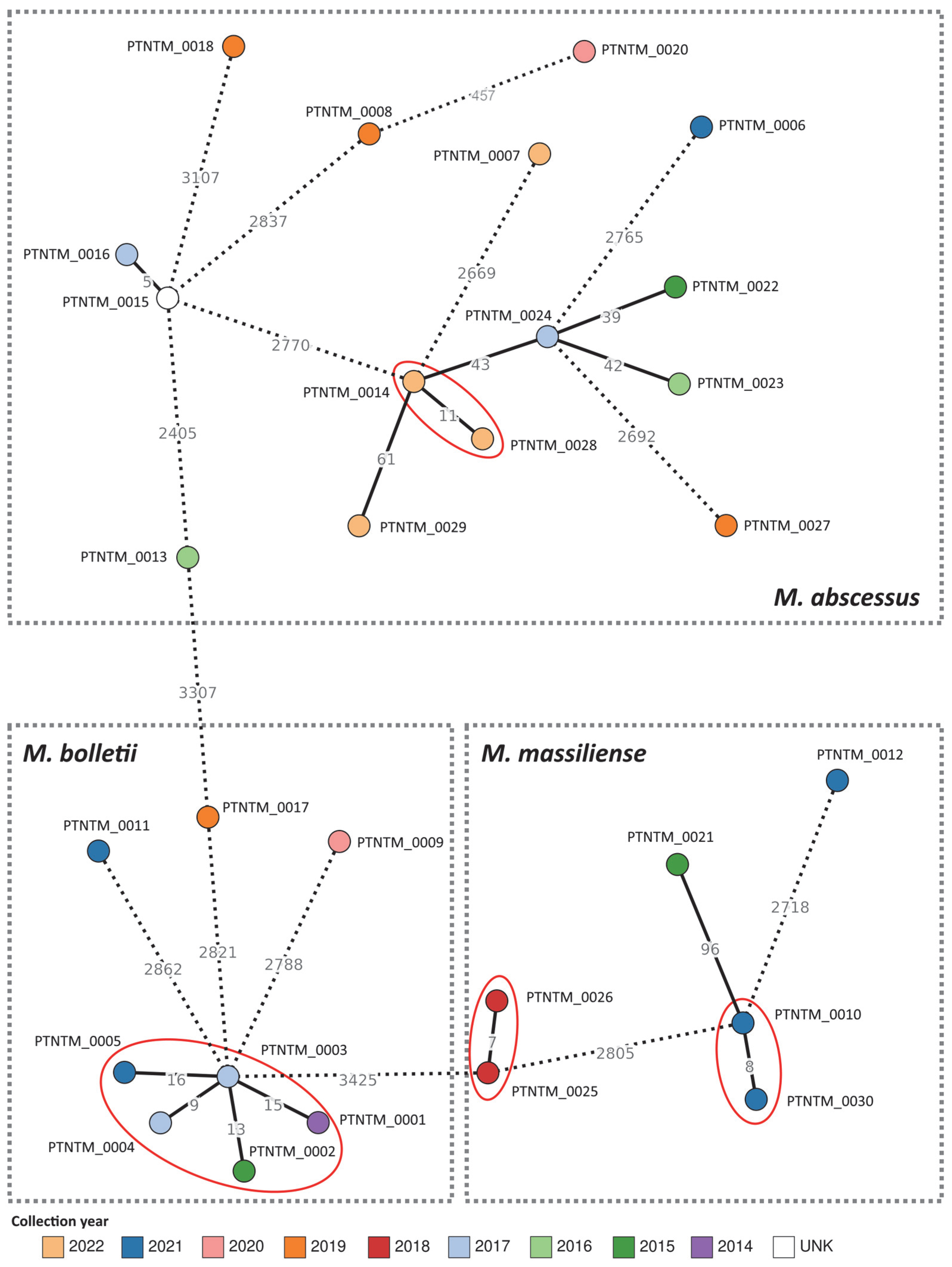
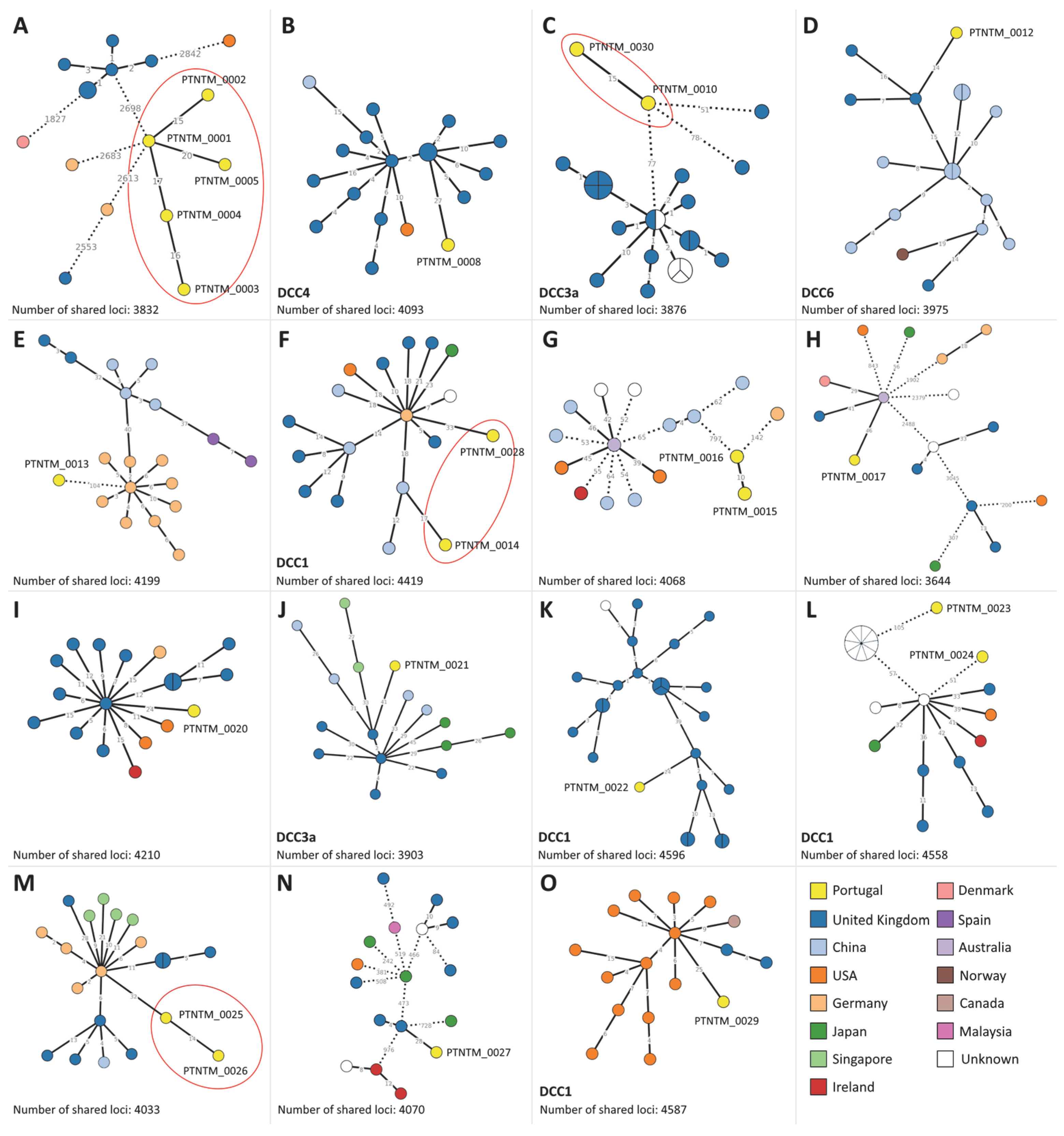
3. Discussion
4. Materials and Methods
4.1. Sample Collection and Characterization
4.2. Whole-Genome Sequencing, Genome Assembly, and Characterization
4.3. Gene-by-Gene Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- To, K.; Cao, R.; Yegiazaryan, A.; Owens, J.; Venketaraman, V. General Overview of Nontuberculous Mycobacteria Opportunistic Pathogens: Mycobacterium avium and Mycobacterium abscessus. J. Clin. Med. 2020, 9, 2541. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.-R.; Sheng, W.-H.; Hung, C.-C.; Yu, C.-J.; Lee, L.-N.; Hsueh, P.-R. Mycobacterium abscessus Complex Infections in Humans. Emerg. Infect. Dis. 2015, 21, 1638. [Google Scholar] [CrossRef] [PubMed]
- Johansen, M.D.; Herrmann, J.-L.; Kremer, L. Non-tuberculous mycobacteria and the rise of Mycobacterium abscessus. Nat. Rev. Microbiol. 2020, 18, 392–407. [Google Scholar] [CrossRef]
- Bryant, J.M.; Grogono, D.M.; Rodriguez-Rincon, D.; Everall, I.; Brown, K.P.; Moreno, P.; Verma, D.; Hill, E.; Drijkoningen, J.; Gilligan, P.; et al. Emergence and spread of a human-transmissible multidrug-resistant nontuberculous mycobacterium. Science 2016, 354, 751–757. [Google Scholar] [CrossRef] [PubMed]
- Falkinham, J.O., III. Surrounded by mycobacteria: Nontuberculous mycobacteria in the human environment. J. Appl. Microbiol. 2009, 107, 356–367. [Google Scholar] [CrossRef]
- Bryant, J.M.; Grogono, D.M.; Greaves, D.; Foweraker, J.; Roddick, I.; Inns, T.; Reacher, M.; Haworth, C.S.; Curran, M.D.; Harris, S.R.; et al. Whole-genome sequencing to identify transmission of Mycobacterium abscessus between patients with cystic fibrosis: A retrospective cohort study. Lancet 2013, 381, 1551–1560. [Google Scholar] [CrossRef]
- Harris, K.A.; Underwood, A.; Kenna, D.T.D.; Brooks, A.; Kavaliunaite, E.; Kapatai, G.; Tewolde, R.; Aurora, P.; Dixon, G. Whole-genome sequencing and epidemiological analysis do not provide evidence for cross-transmission of mycobacterium abscessus in a cohort of pediatric cystic fibrosis patients. Clin. Infect. Dis. 2015, 60, 1007–1016. [Google Scholar] [CrossRef]
- Tortoli, E.; Kohl, T.A.; Trovato, A.; Baldan, R.; Campana, S.; Cariani, L.; Colombo, C.; Costa, D.; Cristadoro, S.; Di Serio, M.C.; et al. Mycobacterium abscessus in patients with cystic fibrosis: Low impact of inter-human transmission in Italy. Eur. Respir. J. 2017, 50, 1602525. [Google Scholar] [CrossRef]
- Aitken, M.L.; Limaye, A.; Pottinger, P.; Whimbey, E.; Goss, C.H.; Tonelli, M.R.; Cangelosi, G.A.; Dirac, M.A.; Olivier, K.N.; Brown-Elliott, B.A.; et al. Respiratory Outbreak of Mycobacterium abscessus Subspecies massiliense in a Lung Transplant and Cystic Fibrosis Center. Am. J. Respir. Crit. Care Med. 2012, 185, 231–232. [Google Scholar] [CrossRef]
- Prevots, D.R.; Shaw, P.A.; Strickland, D.; Jackson, L.A.; Raebel, M.A.; Blosky, M.A.; Montes de Oca, R.; Shea, Y.R.; Seitz, A.E.; Holland, S.M.; et al. Nontuberculous mycobacterial lung disease prevalence at four integrated health care delivery systems. Am. J. Respir. Crit. Care Med. 2010, 182, 970–976. [Google Scholar] [CrossRef]
- Pierre-Audigier, C.; Ferroni, A.; Sermet-Gaudelus, I.; Le Bourgeois, M.; Offredo, C.; Vu-Thien, H.; Fauroux, B.; Mariani, P.; Munck, A.; Bingen, E.; et al. Age-related prevalence and distribution of nontuberculous mycobacterial species among patients with cystic fibrosis. J. Clin. Microbiol. 2005, 43, 3467–3470. [Google Scholar] [CrossRef]
- Nessar, R.; Cambau, E.; Reyrat, J.M.; Murray, A.; Gicquel, B. Mycobacterium abscessus: A new antibiotic nightmare. J. Antimicrob. Chemother. 2012, 67, 810–818. [Google Scholar] [CrossRef] [PubMed]
- Daley, C.L.; Iaccarino, J.M.; Lange, C.; Cambau, E.; Wallace, R.J.; Andrejak, C.; Böttger, E.C.; Brozek, J.; Griffith, D.E.; Guglielmetti, L.; et al. Treatment of Nontuberculous Mycobacterial Pulmonary Disease: An Official ATS/ERS/ESCMID/IDSA Clinical Practice Guideline. Clin. Infect. Dis. 2020, 71, 905–913. [Google Scholar] [CrossRef] [PubMed]
- Nasiri, M.J.; Haeili, M.; Ghazi, M.; Goudarzi, H.; Pormohammad, A.; Imani Fooladi, A.A.; Feizabadi, M.M. New Insights in to the Intrinsic and Acquired Drug Resistance Mechanisms in Mycobacteria. Front. Microbiol. 2017, 8, 681. [Google Scholar] [CrossRef] [PubMed]
- Bronson, R.A.; Gupta, C.; Manson, A.L.; Nguyen, J.A.; Bahadirli-Talbott, A.; Parrish, N.M.; Earl, A.M.; Cohen, K.A. Global phylogenomic analyses of Mycobacterium abscessus provide context for non cystic fibrosis infections and the evolution of antibiotic resistance. Nat. Commun. 2021, 12, 5145. [Google Scholar] [CrossRef]
- Nash, K.A.; Brown-Elliott, B.A.; Wallace, R.J. A Novel Gene, erm(41), Confers Inducible Macrolide Resistance to Clinical Isolates of Mycobacterium abscessus but Is Absent from Mycobacterium chelonae. Antimicrob. Agents Chemother. 2009, 53, 1367–1376. [Google Scholar] [CrossRef]
- Kim, H.-Y.; Kim, B.J.; Kook, Y.; Yun, Y.-J.; Shin, J.H.; Kim, B.-J.; Kook, Y.-H. Mycobacterium massiliense is differentiated from Mycobacterium abscessus and Mycobacterium bolletii by erythromycin ribosome methyltransferase gene (erm) and clarithromycin susceptibility patterns: Characteristic erm(41) of M. massiliense. Microbiol. Immunol. 2010, 54, 347–353. [Google Scholar] [CrossRef]
- Jarand, J.; Levin, A.; Zhang, L.; Huitt, G.; Mitchell, J.D.; Daley, C.L. Clinical and Microbiologic Outcomes in Patients Receiving Treatment for Mycobacterium abscessus Pulmonary Disease. Clin. Infect. Dis. 2011, 52, 565–571. [Google Scholar] [CrossRef]
- Li, B.; Guo, Q.; Mao, Y.; Zou, Y.; Zhang, Y.; Zhang, Z.; Chu, H. Genetic Evolution of Mycobacterium abscessus Conferring Clarithromycin Resistance during Long-Term Antibiotic Therapy. Can. Respir. J. 2020, 2020, 7623828. [Google Scholar] [CrossRef]
- Chew, K.L.; Octavia, S.; Jureen, R.; Ng, O.T.; Marimuthu, K.; Lin, R.T.P.; Teo, J.W.P. Molecular epidemiology and phylogenomic analysis of Mycobacterium abscessus clinical isolates in an Asian population. Microb. Genom. 2021, 7, 000708. [Google Scholar] [CrossRef]
- Jeong, S.H.; Kim, S.-Y.; Huh, H.J.; Ki, C.-S.; Lee, N.Y.; Kang, C.-I.; Chung, D.R.; Peck, K.R.; Shin, S.J.; Koh, W.-J. Mycobacteriological characteristics and treatment outcomes in extrapulmonary Mycobacterium abscessus complex infections. Int. J. Infect. Dis. 2017, 60, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Kasperbauer, S.H.; De Groote, M.A. The Treatment of Rapidly Growing Mycobacterial Infections. Clin. Chest Med. 2015, 36, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Griffith, D.E.; Daley, C.L. Treatment of Mycobacterium abscessus Pulmonary Disease. Chest 2022, 161, 64–75. [Google Scholar] [CrossRef] [PubMed]
- M24-A2; Susceptibility Testing of Mycobacteria, Nocardiae, and Other Aerobic Actinomycetes; Approved Standard—Second Edition; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2019.
- Griffith, D.E. Mycobacterium abscessus subsp abscessus lung disease: ‘Trouble ahead, trouble behind…’. F1000Prime Rep. 2014, 6, 107. [Google Scholar] [CrossRef]
- Dookie, N.; Khan, A.; Padayatchi, N.; Naidoo, K. Application of Next Generation Sequencing for Diagnosis and Clinical Management of Drug-Resistant Tuberculosis: Updates on Recent Developments in the Field. Front. Microbiol. 2022, 13, 775030. [Google Scholar] [CrossRef]
- Jeukens, J.; Freschi, L.; Kukavica-Ibrulj, I.; Emond-Rheault, J.-G.; Tucker, N.P.; Levesque, R.C. Genomics of antibiotic-resistance prediction in Pseudomonas aeruginosa: Predicting antibiotic resistance in Pseudomonas. Ann. N. Y. Acad. Sci. 2019, 1435, 5–17. [Google Scholar] [CrossRef]
- NIHR Global Health Research Unit on Genomic Surveillance of AMR. Whole-genome sequencing as part of national and international surveillance programmes for antimicrobial resistance: A roadmap. BMJ Glob Health 2020, 5, e002244. [Google Scholar] [CrossRef]
- Dal Molin, M.; Gut, M.; Rominski, A.; Haldimann, K.; Becker, K.; Sander, P. Molecular Mechanisms of Intrinsic Streptomycin Resistance in Mycobacterium abscessus. Antimicrob. Agents Chemother. 2018, 62, e01427-17. [Google Scholar] [CrossRef]
- Rominski, A.; Selchow, P.; Becker, K.; Brülle, J.K.; Dal Molin, M.; Sander, P. Elucidation of Mycobacterium abscessus aminoglycoside and capreomycin resistance by targeted deletion of three putative resistance genes. J. Antimicrob. Chemother. 2017, 72, 2191–2200. [Google Scholar] [CrossRef]
- Dubois, V.; Pawlik, A.; Bories, A.; Le Moigne, V.; Sismeiro, O.; Legendre, R.; Varet, H.; Rodríguez-Ordóñez, M.D.P.; Gaillard, J.-L.; Coppée, J.-Y.; et al. Mycobacterium abscessus virulence traits unraveled by transcriptomic profiling in amoeba and macrophages. PLoS Pathog. 2019, 15, e1008069. [Google Scholar] [CrossRef]
- Ung, K.L.; Alsarraf, H.M.A.B.; Olieric, V.; Kremer, L.; Blaise, M. Crystal structure of the aminoglycosides N-acetyltransferase Eis2 from Mycobacterium abscessus. FEBS J. 2019, 286, 4342–4355. [Google Scholar] [CrossRef] [PubMed]
- Alcaide, F.; Pfyffer, G.E.; Telenti, A. Role of embB in natural and acquired resistance to ethambutol in mycobacteria. Antimicrob. Agents Chemother. 1997, 41, 2270–2273. [Google Scholar] [CrossRef] [PubMed]
- Halloum, I.; Viljoen, A.; Khanna, V.; Craig, D.; Bouchier, C.; Brosch, R.; Coxon, G.; Kremer, L. Resistance to Thiacetazone Derivatives Active against Mycobacterium abscessus Involves Mutations in the MmpL5 Transcriptional Repressor MAB_4384. Antimicrob. Agents Chemother. 2017, 61, e02509-16. [Google Scholar] [CrossRef] [PubMed]
- Richard, M.; Gutiérrez, A.V.; Viljoen, A.J.; Ghigo, E.; Blaise, M.; Kremer, L. Mechanistic and Structural Insights Into the Unique TetR-Dependent Regulation of a Drug Efflux Pump in Mycobacterium abscessus. Front. Microbiol. 2018, 9, 649. [Google Scholar] [CrossRef] [PubMed]
- Mizzi, R.; Plain, K.M.; Whittington, R.; Timms, V.J. Global Phylogeny of Mycobacterium avium and Identification of Mutation Hotspots During Niche Adaptation. Front. Microbiol. 2022, 13, 892333. [Google Scholar] [CrossRef]
- Diricks, M.; Merker, M.; Wetzstein, N.; Kohl, T.A.; Niemann, S.; Maurer, F.P. Delineating Mycobacterium abscessus population structure and transmission employing high-resolution core genome multilocus sequence typing. Nat. Commun. 2022, 13, 4936. [Google Scholar] [CrossRef]
- Santos, A.; Pinto, M.; Carneiro, S.; Silva, S.; Rodrigues, I.; Munhá, J.; Gomes, J.P.; Macedo, R. Microevolution of a Mycobacteroides abscessus subsp. bolletii strain in a clinical persistent infection. Infect. Genet. Evol. 2023, 112, 105437. [Google Scholar] [CrossRef]
- Zhou, Z.; Alikhan, N.-F.; Sergeant, M.J.; Luhmann, N.; Vaz, C.; Francisco, A.P.; Carriço, J.A.; Achtman, M. GrapeTree: Visualization of core genomic relationships among 100,000 bacterial pathogens. Genome Res. 2018, 28, 1395–1404. [Google Scholar] [CrossRef]
- Abdelaal, H.F.M.; Chan, E.D.; Young, L.; Baldwin, S.L.; Coler, R.N. Mycobacterium abscessus: It’s Complex. Microorganisms 2022, 10, 1454. [Google Scholar] [CrossRef]
- Koh, W.-J.; Stout, J.E.; Yew, W.-W. Advances in the management of pulmonary disease due to Mycobacterium abscessus complex. Int. J. Tuberc. Lung Dis. 2014, 18, 1141–1148. [Google Scholar] [CrossRef]
- Redondo, N.; Mok, S.; Montgomery, L.; Flanagan, P.R.; McNamara, E.; Smyth, E.G.; O’Sullivan, N.; Schaffer, K.; Rogers, T.R.; Fitzgibbon, M.M. Genomic Analysis of Mycobacterium abscessus Complex Isolates Collected in Ireland between 2006 and 2017. J. Clin. Microbiol. 2020, 58, e00295-20. [Google Scholar] [CrossRef] [PubMed]
- Roux, A.-L.; Catherinot, E.; Ripoll, F.; Soismier, N.; Macheras, E.; Ravilly, S.; Bellis, G.; Vibet, M.-A.; Le Roux, E.; Lemonnier, L.; et al. Multicenter Study of Prevalence of Nontuberculous Mycobacteria in Patients with Cystic Fibrosis in France. J. Clin. Microbiol. 2009, 47, 4124–4128. [Google Scholar] [CrossRef] [PubMed]
- Broda, A.; Jebbari, H.; Beaton, K.; Mitchell, S.; Drobniewski, F. Comparative Drug Resistance of Mycobacterium abscessus and M. chelonae Isolates from Patients with and without Cystic Fibrosis in the United Kingdom. J. Clin. Microbiol. 2013, 51, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Wang, X.; Jin, J.; Wu, J.; Zhang, X.; Chen, J.; Zhang, W. In Vitro Susceptibility of Mycobacterium abscessus and Mycobacterium fortuitum Isolates to 30 Antibiotics. BioMed Res. Int. 2018, 2018, 4902941. [Google Scholar] [CrossRef]
- Guo, Q.; Wei, J.; Zou, W.; Li, Q.; Qian, X.; Zhu, Z. Antimicrobial susceptibility profiles of Mycobacterium abscessus complex isolates from respiratory specimens in Shanghai, China. J. Glob. Antimicrob. Resist. 2021, 25, 72–76. [Google Scholar] [CrossRef]
- Kusuki, M.; Osawa, K.; Arikawa, K.; Tamura, M.; Shigemura, K.; Shirakawa, T.; Nakamura, T.; Nakamachi, Y.; Fujisawa, M.; Saegusa, J.; et al. Determination of the antimicrobial susceptibility and molecular profile of clarithromycin resistance in the Mycobacterium abscessus complex in Japan by variable number tandem repeat analysis. Diagn. Microbiol. Infect. Dis. 2018, 91, 256–259. [Google Scholar] [CrossRef]
- Wang, Y.; He, W.; He, P.; Zheng, H.; Zhao, Y. Comparing genotypes and antibiotic resistance profiles of Mycobacterium abscessus and Mycobacterium massiliense clinical isolates in China. Epidemiol. Infect. 2021, 149, e232. [Google Scholar] [CrossRef]
- Cho, E.H.; Huh, H.J.; Song, D.J.; Lee, S.H.; Kim, C.K.; Shin, S.Y.; Ki, C.-S.; Jhun, B.W.; Moon, S.M.; Kwon, O.J.; et al. Drug susceptibility patterns of Mycobacterium abscessus and Mycobacterium massiliense isolated from respiratory specimens. Diagn. Microbiol. Infect. Dis. 2019, 93, 107–111. [Google Scholar] [CrossRef]
- Bernut, A.; Le Moigne, V.; Lesne, T.; Lutfalla, G.; Herrmann, J.-L.; Kremer, L. In Vivo Assessment of Drug Efficacy against Mycobacterium abscessus Using the Embryonic Zebrafish Test System. Antimicrob. Agents Chemother. 2014, 58, 4054–4063. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, W.; Wang, Y.; Xue, Z.; Li, S.; Pang, Y. Inducible Resistance to Amikacin in Mycobacterium abscessus Isolated in Beijing, China. Infect. Drug Resist. 2022, 15, 2287–2291. [Google Scholar] [CrossRef]
- Griffith, D.E.; Aksamit, T.; Brown-Elliott, B.A.; Catanzaro, A.; Daley, C.; Gordin, F.; Holland, S.M.; Horsburgh, R.; Huitt, G.; Iademarco, M.F.; et al. An Official ATS/IDSA Statement: Diagnosis, Treatment, and Prevention of Nontuberculous Mycobacterial Diseases. Am. J. Respir. Crit. Care Med. 2007, 175, 367–416. [Google Scholar] [CrossRef] [PubMed]
- Philley, J.; Griffith, D. Management of Nontuberculous Mycobacterial (NTM) Lung Disease. Semin Respir. Crit. Care Med. 2013, 34, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Dubée, V.; Bernut, A.; Cortes, M.; Lesne, T.; Dorchene, D.; Lefebvre, A.-L.; Hugonnet, J.-E.; Gutmann, L.; Mainardi, J.-L.; Herrmann, J.-L.; et al. β-Lactamase inhibition by avibactam in Mycobacterium abscessus. J. Antimicrob. Chemother. 2015, 70, 1051–1058. [Google Scholar] [CrossRef] [PubMed]
- Luthra, S.; Rominski, A.; Sander, P. The Role of Antibiotic-Target-Modifying and Antibiotic-Modifying Enzymes in Mycobacterium abscessus Drug Resistance. Front. Microbiol. 2018, 9, 2179. [Google Scholar] [CrossRef] [PubMed]
- Jin, P.; Dai, J.; Guo, Y.; Wang, X.; Lu, J.; Zhu, Y.; Yu, F. Genomic Analysis of Mycobacterium abscessus Complex Isolates from Patients with Pulmonary Infection in China. Microbiol. Spectr. 2022, 10, e00118-22. [Google Scholar] [CrossRef] [PubMed]
- Davidson, R.M.; Hasan, N.A.; Reynolds, P.R.; Totten, S.; Garcia, B.; Levin, A.; Ramamoorthy, P.; Heifets, L.; Daley, C.L.; Strong, M. Genome Sequencing of Mycobacterium abscessus Isolates from Patients in the United States and Comparisons to Globally Diverse Clinical Strains. J. Clin. Microbiol. 2014, 52, 3573–3582. [Google Scholar] [CrossRef]
- Everall, I.; Nogueira, C.L.; Bryant, J.M.; Sánchez-Busó, L.; Chimara, E.; Duarte, R.D.S.; Ramos, J.P.; Lima, K.V.B.; Lopes, M.L.; Palaci, M.; et al. Genomic epidemiology of a national outbreak of post-surgical Mycobacterium abscessus wound infections in Brazil. Microb. Genom. 2017, 3, e000111. [Google Scholar] [CrossRef]
- Ripoll, F.; Pasek, S.; Schenowitz, C.; Dossat, C.; Barbe, V.; Rottman, M.; Macheras, E.; Heym, B.; Herrmann, J.-L.; Daffé, M.; et al. Non Mycobacterial Virulence Genes in the Genome of the Emerging Pathogen Mycobacterium abscessus. PLoS ONE 2009, 4, e5660. [Google Scholar] [CrossRef]
- National Institute for Public Health and the Environment (RIVM). Protocol, Isolation of High Molecular Weight Genomic DNA from Mycobacteria (CTAB Procedure). Available online: https://www.rivm.nl/sites/default/files/2018-11/Protocol%2C%20Isolation%20of%20high%20molecular%20weight%20genomic%20DNA%20%28CTAB%29%20a1a.pdf (accessed on 20 September 2023).
- Hain Lifescience GenoType Mycobacterium CM VER 2.0. Available online: https://www.hain-lifescience.de/en/products/microbiology/mycobacteria/ntm/genotype-mycobacterium-cm.html (accessed on 20 September 2023).
- Hain Lifescience GenoType NTM-DR VER 1.0. Available online: https://www.hain-lifescience.de/en/products/microbiology/mycobacteria/ntm/genotype-ntm-dr.html (accessed on 20 September 2023).
- Saifi, M.; Jabbarzadeh, E.; Bahrmand, A.R.; Karimi, A.; Pourazar, S.; Fateh, A.; Masoumi, M.; Vahidi, E. HSP65-PRA identification of non-tuberculosis mycobacteria from 4892 samples suspicious for mycobacterial infections. Clin. Microbiol. Infect. 2013, 19, 723–728. [Google Scholar] [CrossRef]
- Llarena, A.; Ribeiro-Gonçalves, B.F.; Nuno Silva, D.; Halkilahti, J.; Machado, M.P.; Da Silva, M.S.; Jaakkonen, A.; Isidro, J.; Hämäläinen, C.; Joenperä, J.; et al. INNUENDO: A cross-sectoral platform for the integration of genomics in the surveillance of food-borne pathogens. EFSA Support. Publ. 2018, 15, 1498E. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An Integrated Tool for Comprehensive Microbial Variant Detection and Genome Assembly Improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef] [PubMed]
- Wood, D.E.; Salzberg, S.L. Kraken: Ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014, 15, R46. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.; Machado, M.P.; Silva, D.N.; Rossi, M.; Moran-Gilad, J.; Santos, S.; Ramirez, M.; Carriço, J.A. chewBBACA: A complete suite for gene-by-gene schema creation and strain identification. Microb. Genom. 2018, 4, e000166. [Google Scholar] [CrossRef] [PubMed]
- Mixão, V.; Pinto, M.; Sobral, D.; Di Pasquale, A.; Gomes, J.P.; Borges, V. ReporTree: A surveillance-oriented tool to strengthen the linkage between pathogen genetic clusters and epidemiological data. Genome Med. 2023, 15, 43. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Isolate ID | WGS-Based Taxonomic Classification | DCC Classification | Collection Year | Patient ID | Region of Residence | Gender | Age Group |
|---|---|---|---|---|---|---|---|
| PTNTM_0001 | M. bolletii | Non-DCC | 2014 | Patient 01 | Algarve | Male | [65–70[ |
| PTNTM_0002 | M. bolletii | Non-DCC | 2015 | Patient 01 | Algarve | Male | [65–70[ |
| PTNTM_0003 | M. bolletii | Non-DCC | 2017 | Patient 01 | Algarve | Male | [65–70[ |
| PTNTM_0004 | M. bolletii | Non-DCC | 2017 | Patient 01 | Algarve | Male | [65–70[ |
| PTNTM_0005 | M. bolletii | Non-DCC | 2021 | Patient 01 | Algarve | Male | [70–75[ |
| PTNTM_0006 | M. abscessus | Non-DCC | 2021 | Patient 06 | LMA | Female | [50–55[ |
| PTNTM_0007 | M. abscessus | Non-DCC | 2022 | Patient 17 | LMA | Female | [75–80[ |
| PTNTM_0008 | M. abscessus | DCC4 | 2019 | Patient 11 | LMA | Female | [70–75[ |
| PTNTM_0009 | M. bolletii | Non-DCC | 2020 | Patient 09 | LMA | Female | [65–70[ |
| PTNTM_0010 | M. massiliense | DCC3a | 2021 | Patient 03 | UNK | Male | [40–45[ |
| PTNTM_0011 | M. bolletii | Non-DCC | 2021 | Patient 13 | UNK | Male | UNK |
| PTNTM_0012 | M. massiliense | DCC6 | 2021 | Patient 14 | North | Male | [60–65[ |
| PTNTM_0013 | M. abscessus | Non-DCC | 2016 | Patient 10 | LMA | Female | [65–70[ |
| PTNTM_0014 | M. abscessus | DCC1 | 2022 | Patient 04 | UNK | Male | UNK |
| PTNTM_0015 | M. abscessus | Non-DCC | UNK | UNK | UNK | UNK | UNK |
| PTNTM_0016 | M. abscessus | Non-DCC | 2017 | Patient 21 | LMA | Male | [70–75[ |
| PTNTM_0017 | M. bolletii | Non-DCC | 2019 | Patient 22 | Algarve | Female | [25–30[ |
| PTNTM_0018 | M. abscessus | Non-DCC | 2019 | Patient 08 | Centre | Female | [80–85[ |
| PTNTM_0019 | M. abscessus | Non-DCC | 2020 | Patient 20 | UNK | Female | UNK |
| PTNTM_0020 | M. abscessus | Non-DCC | 2020 | Patient 19 | LMA | Male | [30–35[ |
| PTNTM_0021 | M. massiliense | DCC3a | 2015 | Patient 05 | LMA | Male | [75–80[ |
| PTNTM_0022 | M. abscessus | DCC1 | 2015 | Patient 15 | LMA | Male | [75–80[ |
| PTNTM_0023 | M. abscessus | DCC1 | 2016 | Patient 07 | LMA | Male | [55–60[ |
| PTNTM_0024 | M. abscessus | DCC1 | 2017 | Patient 16 | LMA | Female | [65–70[ |
| PTNTM_0025 | M. massiliense | Non-DCC | 2018 | Patient 02 | LMA | Male | [85–90[ |
| PTNTM_0026 | M. massiliense | Non-DCC | 2018 | Patient 02 | LMA | Male | [85–90[ |
| PTNTM_0027 | M. abscessus | Non-DCC | 2019 | Patient 18 | LMA | Female | [75–80[ |
| PTNTM_0028 | M. abscessus | DCC1 | 2022 | Patient 04 | UNK | Male | UNK |
| PTNTM_0029 | M. abscessus | DCC1 | 2022 | Patient 12 | Algarve | Male | [70–75[ |
| PTNTM_0030 | M. massiliense | DCC3a | 2021 | Patient 03 | UNK | Male | [40–45[ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carneiro, S.; Pinto, M.; Silva, S.; Santos, A.; Rodrigues, I.; Santos, D.; Duarte, S.; Vieira, L.; Gomes, J.P.; Macedo, R. Genome-Scale Characterization of Mycobacterium abscessus Complex Isolates from Portugal. Int. J. Mol. Sci. 2023, 24, 15402. https://doi.org/10.3390/ijms242015402
Carneiro S, Pinto M, Silva S, Santos A, Rodrigues I, Santos D, Duarte S, Vieira L, Gomes JP, Macedo R. Genome-Scale Characterization of Mycobacterium abscessus Complex Isolates from Portugal. International Journal of Molecular Sciences. 2023; 24(20):15402. https://doi.org/10.3390/ijms242015402
Chicago/Turabian StyleCarneiro, Sofia, Miguel Pinto, Sónia Silva, Andrea Santos, Irene Rodrigues, Daniela Santos, Sílvia Duarte, Luís Vieira, João Paulo Gomes, and Rita Macedo. 2023. "Genome-Scale Characterization of Mycobacterium abscessus Complex Isolates from Portugal" International Journal of Molecular Sciences 24, no. 20: 15402. https://doi.org/10.3390/ijms242015402