
Characterization of the First Animal Toxin Acting as an Antagonist on AT1 Receptor
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
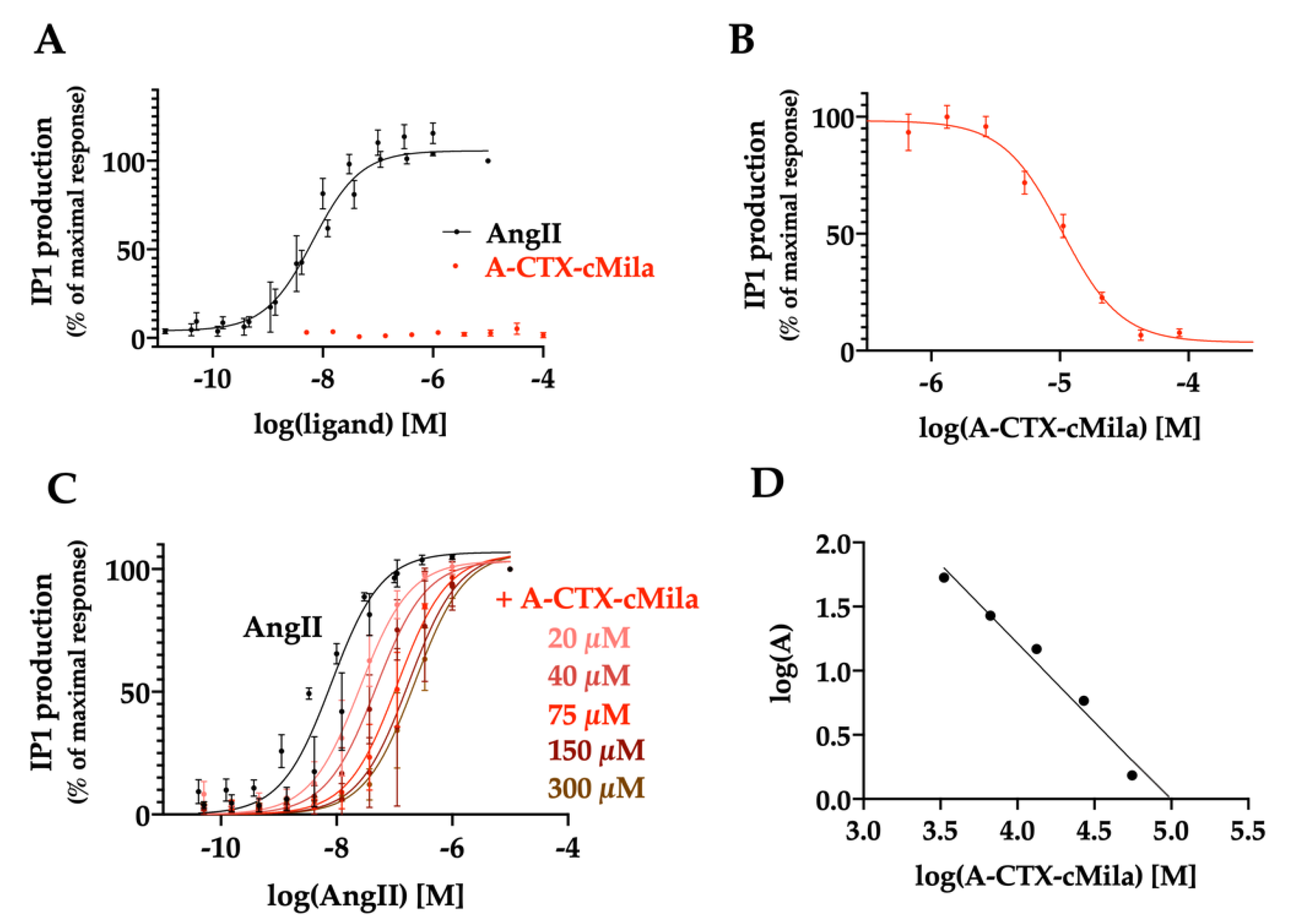
2.1. Screening on Angiotensin II Receptors
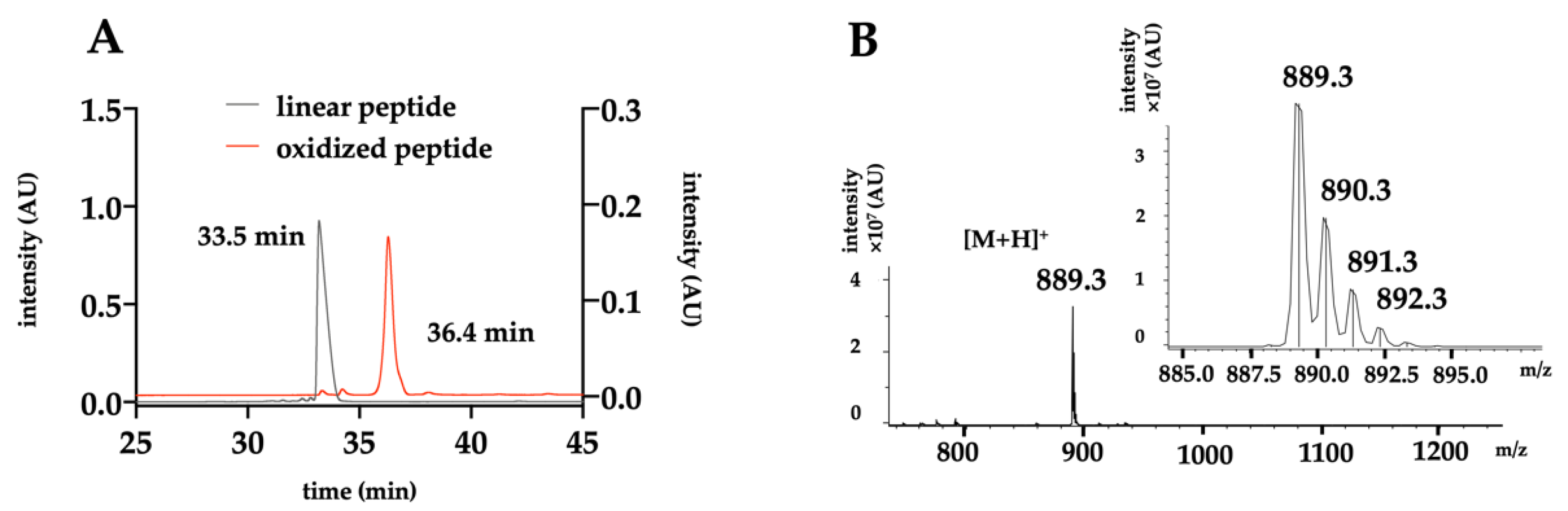
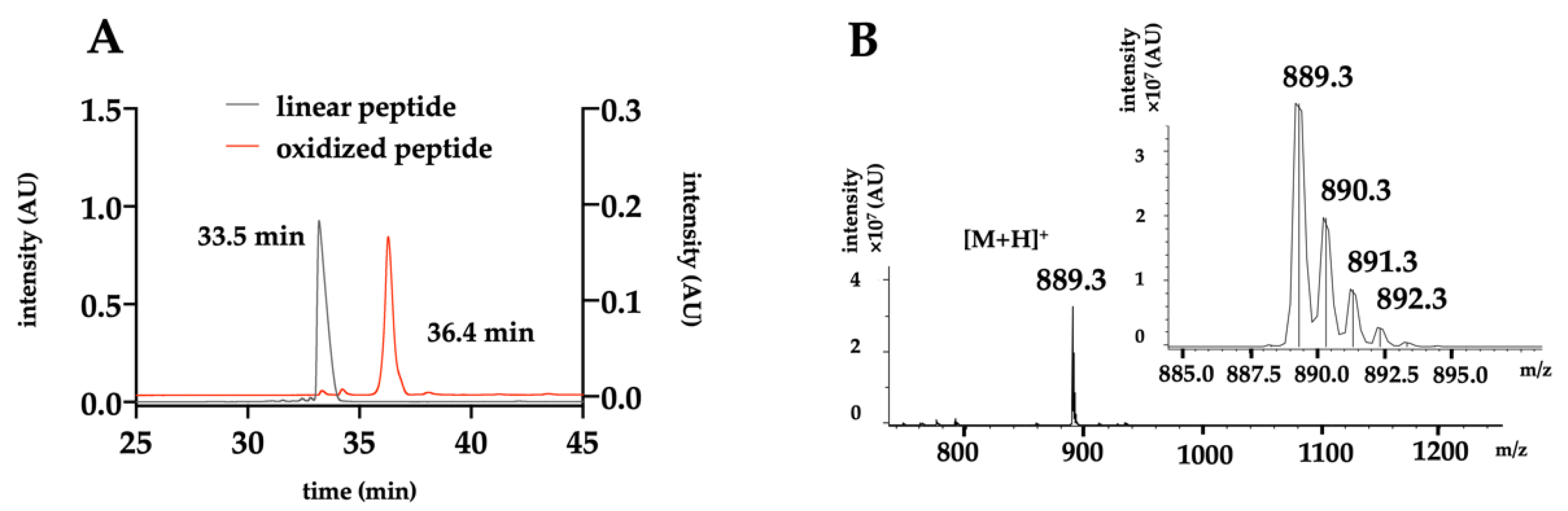
2.2. Chemical Production of the Toxin
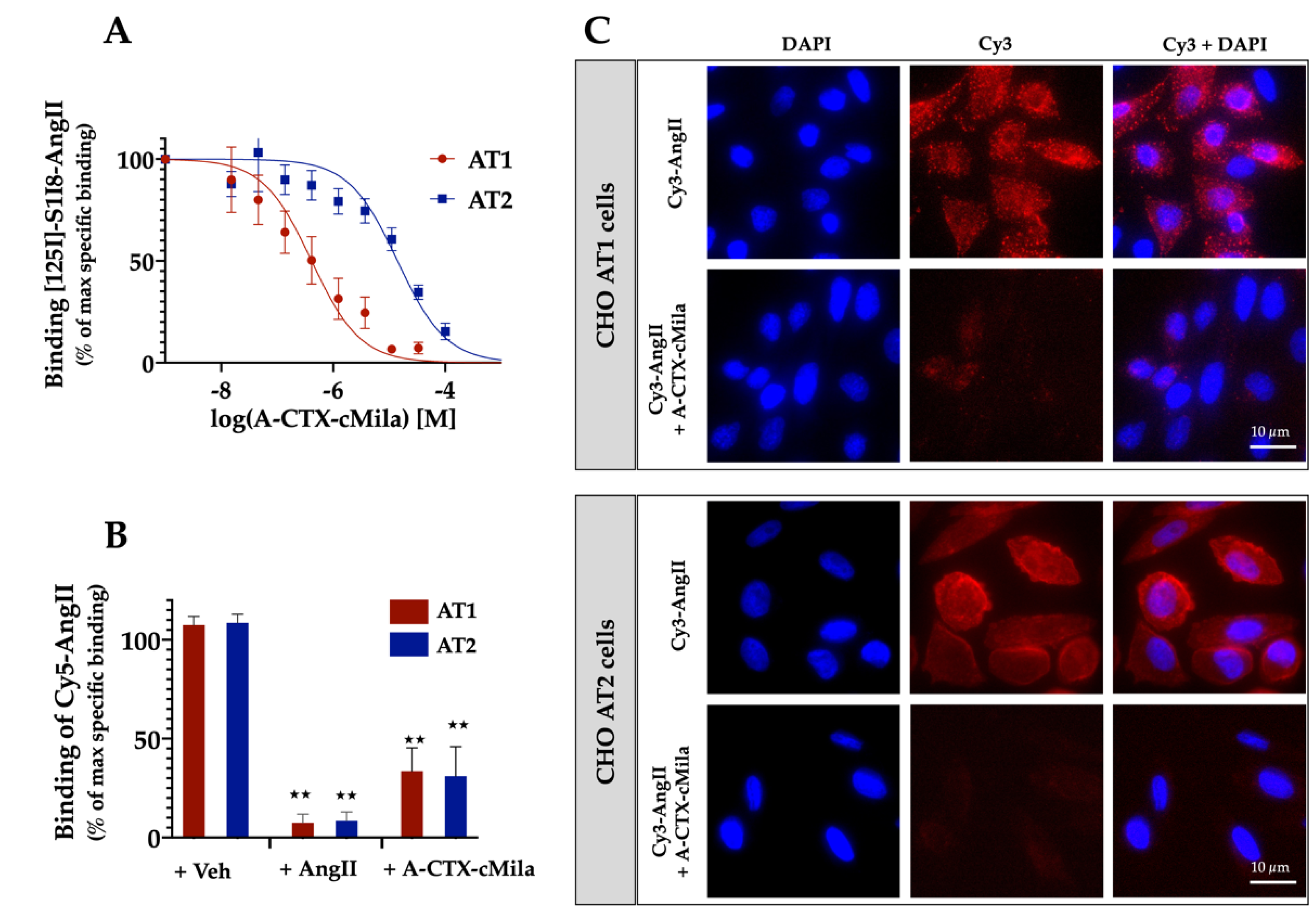
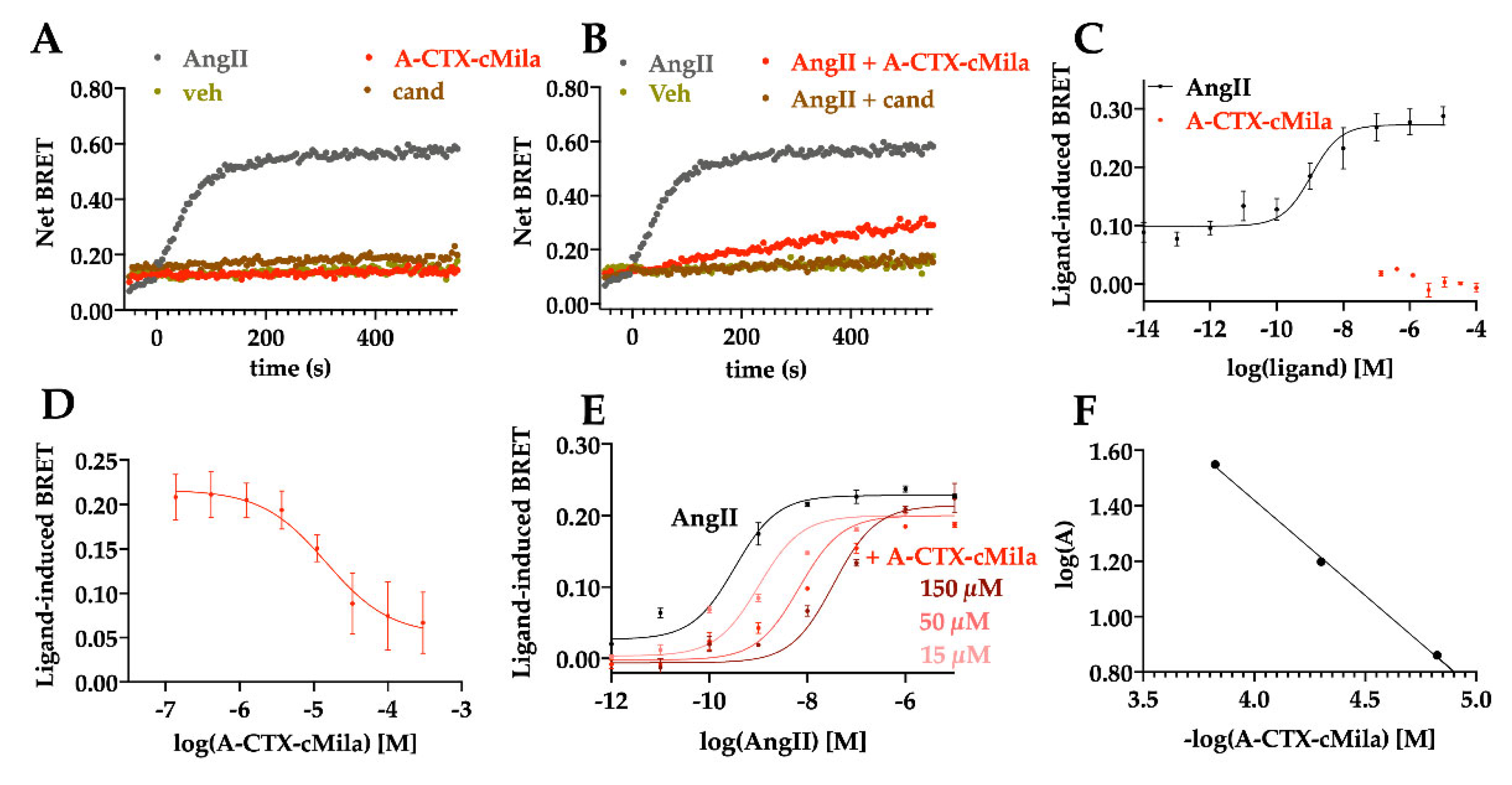
2.3. Binding Properties of A-CTX-cMila on Angiotensin II Receptors
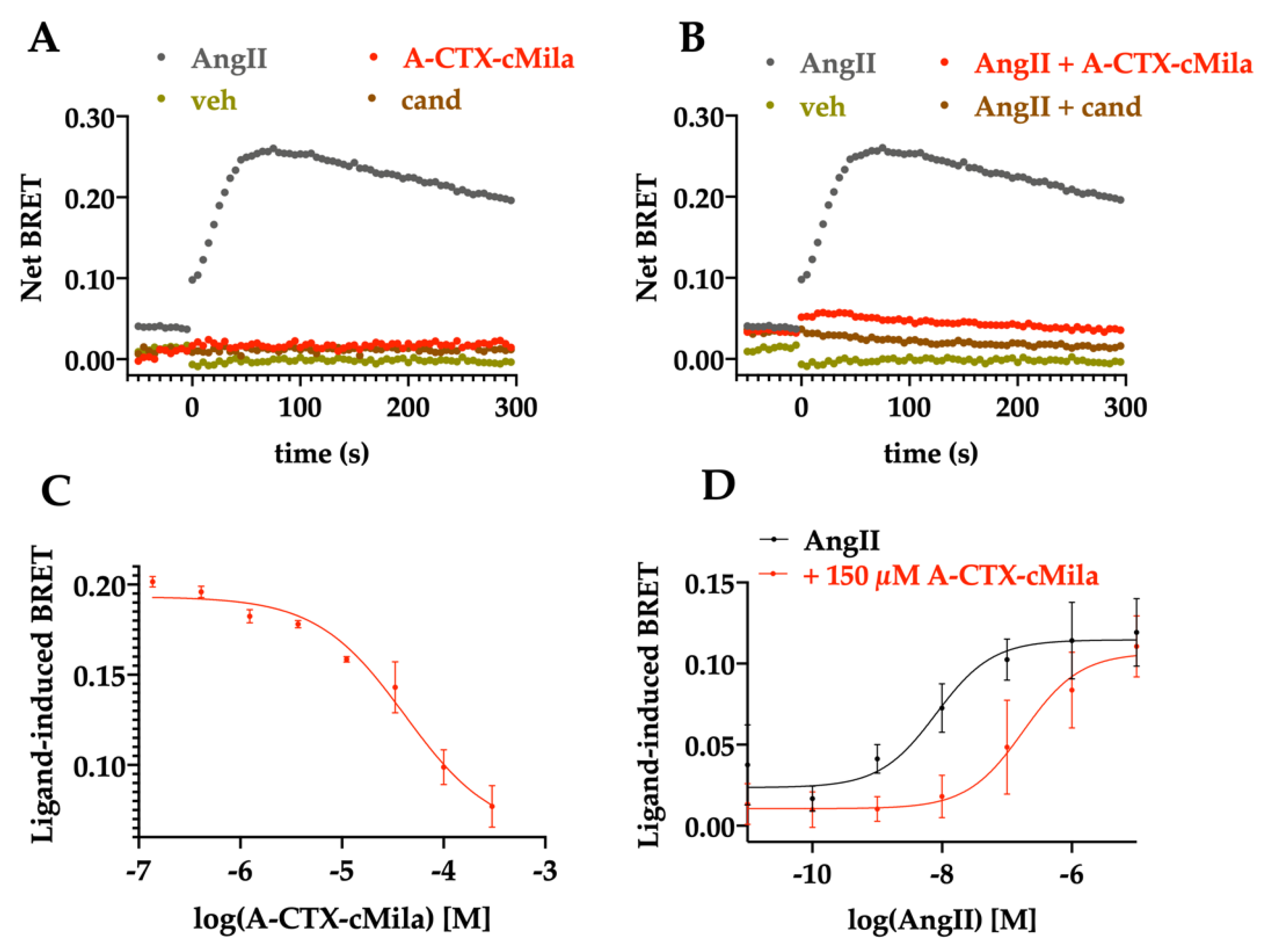
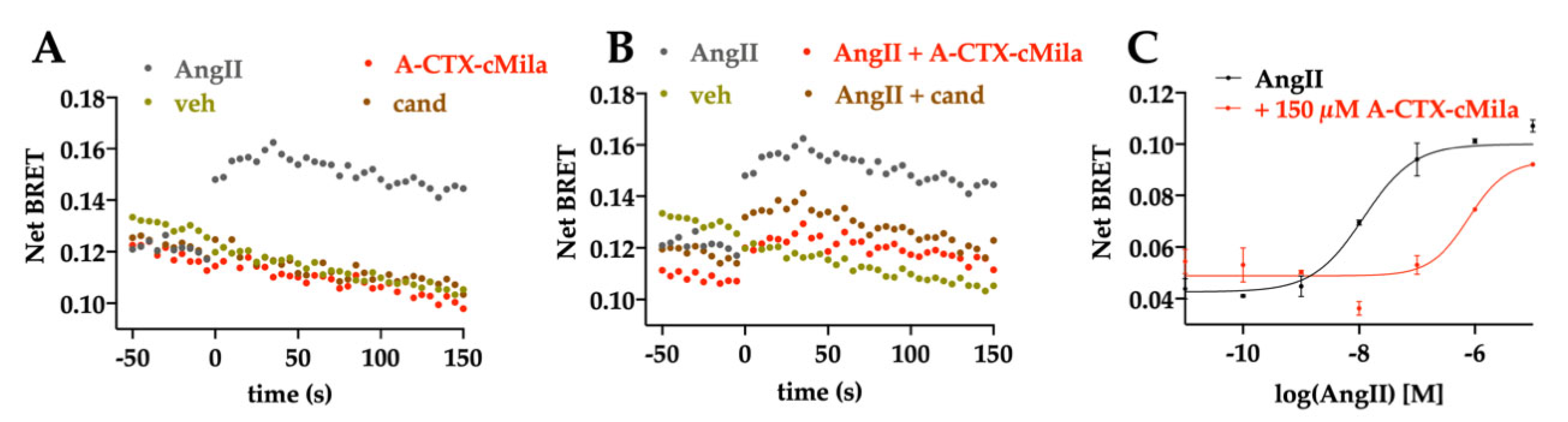
2.4. Pharmacological Characterization of A-CTX-cMila on AT1-Mediated Gαq/PLC Pathway
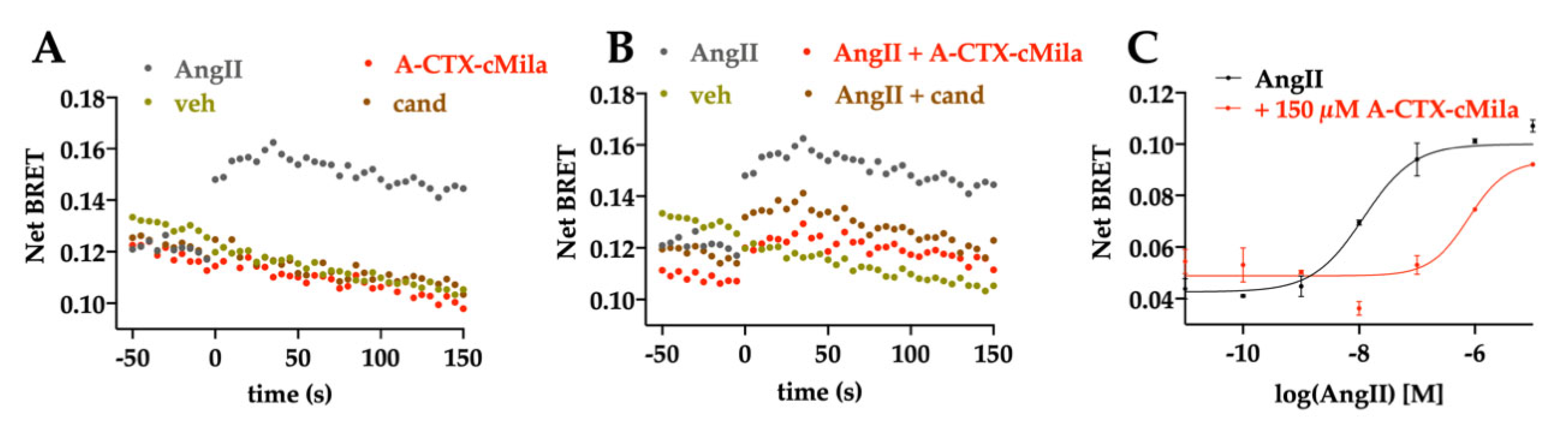
2.5. Pharmacological Characterization of A-CTX-cMila on AT1-Mediated Gαi3 and GαoA Pathways
2.6. Pharmacological Characterization of A-CTX-cMila on AT1-Mediated β-arrestin 2 Recruitment
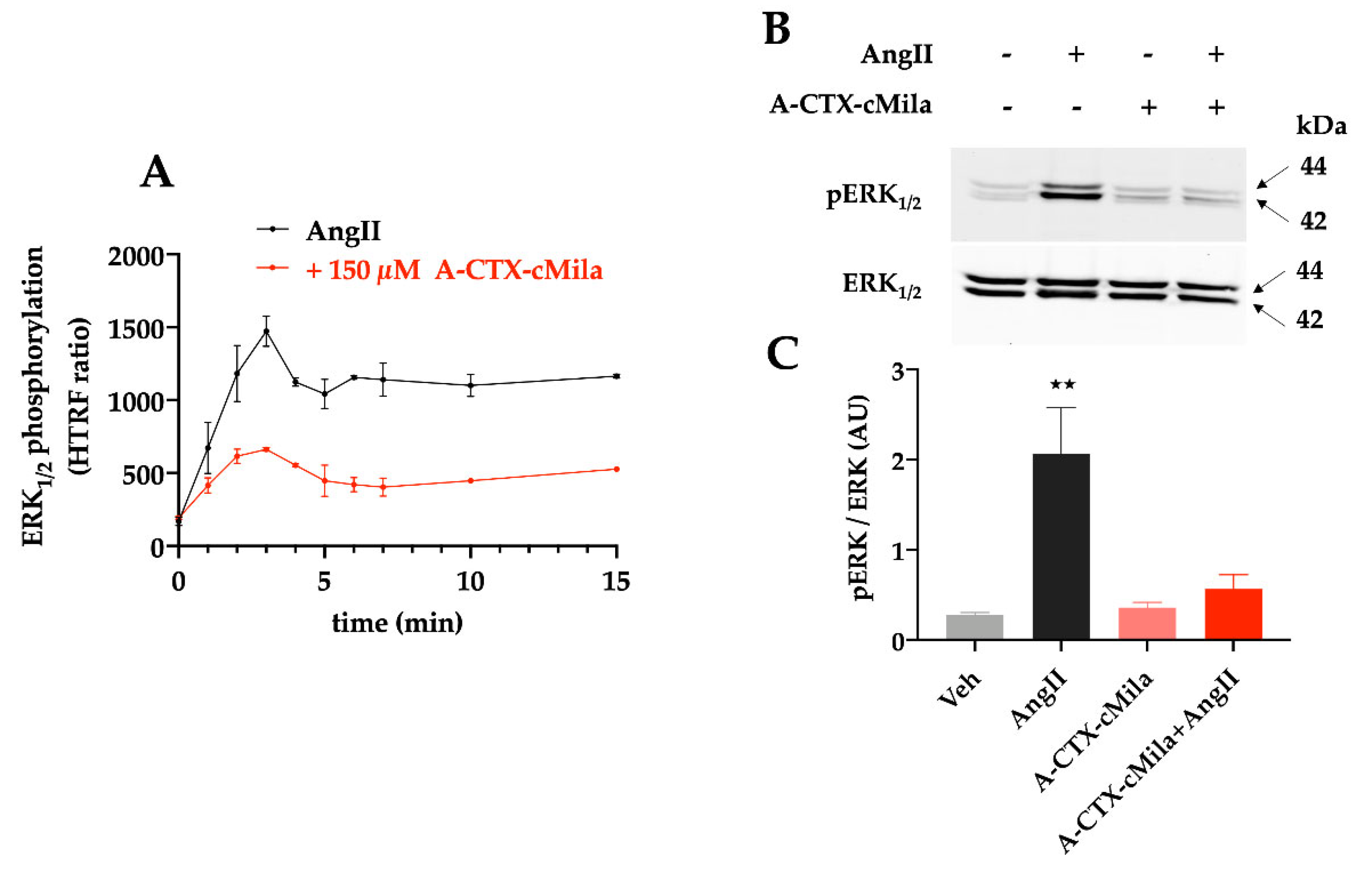
2.7. Pharmacological Characterization of A-CTX-cMila on AT1-Mediated ERK1/2 Activation
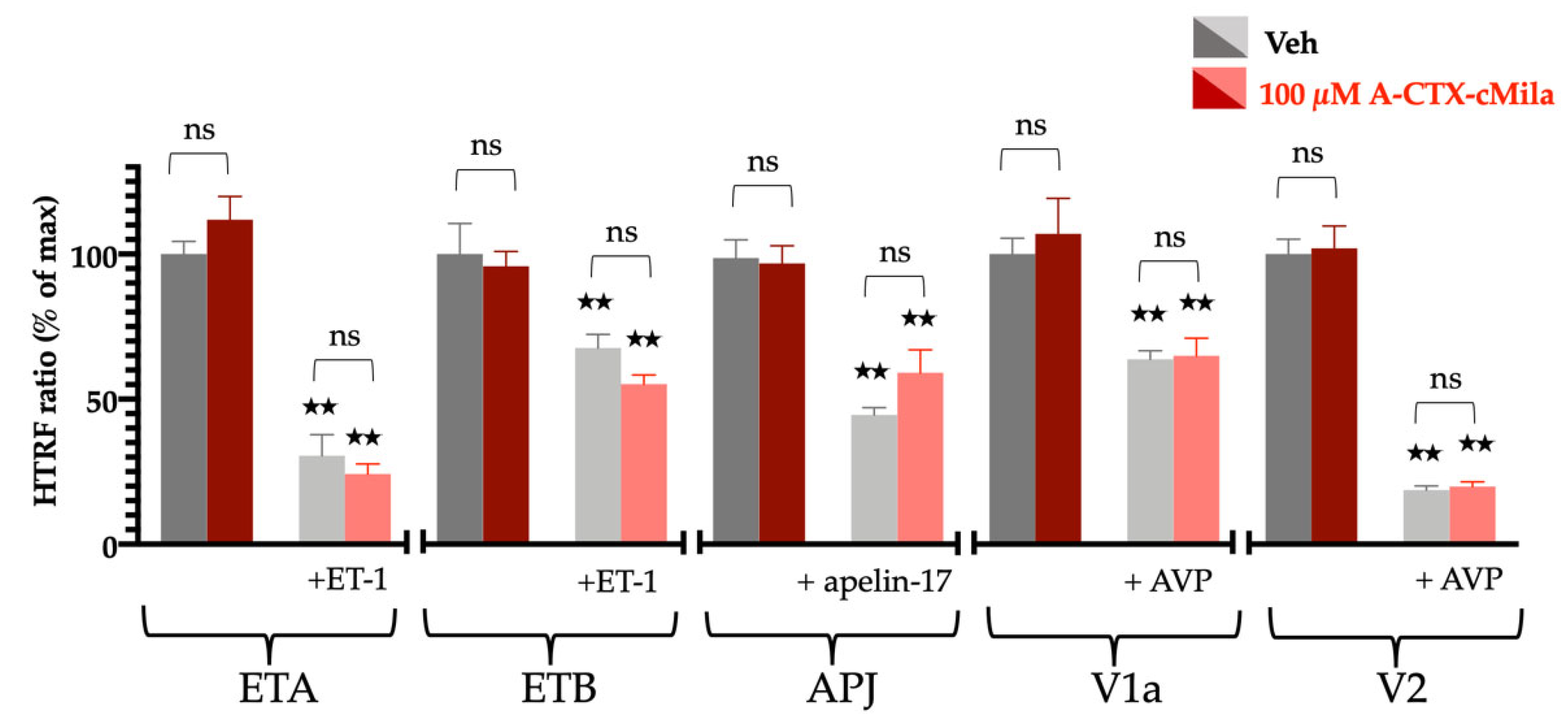
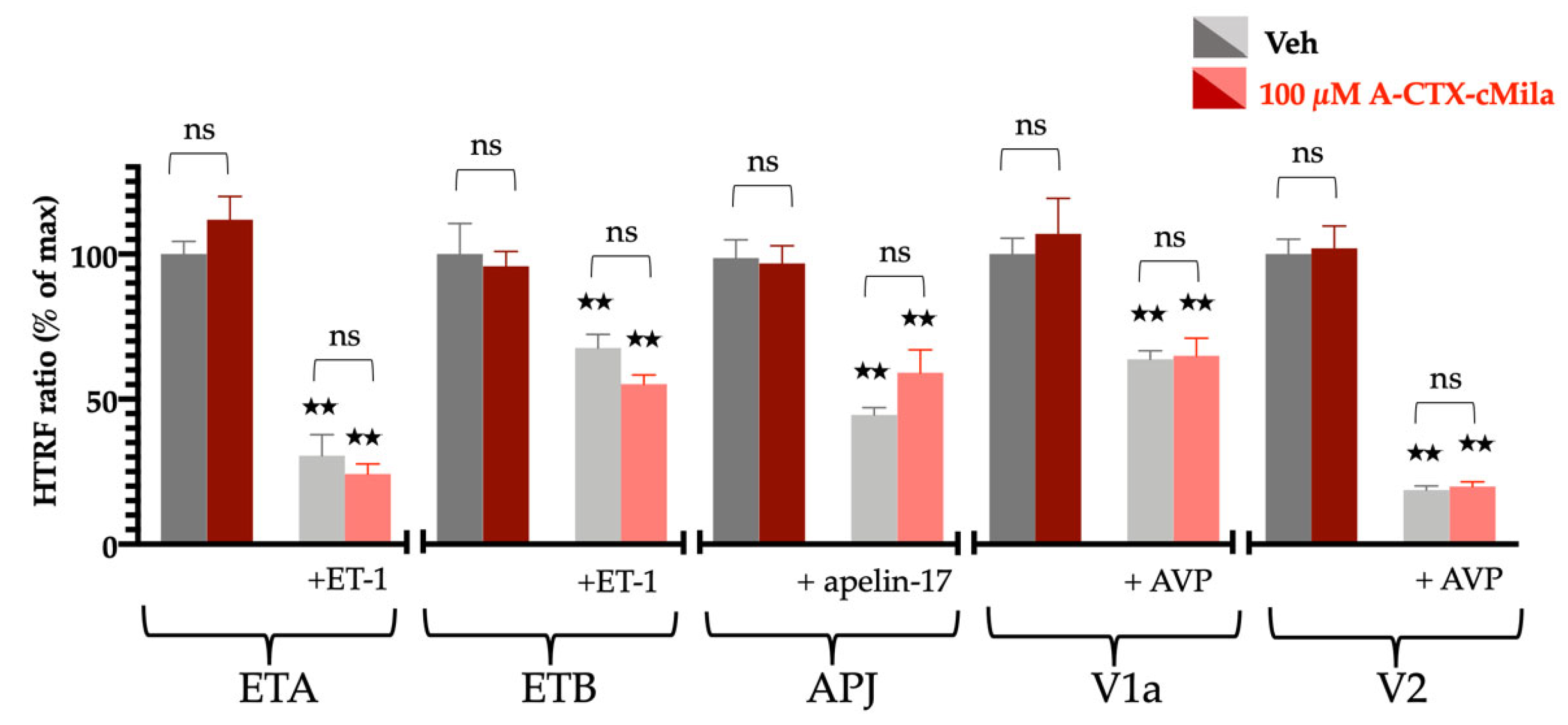
2.8. Selectivity Profile of A-CTX-cMila
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Transfection
4.2. Membrane Preparation
4.3. Radioligand Binding Assays
4.4. Production of A-CTX-cMila
4.5. HPLC Purification and Analysis
4.6. Mass Spectrometry Analysis
4.7. Thermodynamic Oxidation
4.8. Labeling of AngII with Cy3 and Cy5
4.9. Fluorescent-AngII Binding Assays
4.10. Second Messengers Assays
4.10.1. IP1 Assay
4.10.2. cAMP Assay
4.11. β-arrestin 2 Recruitment and Biosensors Assays
4.12. ERK1/2 Activation
4.13. Data Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gilles, N.; Servent, D. The European FP7 Venomics Project. Future Med. Chem. 2014, 6, 1611–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peigneur, S.; Tytgat, J. Toxins in Drug Discovery and Pharmacology. Toxins 2018, 10, 126. [Google Scholar] [CrossRef] [Green Version]
- Williams, J.A.; Day, M.; Heavner, J.E. Ziconotide: An update and review. Expert Opin. Pharmacother. 2008, 9, 1575–1583. [Google Scholar] [CrossRef]
- Van Baelen, A.-C.; Robin, P.; Kessler, P.; Maïga, A.; Gilles, N.; Servent, D. Structural and Functional Diversity of Animal Toxins Interacting with GPCRs. Front. Mol. Biosci. 2022, 9, 811365. [Google Scholar] [CrossRef] [PubMed]
- Ocaranza, M.P.; Riquelme, J.A.; García, L.; Jalil, J.E.; Chiong, M.; Santos, R.A.S.; Lavandero, S. Counter-regulatory renin–angiotensin system in cardiovascular disease. Nat. Rev. Cardiol. 2020, 17, 116–129. [Google Scholar] [CrossRef] [Green Version]
- Freeman, N.E.; Page, I.H. Hypertension produced by constriction of the renal artery in sympathectomized dogs. Am. Heart J. 1937, 14, 405–414. [Google Scholar] [CrossRef]
- Masuyer, G.; Yates, C.J.; Sturrock, E.D.; Acharya, K.R. Angiotensin-I converting enzyme (ACE): Structure, biological roles, and molecular basis for chloride ion dependence. Biol. Chem. 2014, 395, 1135–1149. [Google Scholar] [CrossRef]
- Page, I.H.; Helmer, O.M. A crystalline pressor substance (angiotonin) resulting from the reaction between renin and renin-activator. J. Exp. Med. 1940, 71, 29–42. [Google Scholar] [CrossRef] [Green Version]
- Bottari, S.P.; de Gasparo, M.; Steckelings, U.M.; Levens, N.R. Angiotensin II receptor subtypes: Characterization, signalling mechanisms, and possible physiological implications. Front. Neuroendocrinol. 1993, 14, 123–171. [Google Scholar] [CrossRef]
- Mukoyama, M.; Nakajima, M.; Horiuchi, M.; Sasamura, H.; Pratt, R.E.; Dzau, V.J. Expression cloning of type 2 angiotensin II receptor reveals a unique class of seven-transmembrane receptors. J. Biol. Chem. 1993, 268, 24539–24542. [Google Scholar] [CrossRef] [PubMed]
- Hunyady, L.; Catt, K.J. Pleiotropic AT1 Receptor Signaling Pathways Mediating Physiological and Pathogenic Actions of Angiotensin II. Mol. Endocrinol. 2006, 20, 953–970. [Google Scholar] [CrossRef] [PubMed]
- Paradis, P.; Dali-Youcef, N.; Paradis, F.W.; Thibault, G.; Nemer, M. Overexpression of angiotensin II type I receptor in cardiomyocytes induces cardiac hypertrophy and remodeling. Proc. Natl. Acad. Sci. USA 2000, 97, 931–936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lijnen, P.J.; Petrov, V.V.; Fagard, R.H. Induction of cardiac fibrosis by transforming growth factor-beta(1). Mol. Genet. Metab. 2000, 71, 418–435. [Google Scholar] [CrossRef]
- Bihl, J.C.; Zhang, C.; Zhao, Y.; Xiao, X.; Ma, X.; Chen, Y.; Chen, S.; Zhao, B.; Chen, Y. Angiotensin-(1–7) counteracts the effects of Ang II on vascular smooth muscle cells, vascular remodeling and hemorrhagic stroke: Role of the NFκB inflammatory pathway. Vascul. Pharmacol. 2015, 73, 115–123. [Google Scholar] [CrossRef] [Green Version]
- Nguyen Dinh Cat, A.; Montezano, A.C.; Burger, D.; Touyz, R.M. Angiotensin II, NADPH Oxidase, and Redox Signaling in the Vasculature. Antioxid. Redox Signal. 2013, 19, 1110–1120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibata, T.; Suzuki, C.; Ohnishi, J.; Murakami, K.; Miyazaki, H. Identification of regions in the human angiotensin II receptor type 1 responsible for Gi and Gq coupling by mutagenesis study. Biochem. Biophys. Res. Commun. 1996, 218, 383–389. [Google Scholar] [CrossRef]
- Shenoy, S.K.; Lefkowitz, R.J. β-arrestin-mediated receptor trafficking and signal transduction. Trends Pharmacol. Sci. 2011, 32, 521–533. [Google Scholar] [CrossRef] [Green Version]
- Reiter, E.; Ayoub, M.A.; Pellissier, L.P.; Landomiel, F.; Musnier, A.; Tréfier, A.; Gandia, J.; De Pascali, F.; Tahir, S.; Yvinec, R.; et al. β-arrestin signalling and bias in hormone-responsive GPCRs. Mol. Cell. Endocrinol. 2017, 449, 28–41. [Google Scholar] [CrossRef] [PubMed]
- Carey, R.M.; Howell, N.L.; Jin, X.H.; Siragy, H.M. Angiotensin type 2 receptor-mediated hypotension in angiotensin type-1 receptor-blocked rats. Hypertension 2001, 38, 1272–1277. [Google Scholar] [CrossRef] [Green Version]
- Carey, R.M.; Padia, S.H. Angiotensin AT2 receptors: Control of renal sodium excretion and blood pressure. Trends Endocrinol. Metab. 2008, 19, 84–87. [Google Scholar] [CrossRef]
- Namsolleck, P.; Recarti, C.; Foulquier, S.; Steckelings, U.M.; Unger, T. AT(2) receptor and tissue injury: Therapeutic implications. Curr. Hypertens. Rep. 2014, 16, 416. [Google Scholar] [CrossRef] [Green Version]
- Kemp, B.A.; Howell, N.L.; Gildea, J.J.; Keller, S.R.; Padia, S.H.; Carey, R.M. AT₂ receptor activation induces natriuresis and lowers blood pressure. Circ. Res. 2014, 115, 388–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vieira, T.N.; Saraiva, A.L.L.; Guimarães, R.M.; Luiz, J.P.M.; Pinto, L.G.; de Melo Rodrigues Ávila, V.; Goulart, L.R.; Cunha-Junior, J.P.; McNaughton, P.A.; Cunha, T.M.; et al. Angiotensin type 2 receptor antagonism as a new target to manage gout. Inflammopharmacology 2022, 30, 2399–2410. [Google Scholar] [CrossRef] [PubMed]
- Sehnert, B.; Valero-Esquitino, V.; Schett, G.; Unger, T.; Steckelings, U.M.; Voll, R.E. Angiotensin AT2 Receptor Stimulation Alleviates Collagen-Induced Arthritis by Upregulation of Regulatory T Cell Numbers. Front. Immunol. 2022, 13, 921488. [Google Scholar] [CrossRef]
- Ritter, O.; Schuh, K.; Brede, M.; Röthlein, N.; Burkard, N.; Hein, L.; Neyses, L. AT2 receptor activation regulates myocardial eNOS expression via the calcineurin-NF-AT pathway. FASEB J. 2003, 17, 283–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Pratt, R.E. The AT2 receptor selectively associates with Gialpha2 and Gialpha3 in the rat fetus. J. Biol. Chem. 1996, 271, 15026–15033. [Google Scholar] [CrossRef] [Green Version]
- Hansen, J.L.; Servant, G.; Baranski, T.J.; Fujita, T.; Iiri, T.; Sheikh, S.P. Functional reconstitution of the angiotensin II type 2 receptor and G(i) activation. Circ. Res. 2000, 87, 753–759. [Google Scholar] [CrossRef] [Green Version]
- Connolly, A.; Holleran, B.J.; Simard, É.; Baillargeon, J.-P.; Lavigne, P.; Leduc, R. Interplay between intracellular loop 1 and helix VIII of the angiotensin II type 2 receptor controls its activation. Biochem. Pharmacol. 2019, 168, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Hein, L.; Meinel, L.; Pratt, R.E.; Dzau, V.J.; Kobilka, B.K. Intracellular trafficking of angiotensin II and its AT1 and AT2 receptors: Evidence for selective sorting of receptor and ligand. Mol. Endocrinol. 1997, 11, 1266–1277. [Google Scholar] [CrossRef]
- Zhang, H.; Han, G.W.; Batyuk, A.; Ishchenko, A.; White, K.L.; Patel, N.; Sadybekov, A.; Zamlynny, B.; Rudd, M.T.; Hollenstein, K.; et al. Structural basis for selectivity and diversity in angiotensin II receptors. Nature 2017, 544, 327–332. [Google Scholar] [CrossRef]
- Tipnis, S.R.; Hooper, N.M.; Hyde, R.; Karran, E.; Christie, G.; Turner, A.J. A Human Homolog of Angiotensin-converting Enzyme Cloning and functional expression as a captopril-insensitive carboxypeptidase. J. Biol. Chem. 2000, 275, 33238–33243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organization Guideline for the Pharmacological Treatment of Hypertension in Adults; World Health Organization: Geneva, Switzerland, 2021; ISBN 978-92-4-003398-6.
- King, G.F.; Gentz, M.C.; Escoubas, P.; Nicholson, G.M. A rational nomenclature for naming peptide toxins from spiders and other venomous animals. Toxicon 2008, 52, 264–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaas, Q.; Westermann, J.-C.; Craik, D.J. Conopeptide characterization and classifications: An analysis using ConoServer. Toxicon 2010, 55, 1491–1509. [Google Scholar] [CrossRef] [PubMed]
- Merrifield, R.B. Solid Phase Peptide Synthesis. I. The Synthesis of a Tetrapeptide. J. Am. Chem. Soc. 1963, 85, 2149–2154. [Google Scholar] [CrossRef]
- Maziarz, M.; Park, J.-C.; Leyme, A.; Marivin, A.; Garcia-Lopez, A.; Patel, P.P.; Garcia-Marcos, M. Revealing the Activity of Trimeric G-proteins in Live Cells with a Versatile Biosensor Design. Cell 2020, 182, 770–785.e16. [Google Scholar] [CrossRef]
- Couvineau, P.; Llorens-Cortes, C.; Iturrioz, X. Elabela/Toddler and apelin bind differently to the apelin receptor. FASEB J. 2020, 34, 7989–8000. [Google Scholar] [CrossRef] [Green Version]
- Conklin, B.R.; Farfel, Z.; Lustig, K.D.; Julius, D.; Bourne, H.R. Substitution of three amino acids switches receptor specificity of Gq alpha to that of Gi alpha. Nature 1993, 363, 274–276. [Google Scholar] [CrossRef]
- Hanck, D.A.; Sheets, M.F. Site-3 toxins and cardiac sodium channels. Toxicon 2007, 49, 181–193. [Google Scholar] [CrossRef] [Green Version]
- Cedro, R.C.A.; Menaldo, D.L.; Costa, T.R.; Zoccal, K.F.; Sartim, M.A.; Santos-Filho, N.A.; Faccioli, L.H.; Sampaio, S.V. Cytotoxic and inflammatory potential of a phospholipase A2 from Bothrops jararaca snake venom. J. Venom. Anim. Toxins Incl. Trop. Dis. 2018, 24, 33. [Google Scholar] [CrossRef] [Green Version]
- Ducancel, F. The sarafotoxins. Toxicon 2002, 40, 1541–1545. [Google Scholar] [CrossRef]
- Ciolek, J.; Zoukimian, C.; Dhot, J.; Burban, M.; Triquigneaux, M.; Lauzier, B.; Guimbert, C.; Boturyn, D.; Ferron, M.; Ciccone, L.; et al. MT9, a natural peptide from black mamba venom antagonizes the muscarinic type 2 receptor and reverses the M2R-agonist-induced relaxation in rat and human arteries. Biomed. Pharmacother. 2022, 150, 113094. [Google Scholar] [CrossRef] [PubMed]
- Hodgson, W.C.; Isbister, G.K. The application of toxins and venoms to cardiovascular drug discovery. Curr. Opin. Pharmacol. 2009, 9, 173–176. [Google Scholar] [CrossRef]
- Porrello, E.R.; Delbridge, L.M.D.; Thomas, W.G. The angiotensin II type 2 (AT2) receptor: An enigmatic seven transmembrane receptor. Front. Biosci. (Landmark Ed.) 2009, 14, 958–972. [Google Scholar] [CrossRef] [Green Version]
- Miura, S.; Karnik, S.S. Angiotensin II type 1 and type 2 receptors bind angiotensin II through different types of epitope recognition. J. Hypertens. 1999, 17, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Yamano, Y.; Ohyama, K.; Kikyo, M.; Sano, T.; Nakagomi, Y.; Inoue, Y.; Nakamura, N.; Morishima, I.; Guo, D.F.; Hamakubo, T. Mutagenesis and the molecular modeling of the rat angiotensin II receptor (AT1). J. Biol. Chem. 1995, 270, 14024–14030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noda, K.; Saad, Y.; Karnik, S.S. Interaction of Phe8 of angiotensin II with Lys199 and His256 of AT1 receptor in agonist activation. J. Biol. Chem. 1995, 270, 28511–28514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kufareva, I.; Rueda, M.; Katritch, V.; Stevens, R.C.; Abagyan, R. GPCR Dock 2010 participants Status of GPCR modeling and docking as reflected by community-wide GPCR Dock 2010 assessment. Structure 2011, 19, 1108–1126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Dowd, B.F.; Heiber, M.; Chan, A.; Heng, H.H.; Tsui, L.C.; Kennedy, J.L.; Shi, X.; Petronis, A.; George, S.R.; Nguyen, T. A human gene that shows identity with the gene encoding the angiotensin receptor is located on chromosome 11. Gene 1993, 136, 355–360. [Google Scholar] [CrossRef]
- Marsault, E.; Llorens-Cortes, C.; Iturrioz, X.; Chun, H.J.; Lesur, O.; Oudit, G.Y.; Auger-Messier, M. The apelinergic system: A perspective on challenges and opportunities in cardiovascular and metabolic disorders. Ann. N. Y. Acad. Sci. 2019, 1455, 12–33. [Google Scholar] [CrossRef]
- Shin, K.; Kenward, C.; Rainey, J.K. Apelinergic System Structure and Function. Compr. Physiol. 2017, 8, 407–450. [Google Scholar] [CrossRef]
- Sakurai, T.; Yanagisawa, M.; Masaki, T. Molecular characterization of endothelin receptors. Trends Pharmacol. Sci. 1992, 13, 103–108. [Google Scholar] [CrossRef]
- Barton, M.; Yanagisawa, M. Endothelin: 30 Years from Discovery to Therapy. Hypertension 2019, 74, 1232–1265. [Google Scholar] [CrossRef]
- Urbach, J.; Goldsmith, S.R. Vasopressin antagonism in heart failure: A review of the hemodynamic studies and major clinical trials. Ther. Adv. Cardiovasc. Dis. 2021, 15, 1753944720977741. [Google Scholar] [CrossRef]
- Allison, S. Cryo-EM structure of AVP-V2 receptor complex. Nat. Rev. Nephrol. 2021, 17, 511. [Google Scholar] [CrossRef] [PubMed]
- Sabareesh, V.; Gowd, K.H.; Ramasamy, P.; Sudarslal, S.; Krishnan, K.S.; Sikdar, S.K.; Balaram, P. Characterization of contryphans from Conus loroisii and Conus amadis that target calcium channels. Peptides 2006, 27, 2647–2654. [Google Scholar] [CrossRef]
- Lebbe, E.K.M.; Tytgat, J. In the picture: Disulfide-poor conopeptides, a class of pharmacologically interesting compounds. J. Venom. Anim. Toxins Incl. Trop. Dis. 2016, 22, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dutertre, S.; Croker, D.; Daly, N.L.; Andersson, A.; Muttenthaler, M.; Lumsden, N.G.; Craik, D.J.; Alewood, P.F.; Guillon, G.; Lewis, R.J. Conopressin-T from Conus tulipa reveals an antagonist switch in vasopressin-like peptides. J. Biol. Chem. 2008, 283, 7100–7108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giribaldi, J.; Ragnarsson, L.; Pujante, T.; Enjalbal, C.; Wilson, D.; Daly, N.L.; Lewis, R.J.; Dutertre, S. Synthesis, Pharmacological and Structural Characterization of Novel Conopressins from Conus miliaris. Mar. Drugs 2020, 18, 150. [Google Scholar] [CrossRef] [Green Version]
- Kawamura, T.; Oda, T.; Muramatsu, T. Purification and characterization of a dipeptidyl carboxypeptidase from the polychaete Neanthes virens resembling angiotensin I converting enzyme. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2000, 126, 29–37. [Google Scholar] [CrossRef]
- Satou, R.; Nakagawa, T.; Ido, H.; Tomomatsu, M.; Suzuki, F.; Nakamura, Y. Angiotensin II and III upregulate body fluid volume of the clam worm Perinereis sp. via angiotensin II receptors in different manners. Peptides 2005, 26, 2452–2457. [Google Scholar] [CrossRef] [PubMed]
- Reynaud, S.; Ciolek, J.; Degueldre, M.; Saez, N.J.; Sequeira, A.F.; Duhoo, Y.; Brás, J.L.A.; Meudal, H.; Cabo Díez, M.; Fernández Pedrosa, V.; et al. A Venomics Approach Coupled to High-Throughput Toxin Production Strategies Identifies the First Venom-Derived Melanocortin Receptor Agonists. J. Med. Chem. 2020, 63, 8250–8264. [Google Scholar] [CrossRef] [PubMed]
- Murphy, T.J.; Alexander, R.W.; Griendling, K.K.; Runge, M.S.; Bernstein, K.E. Isolation of a cDNA encoding the vascular type-1 angiotensin II receptor. Nature 1991, 351, 233–236. [Google Scholar] [CrossRef] [PubMed]
- Shanmugam, S.; Llorens-Cortes, C.; Clauser, E.; Corvol, P.; Gasc, J.M. Expression of angiotensin II AT2 receptor mRNA during development of rat kidney and adrenal gland. Am. J. Physiol. 1995, 268, F922–F930. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Van Baelen, A.-C.; Iturrioz, X.; Chaigneau, M.; Kessler, P.; Llorens-Cortes, C.; Servent, D.; Gilles, N.; Robin, P. Characterization of the First Animal Toxin Acting as an Antagonist on AT1 Receptor. Int. J. Mol. Sci. 2023, 24, 2330. https://doi.org/10.3390/ijms24032330
Van Baelen A-C, Iturrioz X, Chaigneau M, Kessler P, Llorens-Cortes C, Servent D, Gilles N, Robin P. Characterization of the First Animal Toxin Acting as an Antagonist on AT1 Receptor. International Journal of Molecular Sciences. 2023; 24(3):2330. https://doi.org/10.3390/ijms24032330
Chicago/Turabian StyleVan Baelen, Anne-Cécile, Xavier Iturrioz, Marion Chaigneau, Pascal Kessler, Catherine Llorens-Cortes, Denis Servent, Nicolas Gilles, and Philippe Robin. 2023. "Characterization of the First Animal Toxin Acting as an Antagonist on AT1 Receptor" International Journal of Molecular Sciences 24, no. 3: 2330. https://doi.org/10.3390/ijms24032330