
Moxonidine Increases Uptake of Oxidised Low-Density Lipoprotein in Cultured Vascular Smooth Muscle Cells and Inhibits Atherosclerosis in Apolipoprotein E-Deficient Mice

 , , , , and
, , , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
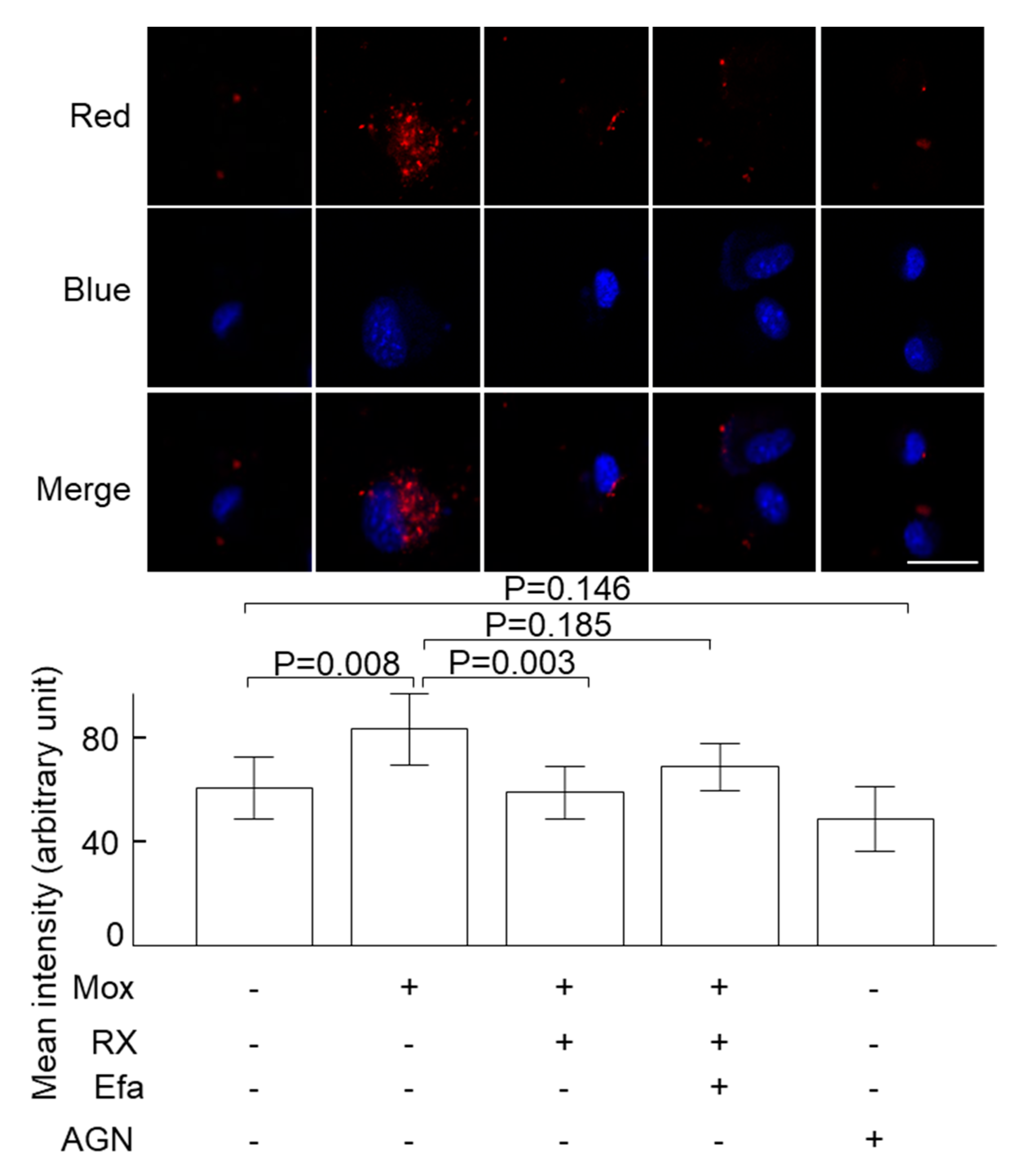
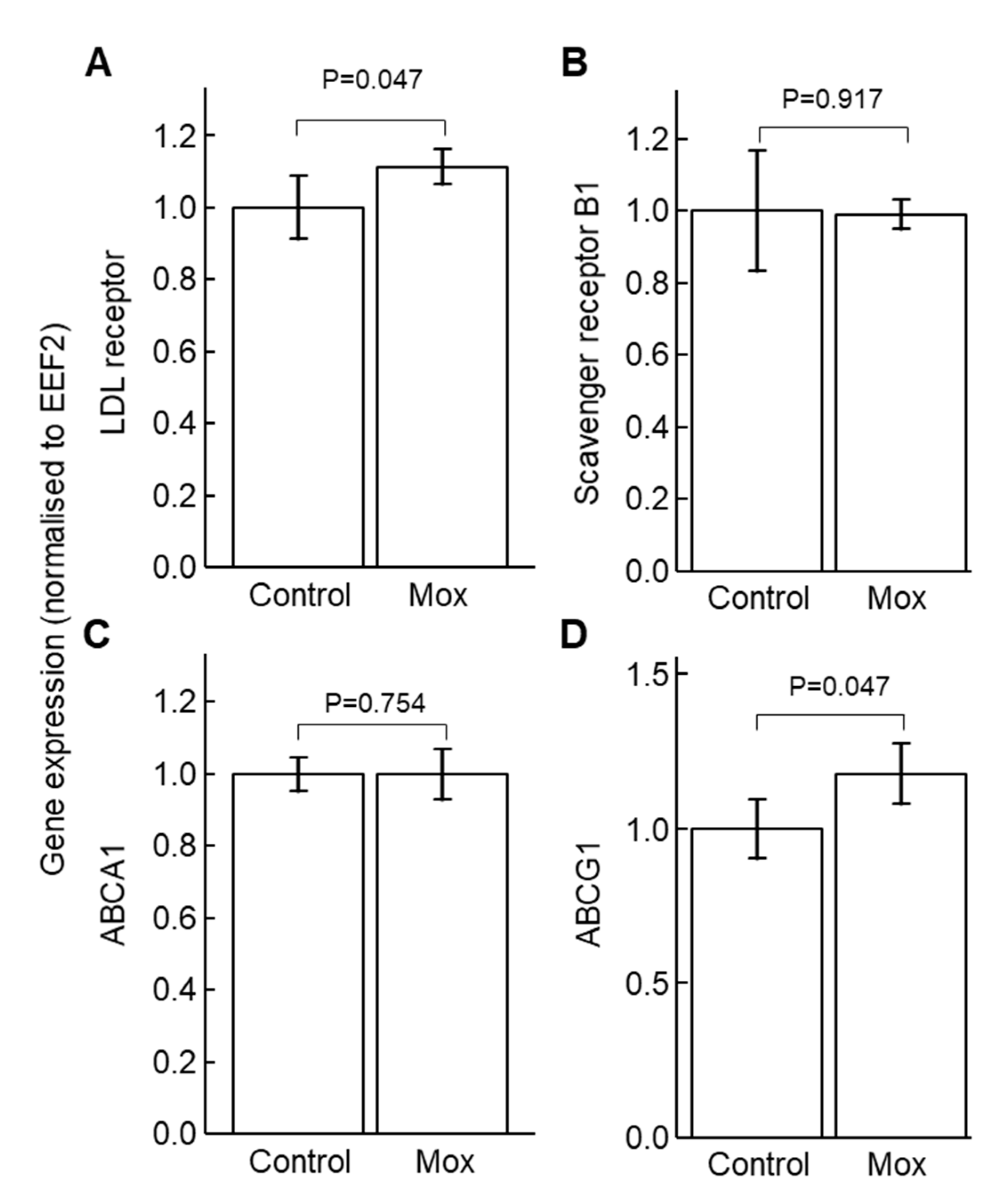
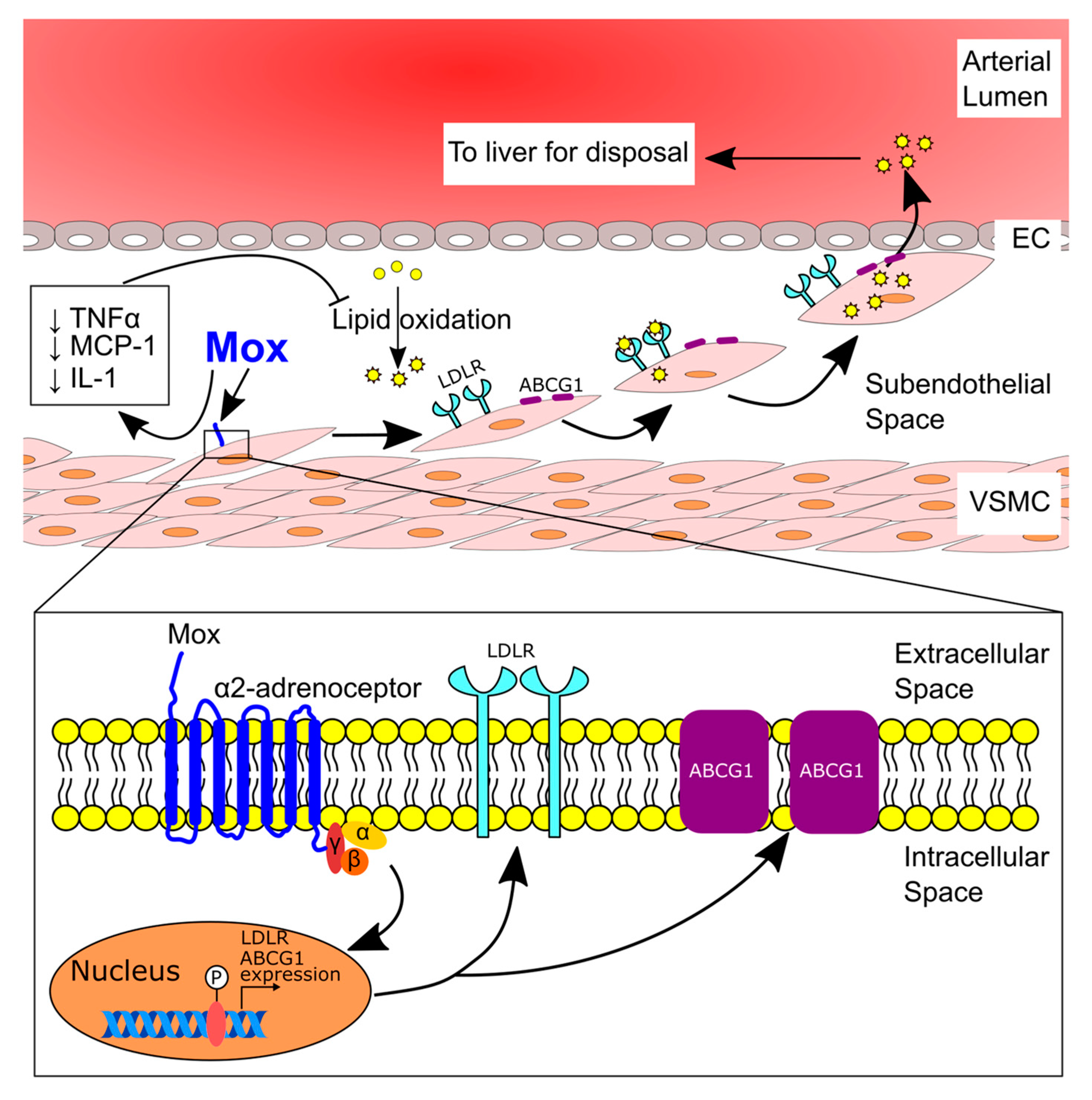
2.1. Moxonidine Increased Oxidised LDL Uptake via α2 Adrenoceptors by VSMCs In Vitro
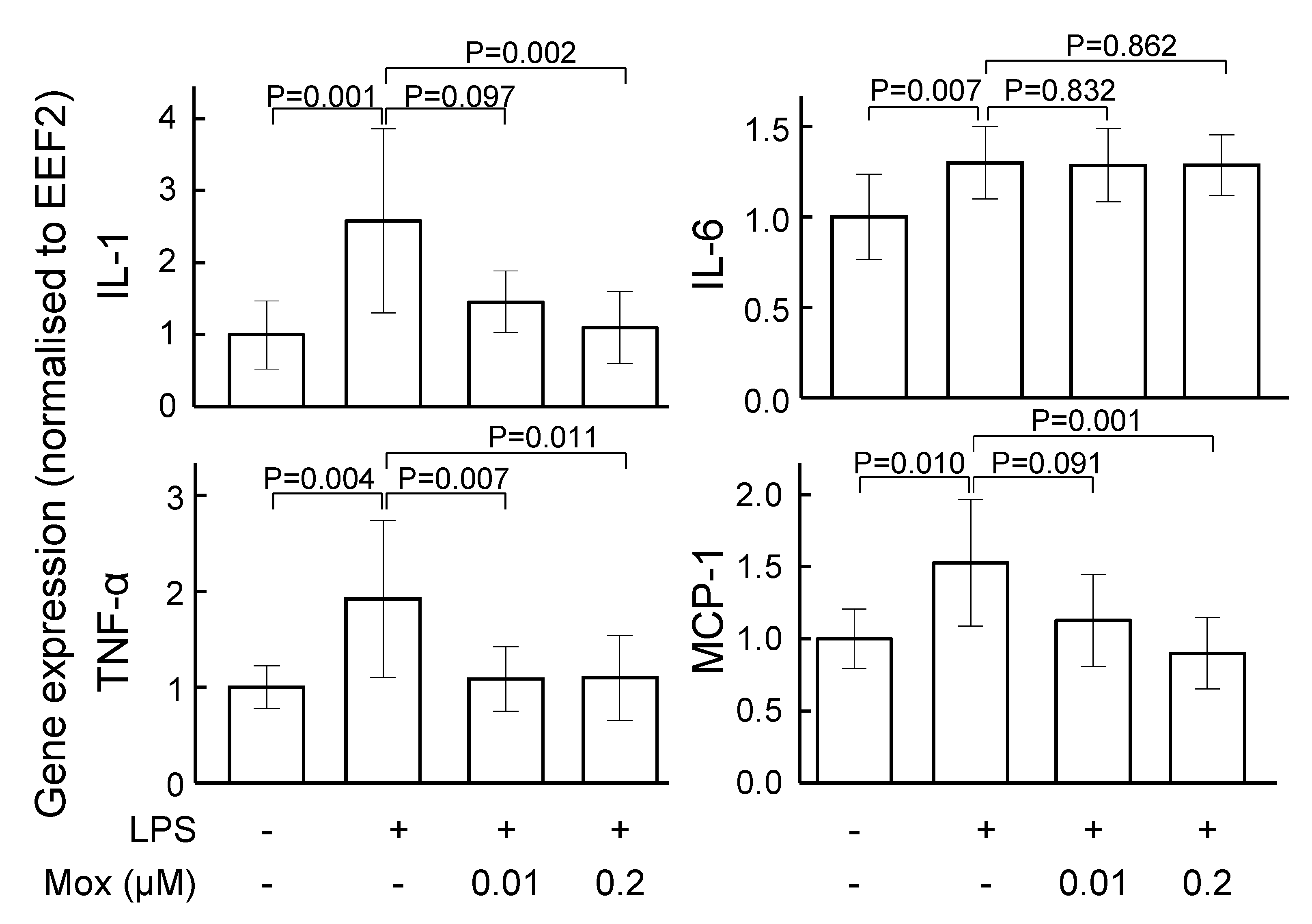
2.2. Moxonidine Inhibited Inflammatory Gene Expression in Lipopolysaccharide-Treated VSMCs and Endothelial Cells In Vitro
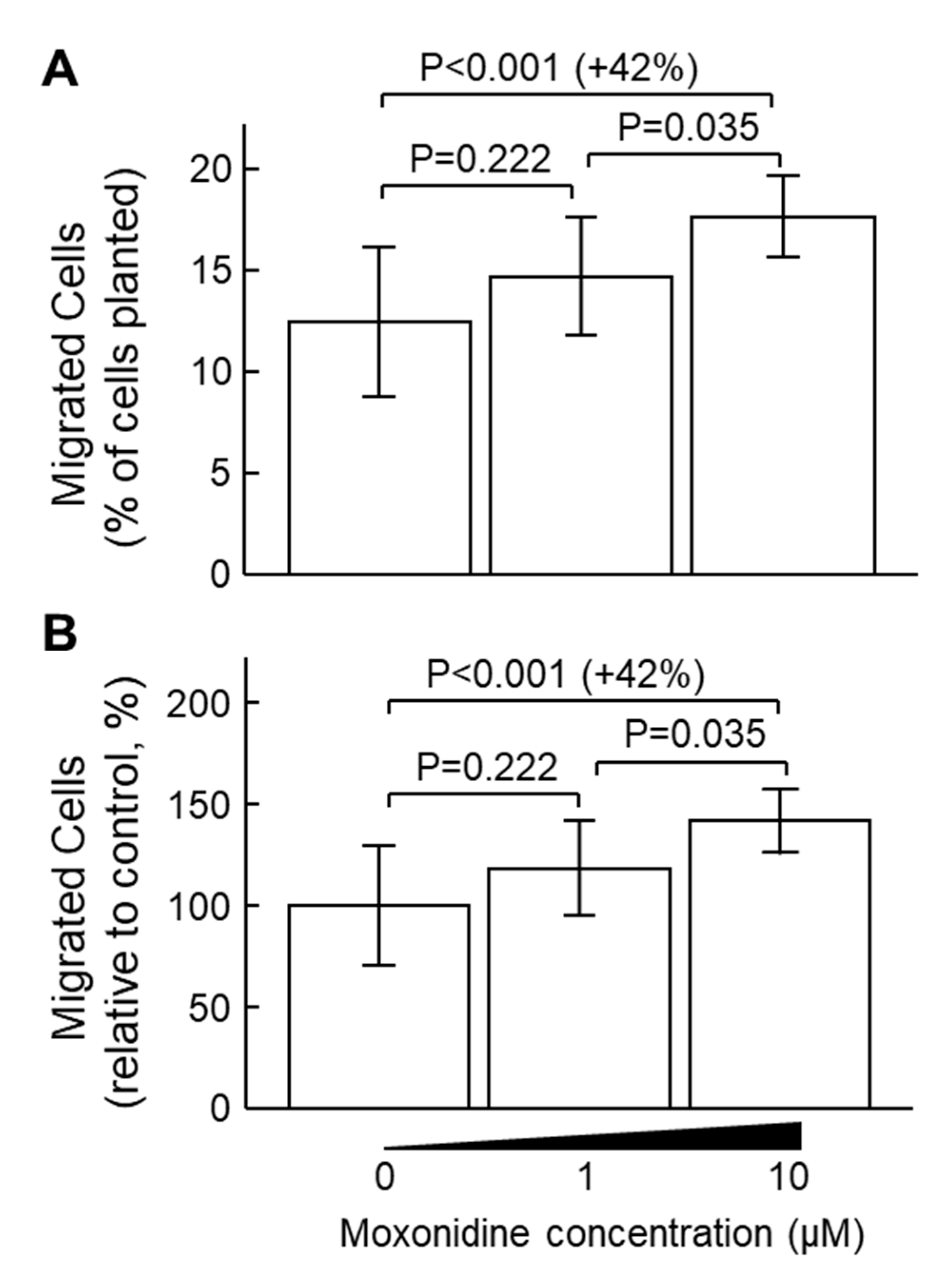
2.3. Moxonidine Enhanced VSMC Migration Without Affecting VSMC Proliferation In Vitro
2.4. Moxonidine Decreased Atherosclerosis in Angiotensin-Infused ApoE−/− Mice
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Experimental Protocol
4.3. Quantification of Atherosclerotic Lesion Area
4.4. Ferrous Oxidation-Xylenol Orange (FOX) Assay
4.5. Cell Culture
4.6. Confocal Microscopy for Oxidised LDL Uptake
4.7. Gene Expression Analysis
4.8. Migration Assay
4.9. Cell Proliferation
4.10. Total Cholesterol
4.11. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Puylaert, P.; Zurek, M.; Rayner, K.J.; De Meyer, G.R.Y.; Martinet, W. Regulated Necrosis in Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2022, 42, 1283–1306. [Google Scholar] [CrossRef] [PubMed]
- Amarenco, P.; Labreuche, J.; Lavallée, P.; Touboul, P.-J. Statins in Stroke Prevention and Carotid Atherosclerosis. Stroke 2004, 35, 2902–2909. [Google Scholar] [CrossRef] [PubMed]
- Gaggini, M.; Gorini, F.; Vassalle, C. Lipids in Atherosclerosis: Pathophysiology and the Role of Calculated Lipid Indices in Assessing Cardiovascular Risk in Patients with Hyperlipidemia. Int. J. Mol. Sci. 2022, 24, 75. [Google Scholar] [CrossRef]
- Alshak, M.N.; Das, J.M. Neuroanatomy, Sympathetic Nervous System. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2019. Available online: https://www.ncbi.nlm.nih.gov/books/NBK542195/ (accessed on 22 November 2022).
- Wang, Y.; Seto, S.W.; Golledge, J. Therapeutic effects of renal denervation on renal failure. Curr. Neurovascular Res. 2013, 10, 172–184. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Golledge, J. Neuronal nitric oxide synthase and sympathetic nerve activity in neurovascular and metabolic systems. Curr. Neurovascular Res. 2013, 10, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Won, E.; Kim, Y.K. Stress, the Autonomic Nervous System, and the Immune-kynurenine Pathway in the Etiology of Depression. Curr. Neuropharmacol. 2016, 14, 665–673. [Google Scholar] [CrossRef]
- Weissman, D.G.; Mendes, W.B. Correlation of sympathetic and parasympathetic nervous system activity during rest and acute stress tasks. Int. J. Psychophysiol. 2021, 162, 60–68. [Google Scholar] [CrossRef]
- Yao, B.C.; Meng, L.B.; Hao, M.L.; Zhang, Y.M.; Gong, T.; Guo, Z.G. Chronic stress: A critical risk factor for atherosclerosis. J. Int. Med. Res. 2019, 47, 1429–1440. [Google Scholar] [CrossRef]
- Vrablik, M.; Corsini, A.; Tůmová, E. Beta-blockers for Atherosclerosis Prevention: A Missed Opportunity? Curr. Atheroscler. Rep. 2022, 24, 161–169. [Google Scholar] [CrossRef]
- Wang, H.; Wang, J.; Guo, C.; Luo, W.; Kleiman, K.; Eitzman, D.T. Renal denervation attenuates progression of atherosclerosis in apolipoprotein E-deficient mice independent of blood pressure lowering. Hypertension 2015, 65, 758–765. [Google Scholar] [CrossRef]
- Wang, Y.; Dinh, T.N.; Nield, A.; Krishna, S.M.; Denton, K.; Golledge, J. Renal Denervation Promotes Atherosclerosis in Hypertensive Apolipoprotein E-Deficient Mice Infused with Angiotensin II. Front. Physiol. 2017, 8, 215. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y. What is the true incidence of renal artery stenosis after sympathetic denervation? Front. Physiol. 2014, 5, 311. [Google Scholar] [CrossRef] [PubMed]
- Pöyhönen-Alho, M.K.; Manhem, K.; Katzman, P.; Kibarskis, A.; Antikainen, R.L.; Erkkola, R.U.; Tuomilehto, J.O.; Ebeling, P.E.; Kaaja, R.J. Central sympatholytic therapy has anti-inflammatory properties in hypertensive postmenopausal women. J. Hypertens. 2008, 26, 2445–2449. [Google Scholar] [CrossRef] [PubMed]
- Mukaddam-Daher, S.; Gutkowska, J. Imidazoline receptors in the heart: A novel target and a novel mechanism of action that involves atrial natriuretic peptides. Braz. J. Med. Biol. Res. 2004, 37, 1239–1245. [Google Scholar] [CrossRef]
- Daugherty, A.; Manning, M.W.; Cassis, L.A. Angiotensin II promotes atherosclerotic lesions and aneurysms in apolipoprotein E-deficient mice. J. Clin. Investig. 2000, 105, 1605–1612. [Google Scholar] [CrossRef]
- Candido, R.; Jandeleit-Dahm, K.A.; Cao, Z.; Nesteroff, S.P.; Burns, W.C.; Twigg, S.M.; Dilley, R.J.; Cooper, M.E.; Allen, T.J. Prevention of Accelerated Atherosclerosis by Angiotensin-Converting Enzyme Inhibition in Diabetic Apolipoprotein E–Deficient Mice. Circulation 2002, 106, 246–253. [Google Scholar] [CrossRef]
- Danchin, N.; Cucherat, M.; Thuillez, C.; Durand, E.; Kadri, Z.; Steg, P.G. Angiotensin-Converting Enzyme Inhibitors in Patients With Coronary Artery Disease and Absence of Heart Failure or Left Ventricular Systolic Dysfunction: An Overview of Long-term Randomized Controlled Trials. Arch. Intern. Med. 2006, 166, 787–796. [Google Scholar] [CrossRef]
- Reid, I.A. Interactions between ANG II, sympathetic nervous system, and baroreceptor reflexes in regulation of blood pressure. Am. J. Physiol. 1992, 262, E763–E778. [Google Scholar] [CrossRef]
- Charkoudian, N.; Rabbitts, J.A. Sympathetic neural mechanisms in human cardiovascular health and disease. Mayo Clin. Proc. 2009, 84, 822–830. [Google Scholar] [CrossRef]
- Nakagawa, T.; Hasegawa, Y.; Uekawa, K.; Ma, M.; Katayama, T.; Sueta, D.; Toyama, K.; Kataoka, K.; Koibuchi, N.; Maeda, M.; et al. Renal denervation prevents stroke and brain injury via attenuation of oxidative stress in hypertensive rats. J. Am. Heart Assoc. 2013, 2, e000375. [Google Scholar] [CrossRef]
- Ganesan, R.; Henkels, K.M.; Wrenshall, L.E.; Kanaho, Y.; Di Paolo, G.; Frohman, M.A.; Gomez-Cambronero, J. Oxidized LDL phagocytosis during foam cell formation in atherosclerotic plaques relies on a PLD2–CD36 functional interdependence. J. Leukoc. Biol. 2018, 103, 867–883. [Google Scholar] [CrossRef]
- Bennett, M.R.; Sinha, S.; Owens, G.K. Vascular Smooth Muscle Cells in Atherosclerosis. Circ. Res. 2016, 118, 692–702. [Google Scholar] [CrossRef]
- Allahverdian, S.; Chehroudi, A.C.; McManus, B.M.; Abraham, T.; Francis, G.A. Contribution of intimal smooth muscle cells to cholesterol accumulation and macrophage-like cells in human atherosclerosis. Circulation 2014, 129, 1551–1559. [Google Scholar] [CrossRef]
- Wang, Y.; Dubland, J.A.; Allahverdian, S.; Asonye, E.; Sahin, B.; Jaw, J.E.; Sin, D.D.; Seidman, M.A.; Leeper, N.J.; Francis, G.A. Smooth Muscle Cells Contribute the Majority of Foam Cells in ApoE (Apolipoprotein E)-Deficient Mouse Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 876–887. [Google Scholar] [CrossRef]
- Prichard, B.N.; Graham, B.R. The use of moxonidine in the treatment of hypertension. J. Hypertens. Suppl. 1997, 15, S47–S55. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, R.R.; Spieker, L.; Qui, S.; Shaw, S.; Luscher, T.F.; Noll, G. I1-imidazoline agonist moxonidine decreases sympathetic nerve activity and blood pressure in hypertensives. Hypertension 1998, 32, 1022–1027. [Google Scholar] [CrossRef] [PubMed]
- Kirch, W.; Hutt, H.J.; Planitz, V. Pharmacodynamic action and pharmacokinetics of moxonidine after single oral administration in hypertension patients. J. Clin. Pharmacol. 1990, 30, 1088–1095. [Google Scholar] [CrossRef]
- Clarke, R.W.; Harris, J. RX 821002 as a tool for physiological investigation of alpha(2)-adrenoceptors. CNS Drug Rev. 2002, 8, 177–192. [Google Scholar] [CrossRef] [PubMed]
- Tolentino-Silva, F.P.; Haxhiu, M.A.; Waldbaum, S.; Dreshaj, I.A.; Ernsberger, P. alpha(2)-adrenergic receptors are not required for central anti-hypertensive action of moxonidine in mice. Brain Res. 2000, 862, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Munk, S.A.; Lai, R.K.; Burke, J.E.; Arasasingham, P.N.; Kharlamb, A.B.; Manlapaz, C.A.; Padillo, E.U.; Wijono, M.K.; Hasson, D.W.; Wheeler, L.A.; et al. Synthesis and pharmacologic evaluation of 2-endo-amino-3-exo- isopropylbicyclo[2.2.1]heptane: A potent imidazoline1 receptor specific agent. J. Med. Chem. 1996, 39, 1193–1195. [Google Scholar] [CrossRef]
- Stringer, M.D.; Görög, P.G.; Freeman, A.; Kakkar, V.V. Lipid peroxides and atherosclerosis. Br. Med. J. 1989, 298, 281–284. [Google Scholar] [CrossRef]
- Mitrovic, V.; Patyna, W.; Huting, J.; Schlepper, M. Hemodynamic and neurohumoral effects of moxonidine in patients with essential hypertension. Cardiovasc. Drugs Ther. 1991, 5, 967–972. [Google Scholar] [CrossRef] [PubMed]
- Harman, J.L.; Jørgensen, H.F. The role of smooth muscle cells in plaque stability: Therapeutic targeting potential. Br. J. Pharmacol. 2019, 176, 3741–3753. [Google Scholar] [CrossRef] [PubMed]
- Hansson, G.K.; Robertson, A.K.; Söderberg-Nauclér, C. Inflammation and atherosclerosis. Annu. Rev. Pathol. 2006, 1, 297–329. [Google Scholar] [CrossRef] [PubMed]
- González, L.; Rivera, K.; Andia, M.E.; Martínez Rodriguez, G. The IL-1 Family and Its Role in Atherosclerosis. Int. J. Mol. Sci. 2022, 24, 17. [Google Scholar] [CrossRef]
- Tedgui, A.; Mallat, Z. Cytokines in atherosclerosis: Pathogenic and regulatory pathways. Physiol. Rev. 2006, 86, 515–581. [Google Scholar] [CrossRef]
- Pober, J.S.; Min, W.; Bradley, J.R. Mechanisms of endothelial dysfunction, injury, and death. Annu. Rev. Pathol. 2009, 4, 71–95. [Google Scholar] [CrossRef]
- Xiao, J.; Li, N.; Xiao, S.; Wu, Y.; Liu, H. Comparison of Selenium Nanoparticles and Sodium Selenite on the Alleviation of Early Atherosclerosis by Inhibiting Endothelial Dysfunction and Inflammation in Apolipoprotein E-Deficient Mice. Int. J. Mol. Sci. 2021, 22, 11612. [Google Scholar] [CrossRef]
- Ruffolo, R.R., Jr.; Nichols, A.J.; Stadel, J.M.; Hieble, J.P. Structure and function of alpha-adrenoceptors. Pharmacol. Rev. 1991, 43, 475–505. [Google Scholar]
- Marieb, E.N.; Hoehn, K. The Peripheral Nervous System and Reflex Activity. Hum. Anat. Physiol. 2018, 13, 521–562. [Google Scholar]
- Carr, A.C.; McCall, M.R.; Frei, B. Oxidation of LDL by Myeloperoxidase and Reactive Nitrogen Species. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1716–1723. [Google Scholar] [CrossRef] [PubMed]
- Elisaf, M.S.; Petris, C.; Bairaktari, E.; Karabina, S.A.; Tzallas, C.; Tselepis, A.; Siamopoulos, K.C. The effect of moxonidine on plasma lipid profile and on LDL subclass distribution. J. Hum. Hypertens. 1999, 13, 781–785. [Google Scholar] [CrossRef] [PubMed]
- Mironova, M.A.; Klein, R.L.; Virella, G.T.; Lopes-Virella, M.F. Anti-modified LDL antibodies, LDL-containing immune complexes, and susceptibility of LDL to in vitro oxidation in patients with type 2 diabetes. Diabetes 2000, 49, 1033–1041. [Google Scholar] [CrossRef] [PubMed]
- Schneider, W.J. The low density lipoprotein receptor. Biochim. Biophys. Acta 1989, 988, 303–317. [Google Scholar] [CrossRef]
- Rupp, H.; Maisch, B.; Brilla, C.G. Drug withdrawal and rebound hypertension: Differential action of the central antihypertensive drugs moxonidine and clonidine. Cardiovasc. Drugs Ther. 1996, 10 (Suppl. 1), 251–262. [Google Scholar] [CrossRef]
- Krishna, S.M.; Li, J.; Wang, Y.; Moran, C.S.; Trollope, A.; Huynh, P.; Jose, R.; Biros, E.; Ma, J.; Golledge, J. Kallistatin limits abdominal aortic aneurysm by attenuating generation of reactive oxygen species and apoptosis. Sci. Rep. 2021, 11, 17451. [Google Scholar] [CrossRef]
- Moran, C.S.; Seto, S.W.; Krishna, S.M.; Sharma, S.; Jose, R.J.; Biros, E.; Wang, Y.; Morton, S.K.; Golledge, J. Parenteral administration of factor Xa/IIa inhibitors limits experimental aortic aneurysm and atherosclerosis. Sci. Rep. 2017, 7, 43079. [Google Scholar] [CrossRef]
- Beck, K.; Wu, B.J.; Ni, J.; Santiago, F.S.; Malabanan, K.P.; Li, C.; Wang, Y.; Khachigian, L.M.; Stocker, R. Interplay Between Heme Oxygenase-1 and the Multifunctional Transcription Factor Yin Yang 1 in the Inhibition of Intimal Hyperplasia. Circ. Res. 2010, 107, 1490–1497. [Google Scholar] [CrossRef]
- Youssef, P.; Chami, B.; Lim, J.; Middleton, T.; Sutherland, G.T.; Witting, P.K. Evidence supporting oxidative stress in a moderately affected area of the brain in Alzheimer’s disease. Sci. Rep. 2018, 8, 11553. [Google Scholar] [CrossRef]
- Wang, Y.; Nguyen, D.T.; Yang, G.; Anesi, J.; Chai, Z.; Charchar, F.; Golledge, J. An Improved 3-(4,5-Dimethylthiazol-2-yl)-5-(3-Carboxymethoxyphenyl)-2-(4-Sulfophenyl)-2H-Tetrazolium Proliferation Assay to Overcome the Interference of Hydralazine. Assay Drug Dev. Technol. 2020, 18, 379–384. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, H.; McKenzie, G.; Witting, P.K.; Stasch, J.P.; Hahn, M.; Changsirivathanathamrong, D.; Wu, B.J.; Ball, H.J.; Thomas, S.R.; et al. Kynurenine is an endothelium-derived relaxing factor produced during inflammation. Nat. Med. 2010, 16, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Nguyen, D.T.; Yang, G.; Anesi, J.; Kelly, J.; Chai, Z.; Ahmady, F.; Charchar, F.; Golledge, J. A Modified MTS Proliferation Assay for Suspended Cells to Avoid the Interference by Hydralazine and β-Mercaptoethanol. Assay Drug Dev Technol 2021, 19, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Wu, J.; Jia, H.; Lu, C.; Liu, J.; Li, Y.; Guo, M. Oncostatin M promotes the ox-LDL-induced activation of NLRP3 inflammasomes via the NF-κB pathway in THP-1 macrophages and promotes the progression of atherosclerosis. Ann. Transl. Med. 2022, 10, 456. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y. Definition, prevalence, and risk factors of low sex hormone-binding globulin in US adults. J. Clin. Endocrinol. Metab. 2021, 106, e3946–e3956. [Google Scholar] [CrossRef]
- Wang, Y.; Charchar, F.J. Establishment of sex difference in circulating uric acid is associated with higher testosterone and lower sex hormone-binding globulin in adolescent boys. Sci. Rep. 2021, 11, 17323. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.; Wen, S.; Wang, Y.; Qian, Z.; Tan, Y.; Li, H.; Hou, Y.; Hu, H.; Golledge, J.; Yang, G. The association between serum uric acid and blood pressure in different age groups in a healthy Chinese cohort. Medicine 2017, 96, e8953. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Nguyen, D.T.; Anesi, J.; Alramahi, A.; Witting, P.K.; Chai, Z.; Khan, A.W.; Kelly, J.; Denton, K.M.; Golledge, J. Moxonidine Increases Uptake of Oxidised Low-Density Lipoprotein in Cultured Vascular Smooth Muscle Cells and Inhibits Atherosclerosis in Apolipoprotein E-Deficient Mice. Int. J. Mol. Sci. 2023, 24, 3857. https://doi.org/10.3390/ijms24043857
Wang Y, Nguyen DT, Anesi J, Alramahi A, Witting PK, Chai Z, Khan AW, Kelly J, Denton KM, Golledge J. Moxonidine Increases Uptake of Oxidised Low-Density Lipoprotein in Cultured Vascular Smooth Muscle Cells and Inhibits Atherosclerosis in Apolipoprotein E-Deficient Mice. International Journal of Molecular Sciences. 2023; 24(4):3857. https://doi.org/10.3390/ijms24043857
Chicago/Turabian StyleWang, Yutang, Dinh Tam Nguyen, Jack Anesi, Ahmed Alramahi, Paul K. Witting, Zhonglin Chai, Abdul Waheed Khan, Jason Kelly, Kate M. Denton, and Jonathan Golledge. 2023. "Moxonidine Increases Uptake of Oxidised Low-Density Lipoprotein in Cultured Vascular Smooth Muscle Cells and Inhibits Atherosclerosis in Apolipoprotein E-Deficient Mice" International Journal of Molecular Sciences 24, no. 4: 3857. https://doi.org/10.3390/ijms24043857