
Genome-Wide Association Analysis of Fruit Shape-Related Traits in Areca catechu

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Genotype Analysis of A. catechu Populations
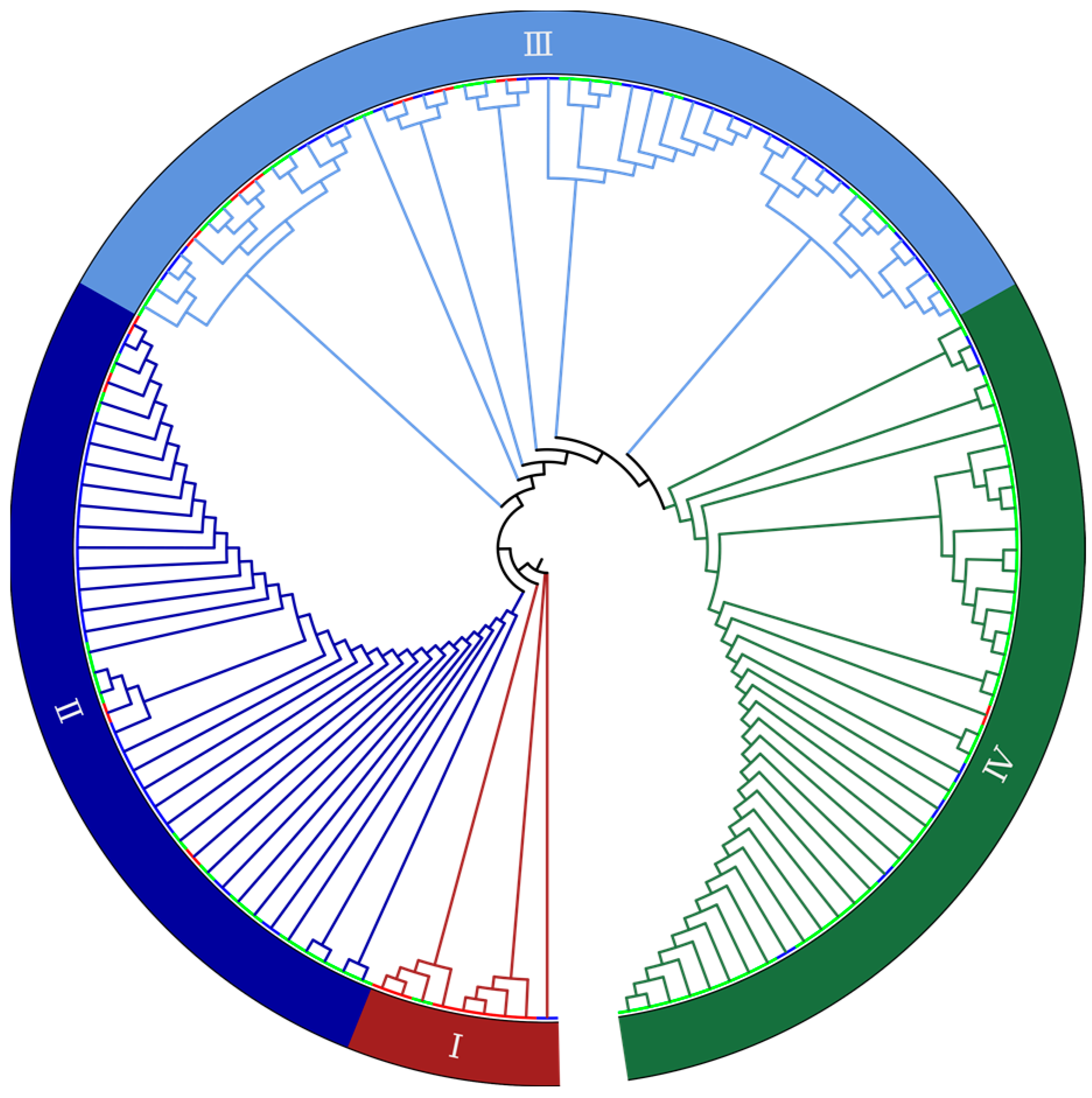
2.2. Genetic Diversity Revealed by SNP Markers
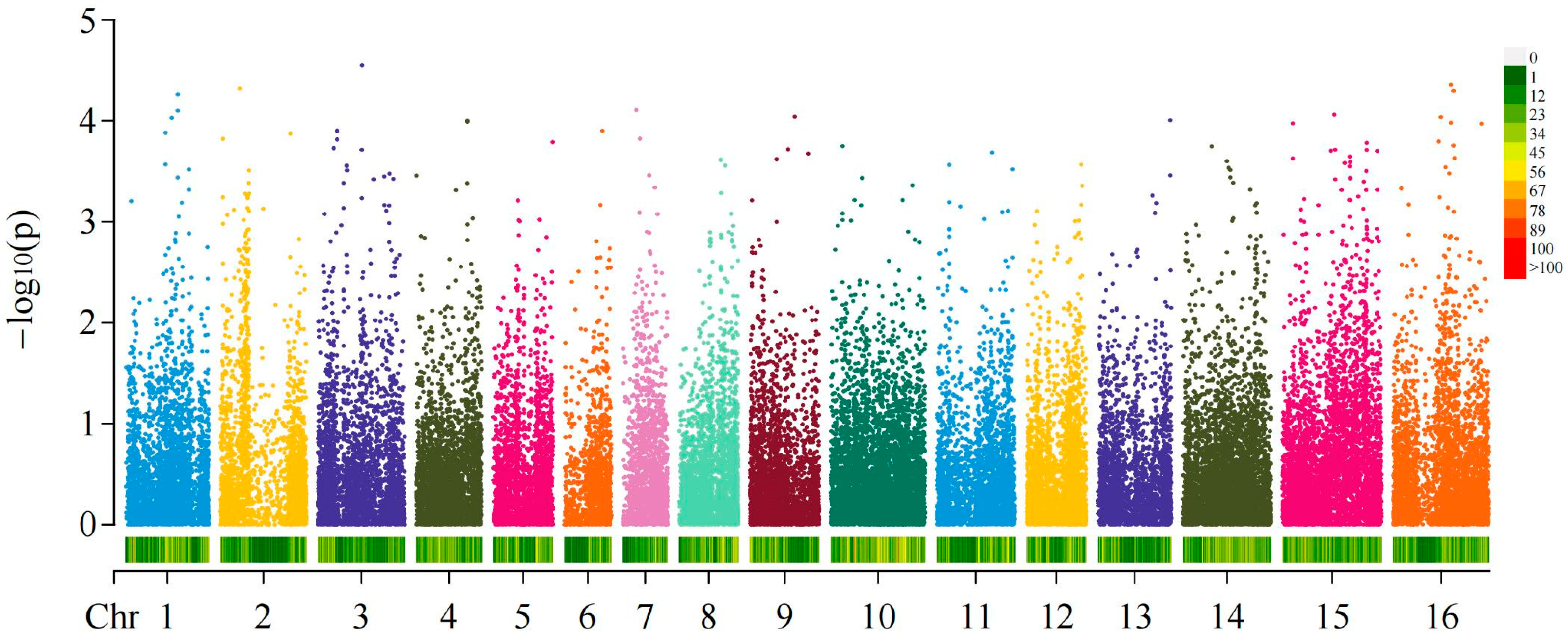
2.3. GWAS of Fruit Shapes and Areca Germplasms
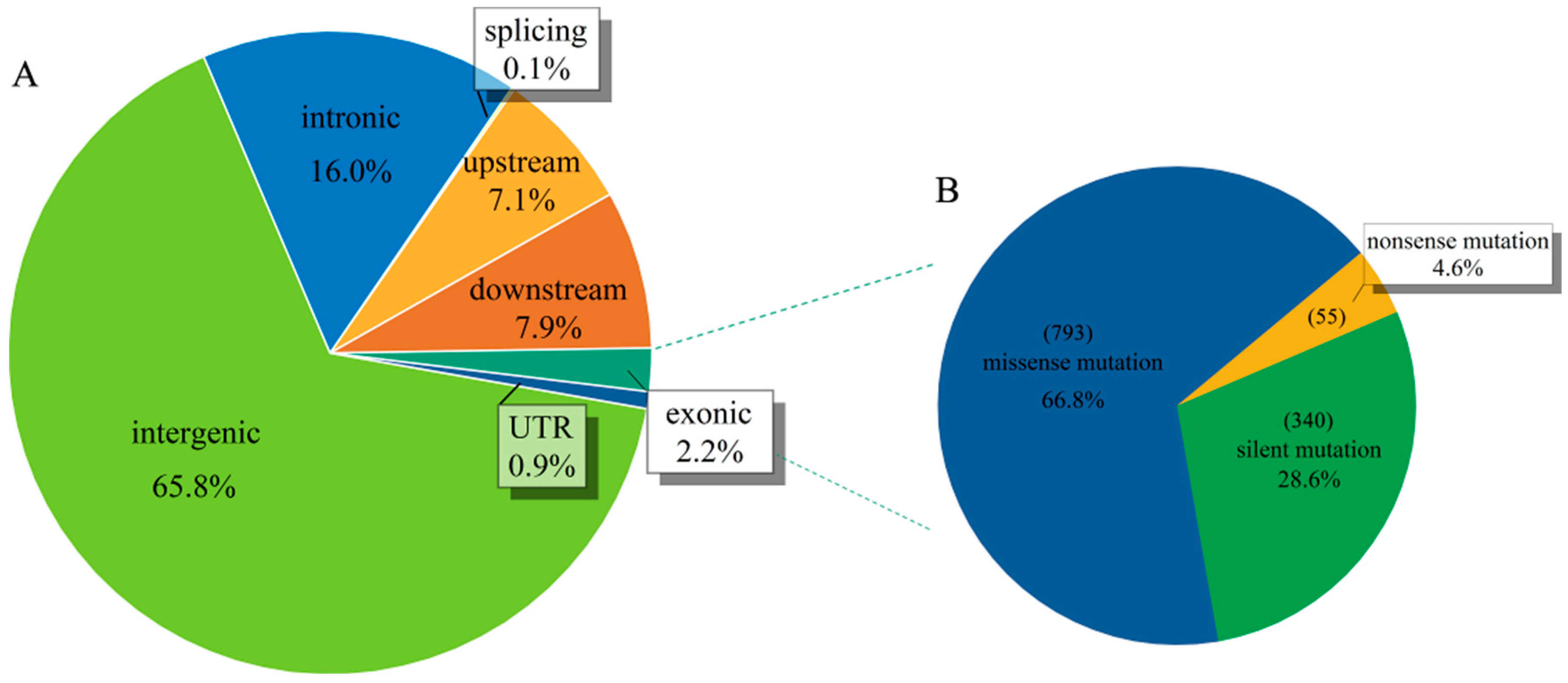
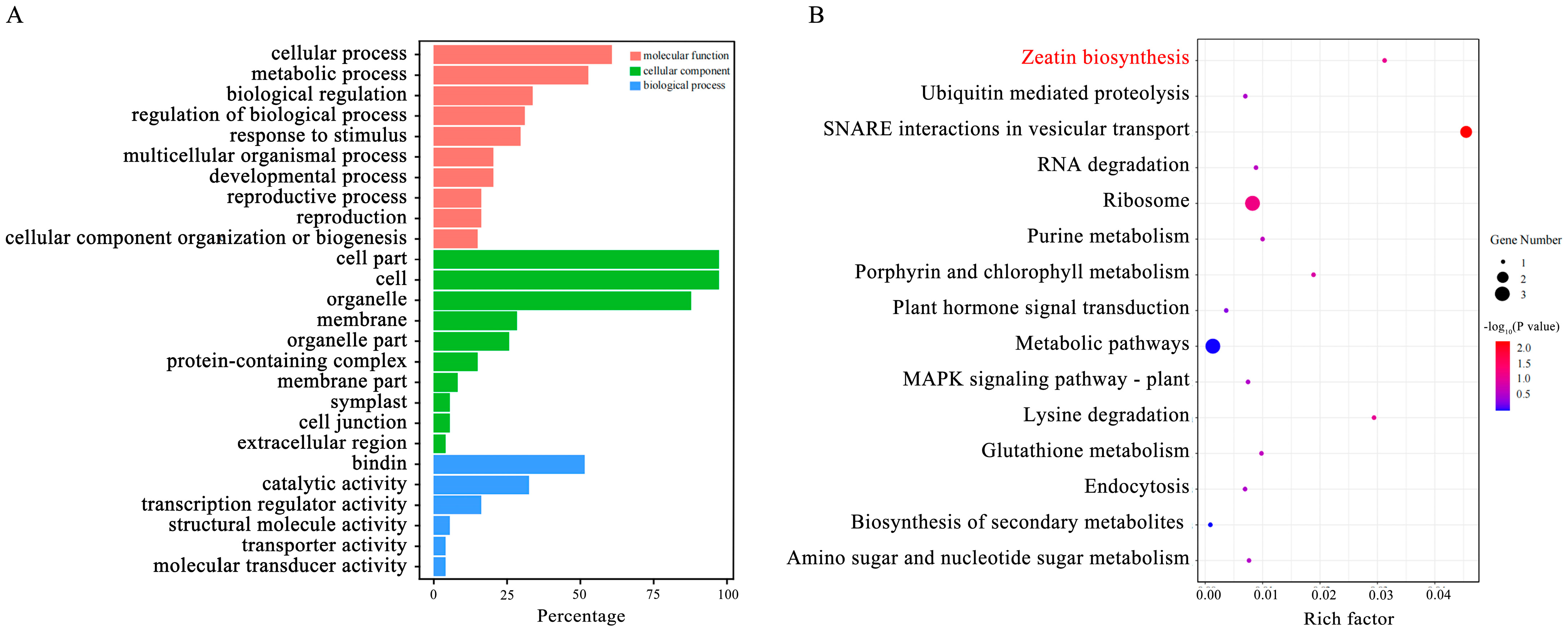
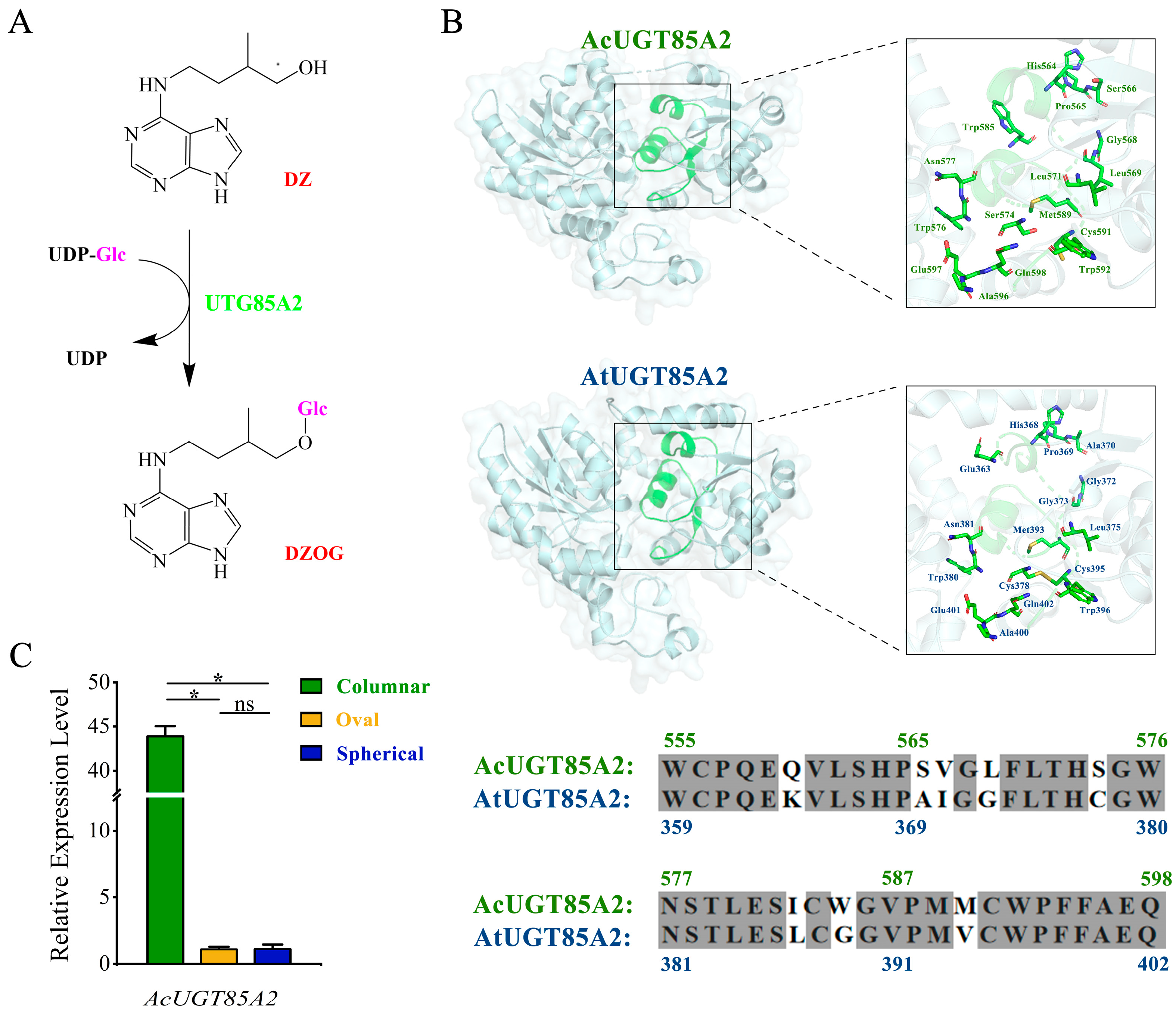
2.4. Candidate Genes
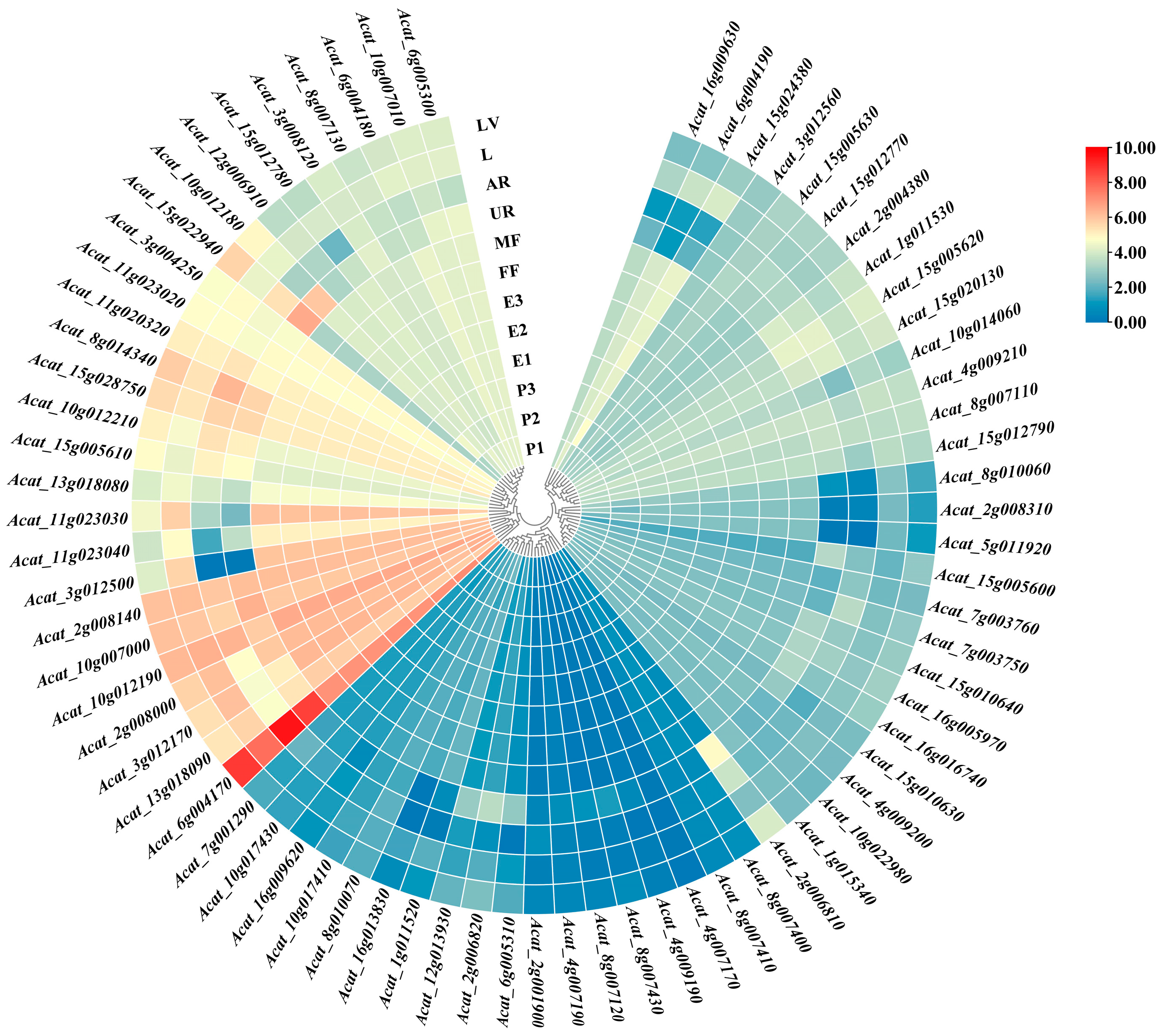
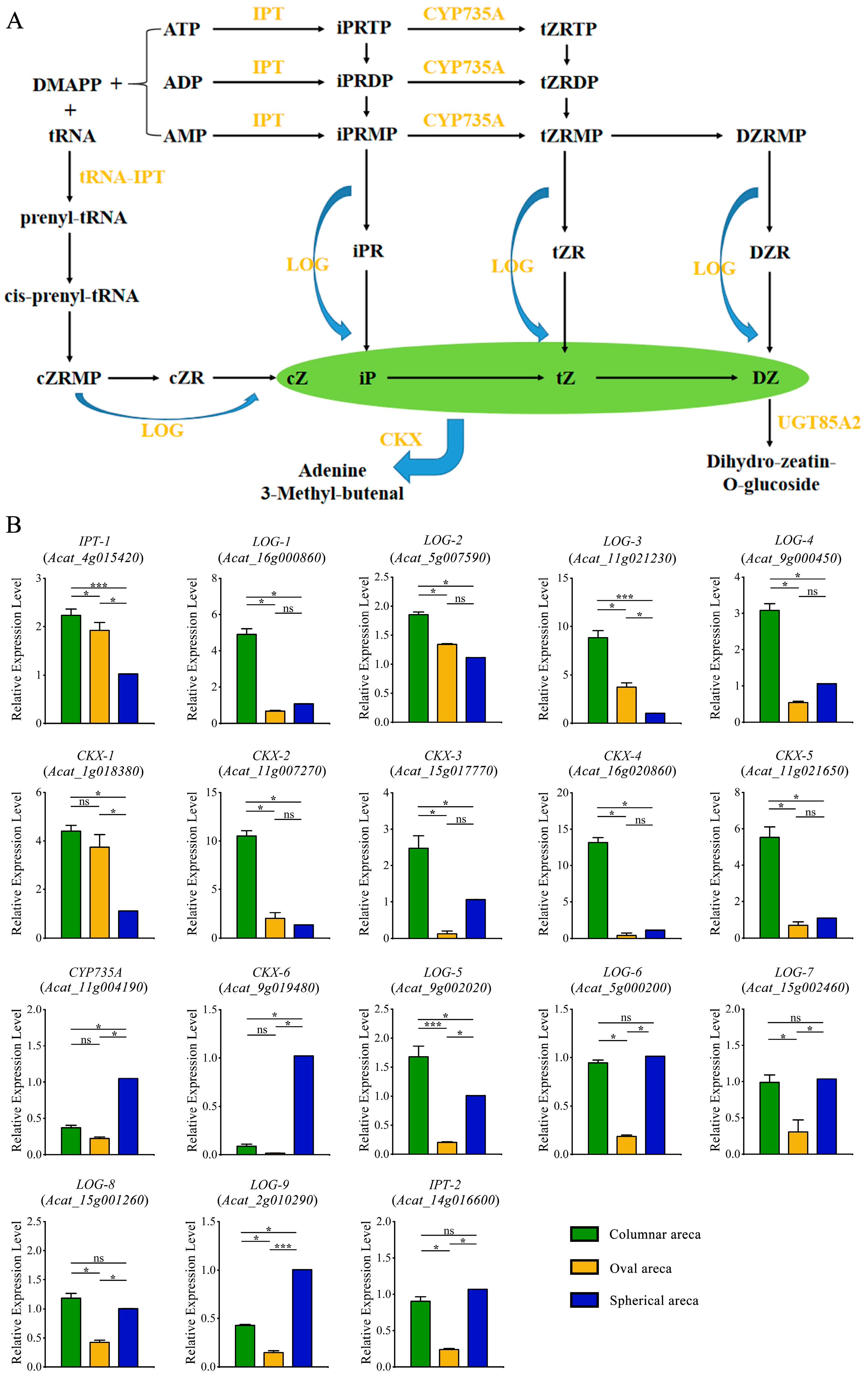
2.5. Expression Patterns of Genes Involved in Zeatin Biosynthesis Pathway
3. Discussion
4. Materials and Methods
4.1. Material Collection and Phenotype Evaluation
4.2. DNA Preparation and Sequencing
4.3. SNP Calling and Annotation
4.4. Genetic Diversity Analysis
4.5. Association Analysis
4.6. Candidate Gene Screening
4.7. Bioinformatics Analysis
4.8. Real-Time Fluorescence Quantitative PCR (qRT-PCR) Verification
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gillaspy, G.; Ben-David, H.; Gruissem, W. Fruits: A Developmental Perspective. Plant Cell 1993, 5, 1439–1451. [Google Scholar] [CrossRef] [Green Version]
- Fenn, M.A.; Giovannoni, J.J. Phytohormones in fruit development and maturation. Plant J. 2021, 105, 446–458. [Google Scholar] [CrossRef]
- Visa, S.; Cao, C.; Gardener, B.M.; van der Knaap, E. Modeling of tomato fruits into nine shape categories using elliptic fourier shape modeling and Bayesian classifcation of contour morphemetric data. Euphytica 2014, 200, 429–439. [Google Scholar] [CrossRef]
- Ziaf, K.; Amjad, M.; Shakeel, A.; Saeed, A. Assessment of genetic diversity in tomato for fruit morphology, composition and yield. Pak. J. Bot. 2016, 48, 2477–2483. [Google Scholar]
- Paran, I.; van der Knaap, E. Genetic and molecular regulation of fruit and plant domestication traits in tomato and pepper. J. Exp. Bot. 2007, 58, 3841–3852. [Google Scholar] [CrossRef] [Green Version]
- Christodoulou, A.; Pool, R.; Weaver, R. Prebloom thinning of Thompson Seedless grapes is feasible when followed by bloom spraying with gibberellin. Calif. Agric. 1966, 20, 8–10. [Google Scholar]
- Beppu, K.; Yanaihara, M.; Ikeda, T.; Kataoka, I. Effects of phytohormones on the occurrence of double pistils in sweet cherry (Prunu savium). Hortic. Res. 2007, 6, 123–125. [Google Scholar]
- Chunthawodtiporn, J.; Hill, T.; Stoffel, K.; Van, D.A. Quantitative trait loci controlling fruit size and other horticultural traits in bell pepper (Capsicum annuum). Plant Genome 2018, 11, 160125. [Google Scholar] [CrossRef] [Green Version]
- Wei, Q.Z.; Fu, W.Y.; Wang, Y.Z.; Qin, X.D.; Wang, J.; Li, J.; Lou, Q.F.; Chen, J.F. Rapid identification of fruit length loci in cucumber (Cucumis sativus L.) using next-generation sequencing (NGS)-based QTL analysis. Sci. Rep. 2016, 6, 27496. [Google Scholar] [CrossRef]
- Dou, J.; Zhao, S.; Lu, X.; He, N.; Zhang, L.; Ali, A.; Kuang, H.; Liu, W. Genetic mapping reveals a candidate gene (ClFS1) for fruit shape in watermelon (Citrullus lanatus L.). Theor. Appl. Genet. 2018, 131, 947–958. [Google Scholar] [CrossRef]
- Xiao, H.; Jiang, N.; Schaffner, E.; Stockinger, E.J.; van der Knaap, E. A retrotransposon-mediated gene duplication underlies morphological variation of tomato fruit. Science 2008, 319, 1527–1530. [Google Scholar] [CrossRef]
- Liu, J.; Van, E.J.; Cong, B.; Tanksley, S.D. A new class of regulatory genes underlying the cause of pear-shaped tomato fruit. Proc. Natl. Acad. Sci. USA 2002, 99, 13302–13306. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Qi, M.; Sun, M.; Liu, Y.; Liu, Y.; Xu, T.; Li, Y.; Li, T. Tomato transcription factor SlWUS plays an important role in tomato flower and locule development. Front. Plant Sci. 2017, 8, 457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, C.; Liberatore, K.L.; MacAlister, C.A.; Huang, Z.; Chu, Y.H.; Jiang, K.; Brooks, C.; Ogawa-Ohnishi, M.; Xiong, G.; Pauly, M.; et al. A cascade of arabinosyltransferases controls shoot meristem size in tomato. Nat. Genet. 2015, 47, 784–792. [Google Scholar] [CrossRef]
- Jiang, N.; Gao, D.; Xiao, H.; van der Knaap, E. Genome organization of the tomato sun locus and characterization of the unusual retrotransposon rider. Plant J. 2009, 60, 181–193. [Google Scholar] [CrossRef]
- Rodríguez, G.R.; Kim, H.J.; van der Knaap, E. Mapping of two suppressors of OVATE (sov) loci in tomato. Heredity 2013, 111, 256–264. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Clevenger, J.P.; Illa-Berenguer, E.; Meulia, T.; van der Knaap, E.; Sun, L. A Comparison of sun, ovate, fs8.1 and auxin application on tomato fruit shape and gene expression. Plant Cell Physiol. 2019, 60, 1067–1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez, G.R.; Muños, S.; Anderson, C.; Sim, S.C.; Michel, A.; Causse, M.; Gardener, B.B.; Francis, D.; van der Knaap, E. Distribution of SUN, OVATE, LC, and FAS in the tomato germplasm and the relationship to fruit shape diversity. Plant Physiol. 2011, 156, 275–285. [Google Scholar] [CrossRef] [Green Version]
- Cong, B.; Barrero, L.S.; Tanksley, S.D. Regulatory change in YABBY-like transcription factor led to evolution of extreme fruit size during tomato domestication. Nat. Genet. 2008, 40, 800–804. [Google Scholar] [CrossRef] [PubMed]
- Muños, S.; Ranc, N.; Botton, E.; Bérard, A.; Rolland, S.; Duffé, P.; Carretero, Y.; Le, P.M.C.; Delalande, C.; Bouzayen, M.; et al. Increase in tomato locule number is controlled by two single-nucleotide polymorphisms located near WUSCHEL. Plant Physiol. 2011, 156, 2244–2254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Zhang, J.; Wang, J.; Zhang, J.; Miao, H.; Jia, C.; Wang, Z.; Xu, B.; Jin, Z. MuMADS1 and MaOFP1 regulate fruit quality in a tomato ovate mutant. Plant Biotechnol. J. 2016, 16, 989–1001. [Google Scholar] [CrossRef] [Green Version]
- Yao, J.L.; Xu, J.; Tomes, S.; Cui, W.; Luo, Z.; Deng, C.; Ireland, H.S.; Schaffer, R.J.; Gleave, A.P. Ectopic expression of the PISTILLATA homologous MdPI inhibits fruit tissue growth and changes fruit shape in apple. Plant Direct 2018, 2, e00051. [Google Scholar] [CrossRef]
- Nordborg, M.; Weigel, D. Next-generation genetics in plants. Nature 2008, 456, 720–723. [Google Scholar] [CrossRef]
- Korte, A.; Farlow, A. The advantages and limitations of trait analysis with GWAS: A review. Plant Methods 2013, 9, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.; Cao, K.; Deng, C.; Li, Y.; Zhu, G.; Fang, W.; Chen, C.; Wang, X.; Wu, J.; Guan, L.; et al. An integrated peach genome structural variation map uncovers genes associated with fruit traits. Genome Biol. 2020, 21, 258. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.; Duan, S.; Sheng, J.; Zhu, S.; Ni, X.; Shao, J.; Liu, C.; Nick, P.; Du, F.; Fan, P.; et al. Whole-genome resequencing of 472 Vitis accessions for grapevine diversity and demographic history analyses. Nat. Commun. 2019, 10, 1190. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Cui, L.; Fang, J. Genome-wide association study of the candidate genes for grape berry shape-related traits. BMC Plant Biol. 2022, 22, 42. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Gao, P.; Zhu, Q.; Zhu, Z.; Liu, H.; Wang, X.; Weng, Y.; Gao, M.; Luan, F. Resequencing of 297 melon accessions reveals the genomic history of improvement and loci related to fruit traits in melon. Plant Biotechnol. J. 2020, 18, 2545–2558. [Google Scholar] [CrossRef]
- Amudhan, M.S.; Begum, V.H.; Hebbar, K.B. A review on phytochemical and pharmacological potential of Areca catechu L. Seed. Int. J. Pharm. Sci. Res. 2012, 3, 4151–4157. [Google Scholar]
- Meng, K.; Mei, F.; Zhu, L.; Chen, H. Arecanut (Areca catechu L.) seed polyphenol improves osteoporosis via gut-serotonin mediated Wnt/β-catenin pathway in ovariectomized rats. J. Funct. Foods 2021, 84, 104598. [Google Scholar] [CrossRef]
- Li, J.; Jia, X.; Liu, L.; Cao, X.; Xiong, Y.; Yang, Y.; Zhou, H.; Yi, M.; Li, M. Comparative biochemical and transcriptome analysis provides insights into the regulatory mechanism of striped leaf albinism in arecanut (Areca catechu L.). Ind. Crop. Prod. 2020, 154, 112734. [Google Scholar] [CrossRef]
- Pattison, R.J.; Catalá, C. Evaluating auxin distribution in tomato (Solanum lycopersicum) through an analysis of the PIN and AUX/LAX gene families. Plant J. 2012, 70, 585–598. [Google Scholar] [CrossRef]
- Ripoll, J.J.; Zhu, M.; Brocke, S.; Hon, C.T.; Yanofsky, M.F.; Boudaoud, A.; Roeder, A.H.K. Growth dynamics of the Arabidopsis fruit is mediated by cell expansion. Proc. Natl. Acad. Sci. USA 2019, 116, 25333–25342. [Google Scholar] [CrossRef]
- Devoghalaere, F.; Doucen, T.; Guitton, B.; Keeling, J.; Payne, W.; Ling, T.J.; Ross, J.J.; Hallett, I.C.; Gunaseelan, K.; Dayatilake, G.A.; et al. A genomics approach to understanding the role of auxin in apple (Malusx domestica) fruit size control. BMC Plant Biol. 2012, 12, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Xu, T.; Dong, X.; Liu, Y.; Liu, Z.; Shi, Z.; Wang, Y.; Qi, M.; Li, T. The role of gibberellins and auxin on the tomato cell layers in pericarp via the expression of ARFs regulated by miRNAs in fruit set. Acta Physiol. Plant 2016, 38, 77. [Google Scholar] [CrossRef]
- He, H.; Yamamuro, C. Interplays between auxin and GA signaling coordinate early fruit development. Hortic. Res. 2022, 9, uhab078. [Google Scholar] [CrossRef]
- McAtee, P.; Karim, S.; Schaffer, R.; David, K. A dynamic interplay between phytohormones is required for fruit development, maturation, and ripening. Front. Plant Sci. 2013, 4, 79. [Google Scholar] [CrossRef] [Green Version]
- Cong, L.; Yue, R.; Wang, H.; Liu, J.; Zhai, R.; Yang, J.; Wu, M.; Si, M.; Zhang, H.; Yang, C.; et al. 2,4-D-induced parthenocarpy in pear is mediated by enhancement of GA4 biosynthesis. Physiol. Plant 2019, 166, 812–820. [Google Scholar] [CrossRef]
- Zhang, H.; Tan, J.; Zhang, M.; Huang, S.; Chen, X. Comparative transcriptomic analysis of two bottle gourd accessions differing in fruit size. Genes 2020, 11, 359. [Google Scholar] [CrossRef] [Green Version]
- Hussain, Q.; Shi, J.; Scheben, A.; Zhan, J.; Wang, X.; Liu, G.; Yan, G.; King, G.J.; Edwards, D.; Wang, H. Genetic and signalling pathways of dry fruit size: Targets for genome editing-based crop improvement. Plant Biotechnol. J. 2020, 18, 1124–1140. [Google Scholar] [CrossRef]
- Cucinotta, M.; Manrique, S.; Cuesta, C.; Benkova, E.; Novak, O.; Colombo, L. CUP-SHAPED COTYLEDON1 (CUC1) and CUC2 regulate cytokinin homeostasis to determine ovule number in Arabidopsis. J. Exp. Bot. 2018, 69, 5169–5176. [Google Scholar] [CrossRef]
- Li, X.; Cui, H.; Song, X.; Sun, C.; Ding, Z.; Zhu, X.; Liu, X.; Yan, L. CsIPT1b and CsUGT85A2 delay female corolla opening in cucumber by regulating the content of cytokinins. Hortic. Plant J. 2022; pre-proof. [Google Scholar] [CrossRef]
- Shu, K.; Yang, W. E3 Ubiquitin Ligases: Ubiquitous actors in plant development and abiotic stress responses. Plant Cell Physiol. 2017, 58, 1461–1476. [Google Scholar] [CrossRef] [Green Version]
- Trujillo, M. News from the PUB: Plant U-box type E3 ubiquitin ligases. J. Exp. Bot. 2018, 69, 371–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Girona, E.; Zhang, Y.; Eduardo, I.; Mora, J.R.H.; Alexiou, K.G.; Arús, P.; Aranzana, M.J. A deletion affecting an LRR-RLK gene co-segregates with the fruit flat shape trait in peach. Sci. Rep. 2017, 7, 6714. [Google Scholar] [CrossRef] [Green Version]
- Stewart, C.N., Jr.; Via, L.E. A rapid CTAB DNA isolation technique useful for RAPD fingerprinting and other PCR applications. Biotechniques 1993, 14, 748–750. [Google Scholar]
- Zou, M.; Xia, Z. Hyper-seq: A novel, effective, and flexible marker-assisted selection and genotyping approach. Innovation 2022, 3, 100254. [Google Scholar] [CrossRef]
- Vashishtha, K.; Gaud, C.; Andrews, S.; Krueger, C. Librarian: A quality control tool to analyse sequencing library compositions. F1000Research 2022, 11, 1122. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Huang, L.; Xu, C.; Qi, L.; Wu, Z.; Li, J.; Chen, H.; Wu, Y.; Fu, T.; Zhu, H.; et al. Chromosome-scale genome assembly of areca palm (Areca catechu). Mol. Ecol. Resour. 2021, 21, 2504–2519. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [Green Version]
- Retief, J.D. Phylogenetic analysis using PHYLIP. Methods Mol. Biol. 2000, 132, 243–258. [Google Scholar]
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2011, 88, 76–82. [Google Scholar] [CrossRef] [Green Version]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ding, H.; Zhou, G.; Zhao, L.; Li, X.; Wang, Y.; Xia, C.; Xia, Z.; Wan, Y. Genome-Wide Association Analysis of Fruit Shape-Related Traits in Areca catechu. Int. J. Mol. Sci. 2023, 24, 4686. https://doi.org/10.3390/ijms24054686
Ding H, Zhou G, Zhao L, Li X, Wang Y, Xia C, Xia Z, Wan Y. Genome-Wide Association Analysis of Fruit Shape-Related Traits in Areca catechu. International Journal of Molecular Sciences. 2023; 24(5):4686. https://doi.org/10.3390/ijms24054686
Chicago/Turabian StyleDing, Hao, Guangzhen Zhou, Long Zhao, Xinyu Li, Yicheng Wang, Chengcai Xia, Zhiqiang Xia, and Yinglang Wan. 2023. "Genome-Wide Association Analysis of Fruit Shape-Related Traits in Areca catechu" International Journal of Molecular Sciences 24, no. 5: 4686. https://doi.org/10.3390/ijms24054686