
Genome-Wide Analysis of Antigen 43 (Ag43) Variants: New Insights in Their Diversity, Distribution and Prevalence in Bacteria
 , , , , ,
, , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
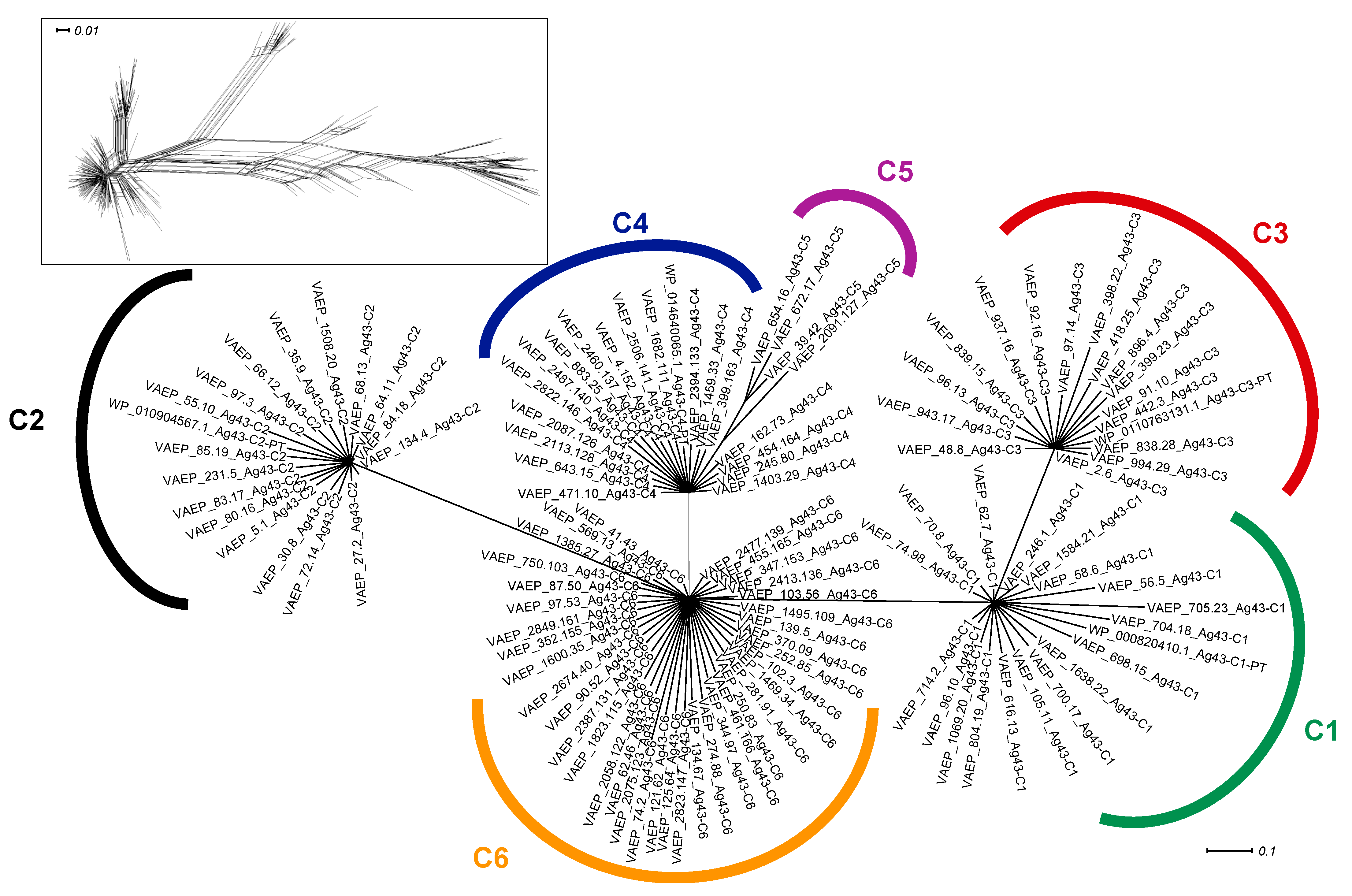
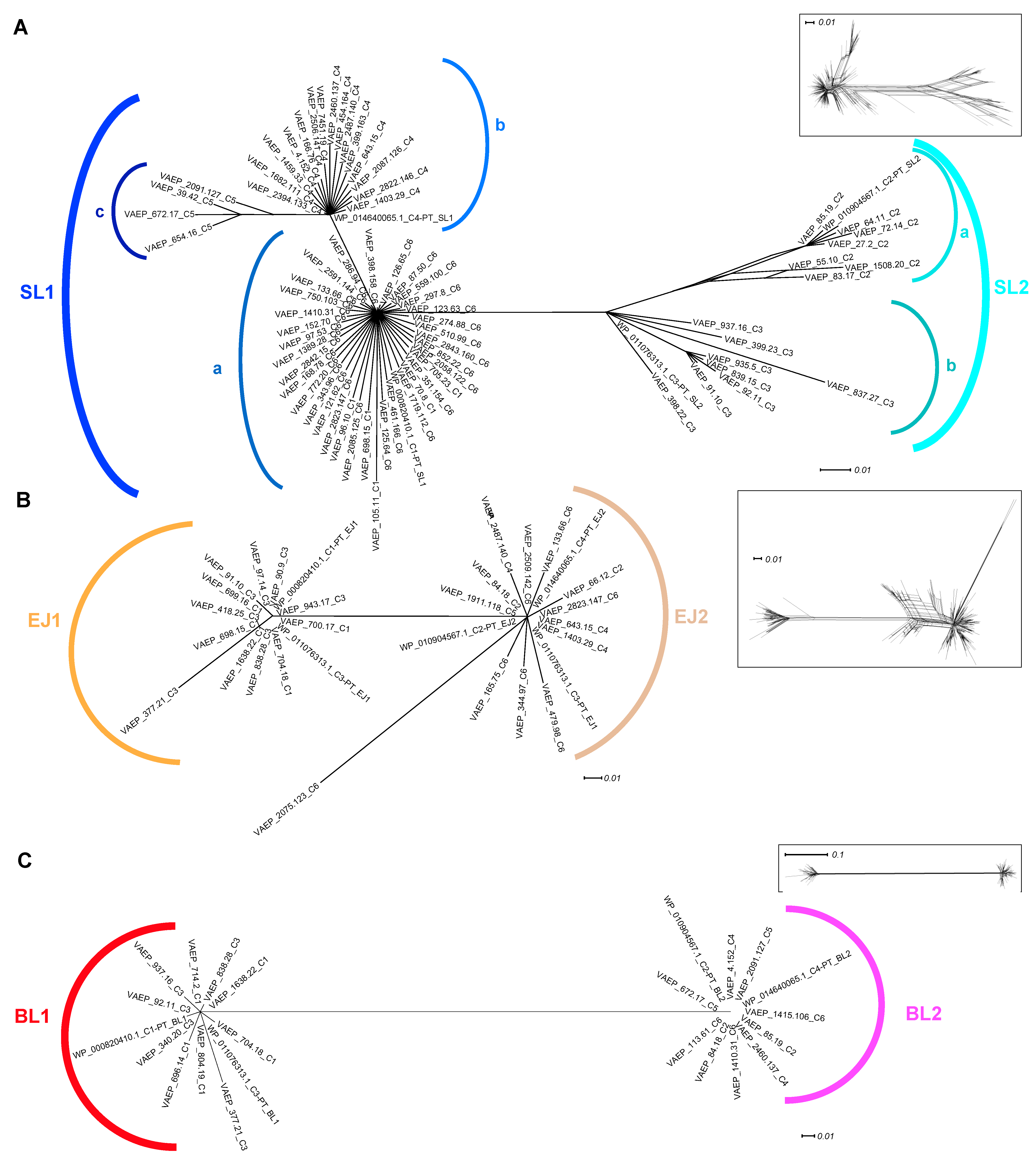
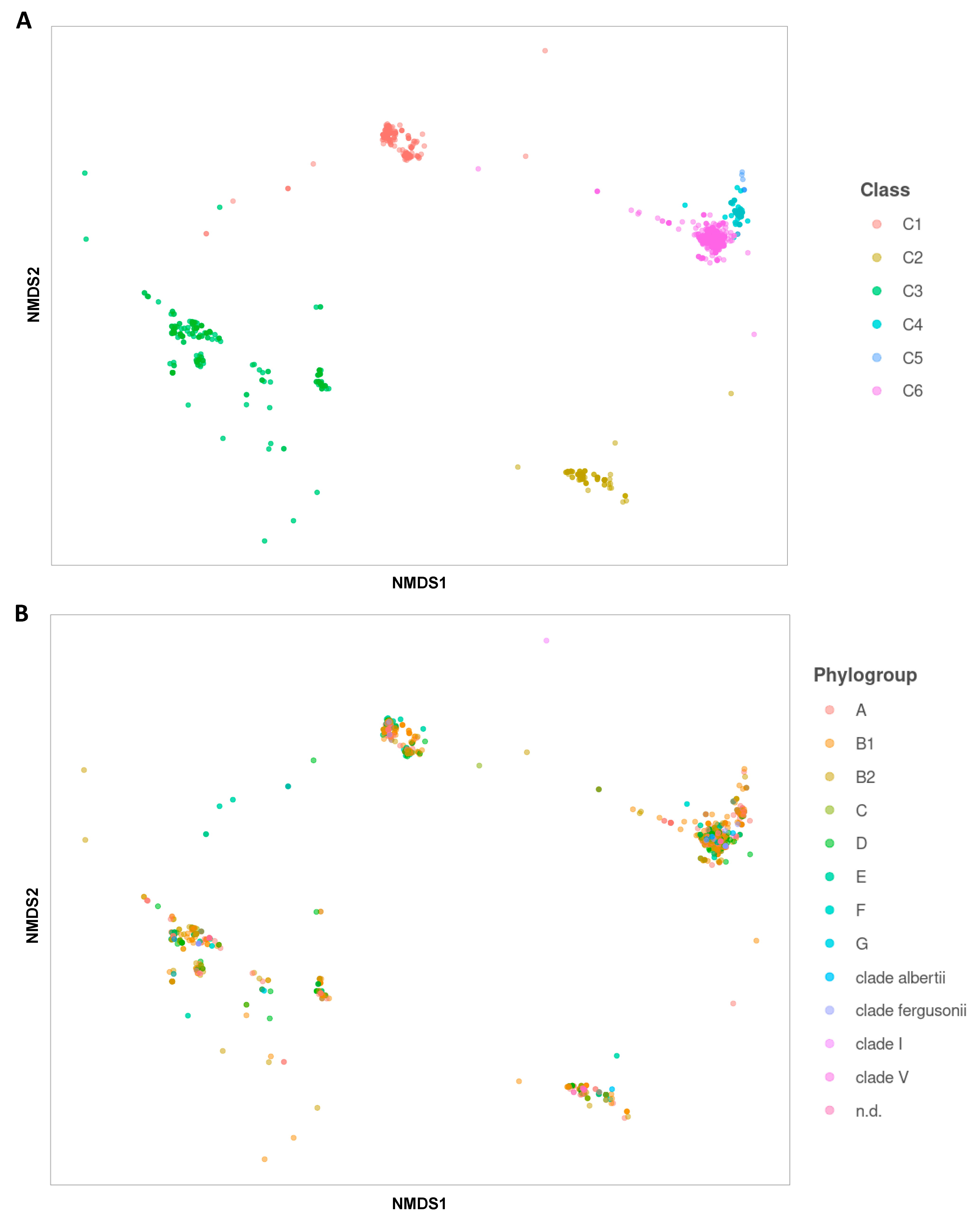
2.1. Phylogenetic Analyses of Ag43 Passenger Domains
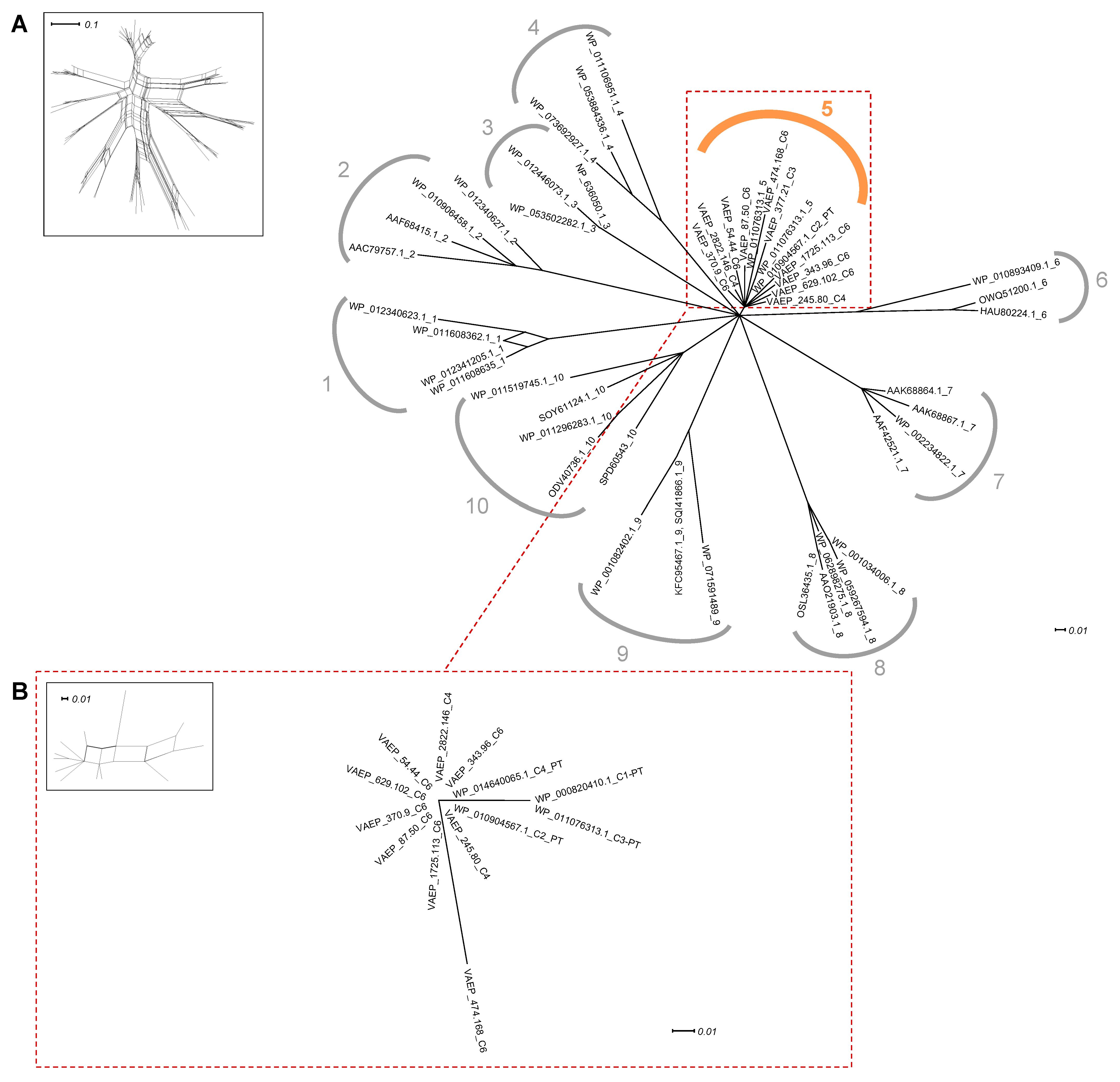
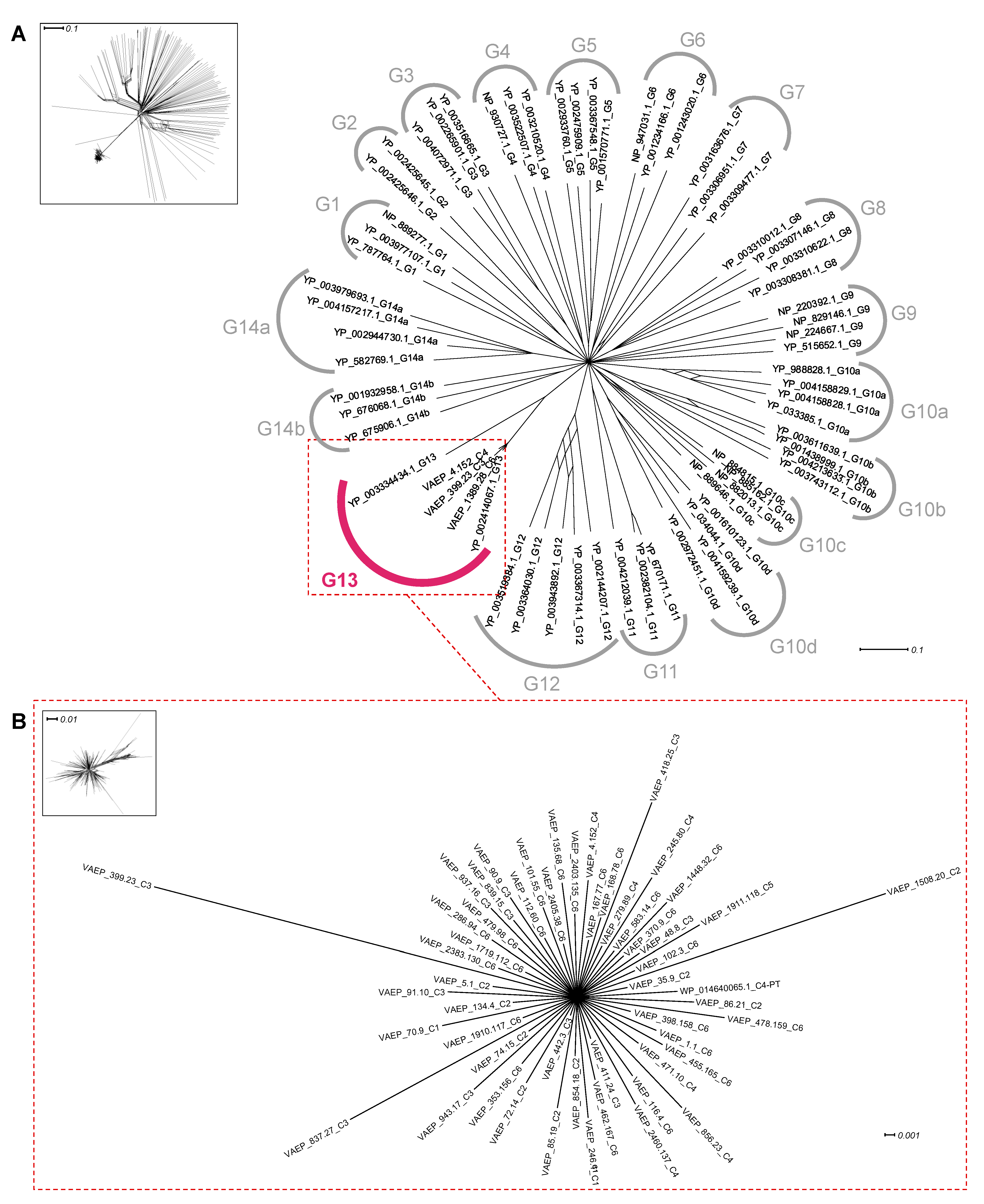
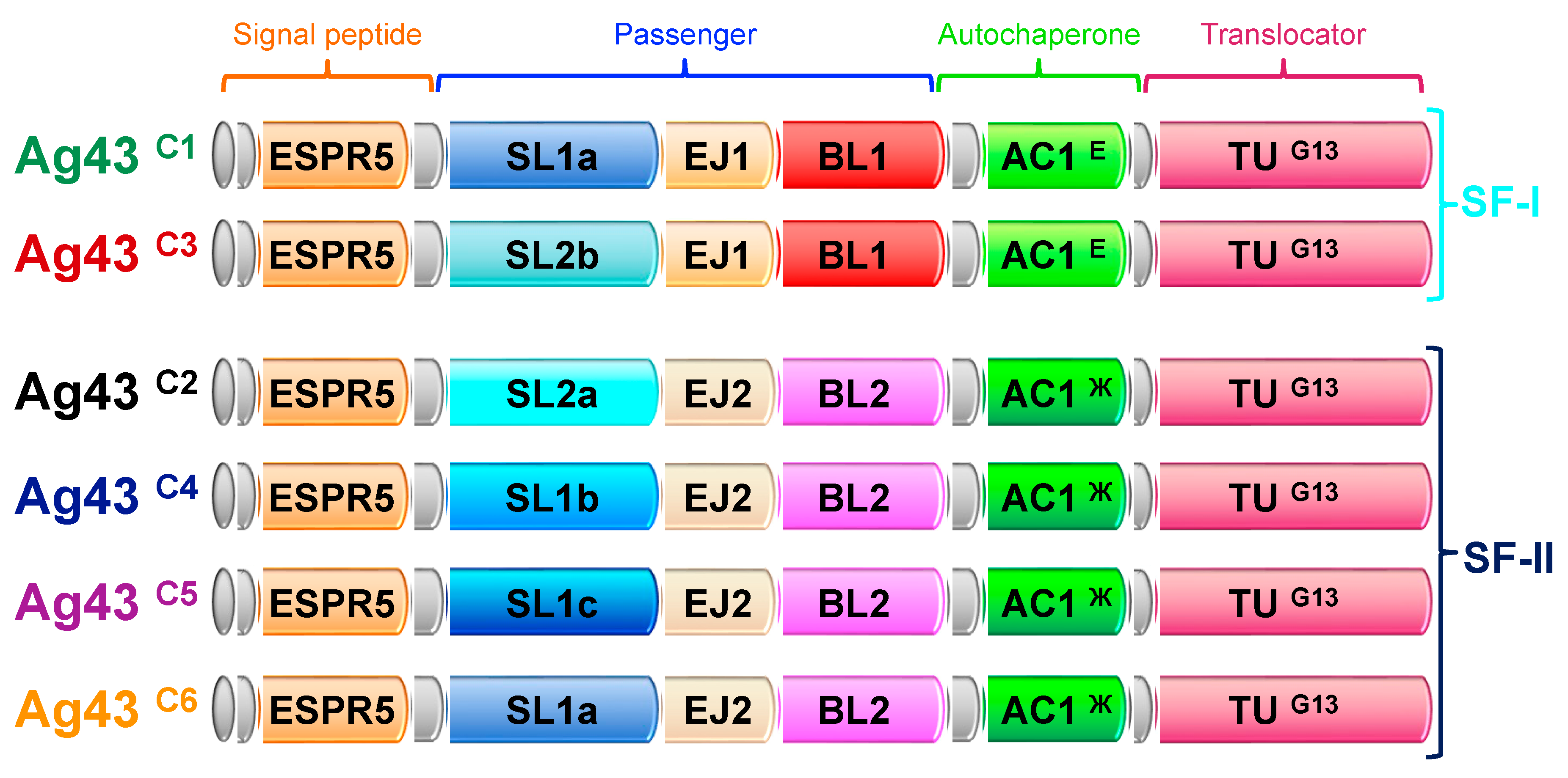
2.2. Identification of Conserved Domain Modules in Ag43
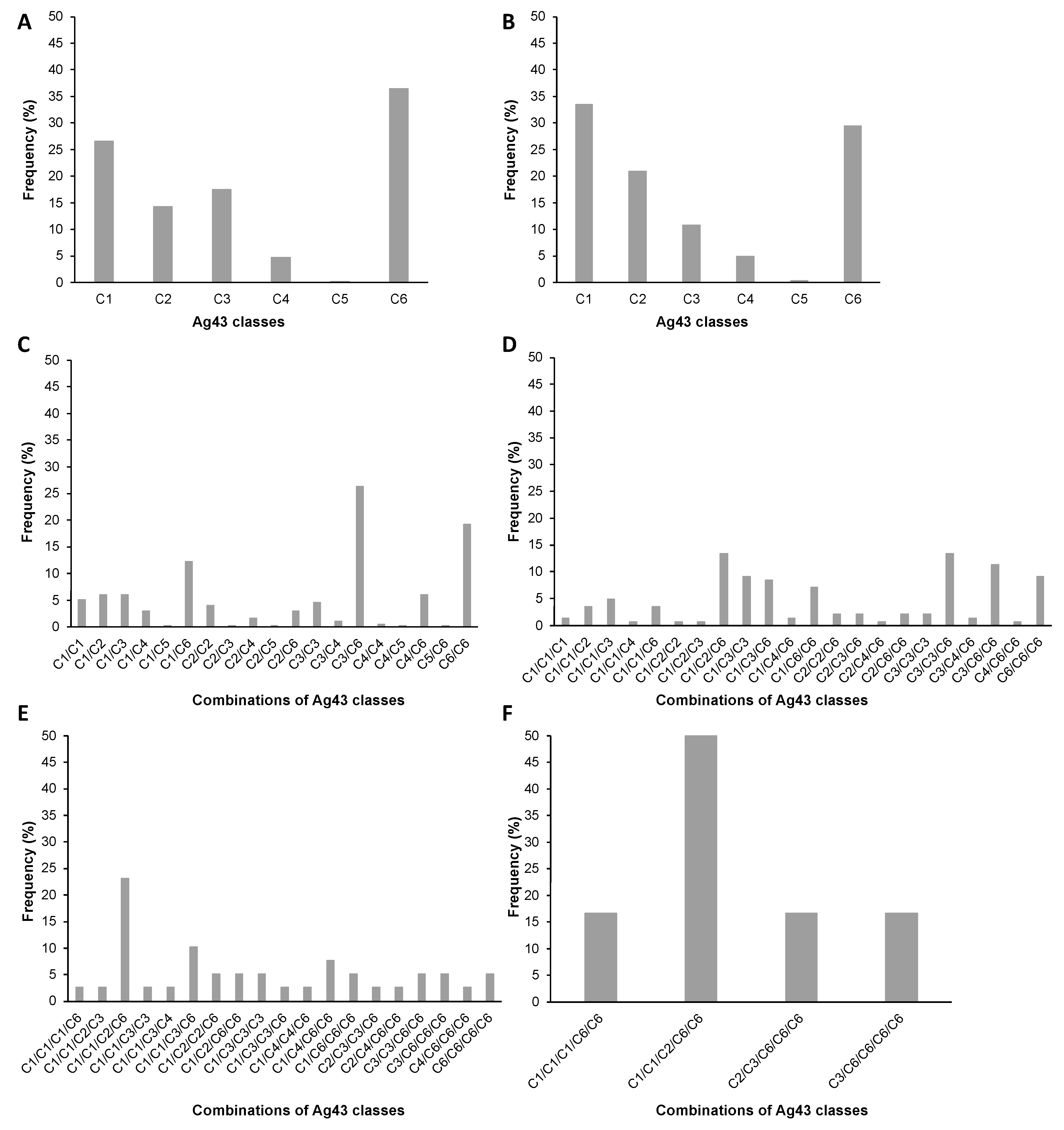
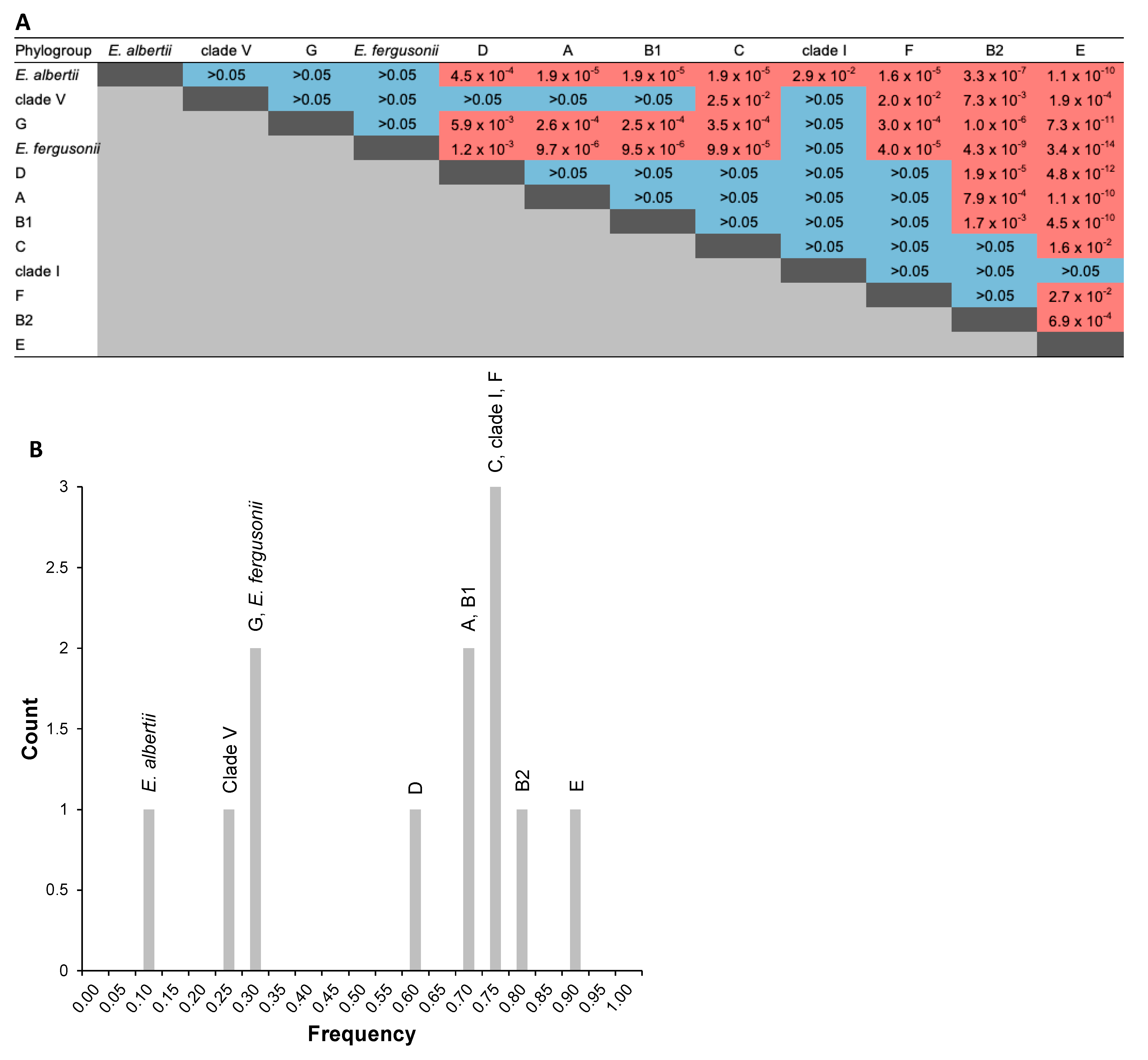
2.3. Distribution of Ag43 Paralogs within E. coli
3. Discussion
4. Materials and Methods
4.1. Data Mining for Identification of Ag43 Protein Sequences
4.2. Phylogenetic Network Analysis of Protein Sequences
4.3. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Desvaux, M.; Parham, N.J.; Henderson, I.R. Le Système de Sécrétion de Type V Chez Les Bactéries Gram-Négatives (Type V Secretion System in Gram-Negative Bacteria). Biofutur 2003, 237, 34–37. [Google Scholar]
- Desvaux, M.; Parham, N.J.; Henderson, I.R. The Autotransporter Secretion System. Res. Microbiol. 2004, 155, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Leo, J.C.; Grin, I.; Linke, D. Type V Secretion: Mechanism(s) of Autotransport through the Bacterial Outer Membrane. Philos. Trans. R. Soc. B-Biol. Sci. 2012, 367, 1088–1101. [Google Scholar] [CrossRef]
- Henderson, I.R.; Navarro-Garcia, F.; Desvaux, M.; Fernandez, R.C.; Ala’Aldeen, D. Type V Protein Secretion Pathway: The Autotransporter Story. Microbiol. Mol. Biol. Rev. 2004, 68, 692–744. [Google Scholar] [CrossRef]
- Drobnak, I.; Braselmann, E.; Chaney, J.L.; Leyton, D.L.; Bernstein, H.D.; Lithgow, T.; Luirink, J.; Nataro, J.P.; Clark, P.L. Of Linkers and Autochaperones: An Unambiguous Nomenclature to Identify Common and Uncommon Themes for Autotransporter Secretion. Mol. Microbiol. 2015, 95, 1–16. [Google Scholar] [CrossRef]
- Clarke, K.R.; Hor, L.; Pilapitiya, A.; Luirink, J.; Paxman, J.J.; Heras, B. Phylogenetic Classification and Functional Review of Autotransporters. Front. Immunol. 2022, 13, 921272. [Google Scholar] [CrossRef] [PubMed]
- Klemm, P.; Vejborg, R.M.; Sherlock, O. Self-Associating Autotransporters, SAATs: Functional and Structural Similarities. Int. J. Med. Microbiol. 2006, 296, 187–195. [Google Scholar] [CrossRef]
- Henderson, I.R.; Meehan, M.; Owen, P. Antigen 43, a Phase-Variable Bipartite Outer Membrane Protein, Determines Colony Morphology and Autoaggregation in Escherichia coli K-12. FEMS Microbiol. Lett. 1997, 149, 115–120. [Google Scholar] [CrossRef]
- Trunk, T.; Salah Khalil, H.; Leo, J.C. Bacterial Autoaggregation. AIMS Microbiol. 2018, 4, 140–164. [Google Scholar] [CrossRef]
- Paxman, J.J.; Lo, A.W.; Sullivan, M.J.; Panjikar, S.; Kuiper, M.; Whitten, A.E.; Wang, G.; Luan, C.-H.; Moriel, D.G.; Tan, L.; et al. Unique Structural Features of a Bacterial Autotransporter Adhesin Suggest Mechanisms for Interaction with Host Macromolecules. Nat. Commun. 2019, 10, 1967. [Google Scholar] [CrossRef]
- Meng, G.; Spahich, N.; Kenjale, R.; Waksman, G.; St Geme, J.W., III. Crystal Structure of the Haemophilus Influenzae Hap Adhesin Reveals an Intercellular Oligomerization Mechanism for Bacterial Aggregation. EMBO J 2011, 30, 3864–3874. [Google Scholar] [CrossRef] [PubMed]
- Arenas, J.; Cano, S.; Nijland, R.; van Dongen, V.; Rutten, L.; van der Ende, A.; Tommassen, J. The Meningococcal Autotransporter AutA Is Implicated in Autoaggregation and Biofilm Formation. Environ. Microbiol. 2015, 17, 1321–1337. [Google Scholar] [CrossRef]
- Vo, J.L.; Martinez Ortiz, G.C.; Subedi, P.; Keerthikumar, S.; Mathivanan, S.; Paxman, J.J.; Heras, B. Autotransporter Adhesins in Escherichia coli Pathogenesis. Proteomics 2017, 296, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Wells, T.J.; Sherlock, O.; Rivas, L.; Mahajan, A.; Beatson, S.A.; Torpdahl, M.; Webb, R.I.; Allsopp, L.P.; Gobius, K.S.; Gally, D.L.; et al. EhaA Is a Novel Autotransporter Protein of Enterohemorrhagic Escherichia coli O157:H7 That Contributes to Adhesion and Biofilm Formation. Environ. Microbiol. 2008, 10, 589–604. [Google Scholar] [CrossRef] [PubMed]
- Dorsey, C.W.; Laarakker, M.C.; Humphries, A.D.; Weening, E.H.; Baumler, A.J. Salmonella Enterica Serotype Typhimurium MisL Is an Intestinal Colonization Factor That Binds Fibronectin. Mol. Microbiol. 2005, 57, 196–211. [Google Scholar] [CrossRef]
- Goldberg, M.B.; Theriot, J.A. Shigella Flexneri Surface Protein IcsA Is Sufficient to Direct Actin-Based Motility. Proc. Natl. Acad. Sci. USA 1995, 92, 6572–6576. [Google Scholar] [CrossRef]
- Owen, P.; Meehan, M.; de Loughry-Doherty, H.; Henderson, I. Phase-Variable Outer Membrane Proteins in Escherichia coli. FEMS Immunol. Med. Microbiol. 1996, 16, 63–76. [Google Scholar] [CrossRef]
- van der Woude, M.W.; Henderson, I.R. Regulation and Function of Ag43 (Flu). Annu. Rev. Microbiol. 2008, 62, 153–169. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Vo, J.L.; Ortiz, G.C.M.; Totsika, M.; Lo, A.W.; Hancock, S.J.; Whitten, A.E.; Hor, L.; Peters, K.M.; Ageorges, V.; Caccia, N.; et al. Variation of Antigen 43 Self-Association Modulates Bacterial Compacting within Aggregates and Biofilms. npj Biofilms Microbiomes 2022, 8, 20. [Google Scholar] [CrossRef]
- Heras, B.; Totsika, M.; Peters, K.M.; Paxman, J.J.; Gee, C.L.; Jarrott, R.J.; Perugini, M.A.; Whitten, A.E.; Schembri, M.A. The Antigen 43 Structure Reveals a Molecular Velcro-like Mechanism of Autotransporter-Mediated Bacterial Clumping. Proc. Natl. Acad. Sci. USA 2014, 111, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J. The Phyre2 Web Portal for Protein Modeling, Prediction and Analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [PubMed]
- Gawarzewski, I.; DiMaio, F.; Winterer, E.; Tschapek, B.; Smits, S.H.J.; Jose, J.; Schmitt, L. Crystal Structure of the Transport Unit of the Autotransporter Adhesin Involved in Diffuse Adherence from Escherichia coli. J. Struct. Biol. 2014, 187, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Desvaux, M.; Scott-Tucker, A.; Turner, S.M.; Cooper, L.M.; Huber, D.; Nataro, J.P.; Henderson, I.R. A Conserved Extended Signal Peptide Region Directs Posttranslational Protein Translocation via a Novel Mechanism. Microbiology 2007, 153, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Jong, W.S.; ten Hagen-Jongman, C.M.; Ruijter, E.; Orru, R.V.; Genevaux, P.; Luirink, J. YidC Is Involved in the Biogenesis of the Secreted Autotransporter Hemoglobin Protease. J. Biol. Chem. 2010, 285, 39682–39690. [Google Scholar] [CrossRef]
- Rojas-Lopez, M.; Zorgani, M.A.; Kelley, L.A.; Bailly, X.; Kajava, A.V.; Henderson, I.R.; Polticelli, F.; Pizza, M.; Rosini, R.; Desvaux, M. Identification of the Autochaperone Domain in the Type Va Secretion System (T5aSS): Prevalent Feature of Autotransporters with a β-Helical Passenger. Front. Microbiol. 2018, 8, 2607. [Google Scholar] [CrossRef]
- Oliver, D.C.; Huang, G.; Nodel, E.; Pleasance, S.; Fernandez, R.C. A Conserved Region within the Bordetella Pertussis Autotransporter BrkA Is Necessary for Folding of Its Passenger Domain. Mol. Microbiol. 2003, 47, 1367–1383. [Google Scholar] [CrossRef]
- Reidl, S.; Lehmann, A.; Schiller, R.; Salam Khan, A.; Dobrindt, U. Impact of O-Glycosylation on the Molecular and Cellular Adhesion Properties of the Escherichia coli Autotransporter Protein Ag43. Int. J. Med. Microbiol. 2009, 299, 389–401. [Google Scholar] [CrossRef]
- Ageorges, V.; Schiavone, M.; Jubelin, G.; Caccia, N.; Ruiz, P.; Chafsey, I.; Bailly, X.; Dague, E.; Leroy, S.; Paxman, J.; et al. Differential Homotypic and Heterotypic Interactions of Antigen 43 (Ag43) Variants in Autotransporter-Mediated Bacterial Autoaggregation. Sci. Rep. 2019, 9, 11100. [Google Scholar] [CrossRef]
- Roche, A.; McFadden, J.; Owen, P. Antigen 43, the Major Phase-Variable Protein of the Escherichia coli Outer Membrane, Can Exist as a Family of Proteins Encoded by Multiple Alleles. Microbiology 2001, 147, 161–169. [Google Scholar] [CrossRef]
- Restieri, C.; Garriss, G.; Locas, M.C.; Dozois, C.M. Autotransporter-Encoding Sequences Are Phylogenetically Distributed among Escherichia coli Clinical Isolates and Reference Strains. Appl Environ. Microbiol. 2007, 73, 1553–1562. [Google Scholar] [CrossRef] [PubMed]
- Armbruster, K.M.; Meredith, T.C. Identification of the Lyso-Form N-Acyl Intramolecular Transferase in Low-GC Firmicutes. J. Bacteriol. 2017, 199, e00099-17. [Google Scholar] [CrossRef] [PubMed]
- Desvaux, M.; Cooper, L.M.; Filenko, N.A.; Scott-Tucker, A.; Turner, S.M.; Cole, J.A.; Henderson, I.R. The Unusual Extended Signal Peptide Region of the Type V Secretion System Is Phylogenetically Restricted. FEMS Microbiol. Lett. 2006, 264, 22–30. [Google Scholar] [CrossRef]
- Celik, N.; Webb, C.T.; Leyton, D.L.; Holt, K.E.; Heinz, E.; Gorrell, R.; Kwok, T.; Naderer, T.; Strugnell, R.A.; Speed, T.P.; et al. A Bioinformatic Strategy for the Detection, Classification and Analysis of Bacterial Autotransporters. PLoS ONE 2012, 7, e43245. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, R.R.; Henderson, I.R. The Evolution of the Escherichia coli Phylogeny. Infect Genet Evol 2012, 12, 214–226. [Google Scholar] [CrossRef] [PubMed]
- Oksanen, J.; Blanchet, F.G.; Kindt, R.; Legendre, P.; Minchin, P.R.; O’hara, R.; Simpson, G.L.; Solymos, P.; Stevens, M.H.H.; Wagner, H. Package ‘Vegan’. Community Ecol. Package Version 2022, 2, 1–295. [Google Scholar]
- Schliep, K.P. Phangorn: Phylogenetic Analysis in R. Bioinformatics 2011, 27, 592–593. [Google Scholar] [CrossRef]
- Loveless, B.J.; Saier, M.H. A Novel Family of Channel-Forming, Autotransporting, Bacterial Virulence Factors. Mol. Membr. Biol. 1997, 14, 113–123. [Google Scholar] [CrossRef]
- Torres, A.G.; Perna, N.T.; Burland, V.; Ruknudin, A.; Blattner, F.R.; Kaper, J.B. Characterization of Cah, a Calcium-Binding and Heat-Extractable Autotransporter Protein of Enterohaemorrhagic Escherichia coli. Mol. Microbiol. 2002, 45, 951–966. [Google Scholar] [CrossRef]
- Maheux, A.F.; Boudreau, D.K.; Bergeron, M.G.; Rodriguez, M.J. Characterization of Escherichia Fergusonii and Escherichia Albertii Isolated from Water. J. Appl. Microbiol. 2014, 117, 597–609. [Google Scholar] [CrossRef]
- Wattam, A.R.; Davis, J.J.; Assaf, R.; Boisvert, S.; Brettin, T.; Bun, C.; Conrad, N.; Dietrich, E.M.; Disz, T.; Gabbard, J.L.; et al. Improvements to PATRIC, the All-Bacterial Bioinformatics Database and Analysis Resource Center. Nucleic Acids Res. 2017, 45, D535–D542. [Google Scholar] [CrossRef] [PubMed]
- Beghain, J.; Bridier-Nahmias, A.; Le Nagard, H.; Denamur, E.; Clermont, O. ClermonTyping: An Easy-to-Use and Accurate in Silico Method for Escherichia Genus Strain Phylotyping. Microb. Genom. 2018, 4, e000192. [Google Scholar] [CrossRef] [PubMed]
- Denamur, E.; Clermont, O.; Bonacorsi, S.; Gordon, D. The Population Genetics of Pathogenic Escherichia coli. Nat. Rev. Microbiol. 2021, 19, 37–54. [Google Scholar] [CrossRef] [PubMed]
- Desvaux, M.; Dalmasso, G.; Beyrouthy, R.; Barnich, N.; Delmas, J.; Bonnet, R. Pathogenicity Factors of Genomic Islands in Intestinal and Extraintestinal Escherichia coli. Front. Microbiol. 2020, 11, 2065. [Google Scholar] [CrossRef]
- Nwoko, E.Q.A.; Okeke, I.N. Bacteria Autoaggregation: How and Why Bacteria Stick Together. Biochem. Soc. Trans. 2021, 49, 1147–1157. [Google Scholar] [CrossRef]
- Ulett, G.C.; Valle, J.; Beloin, C.; Sherlock, O.; Ghigo, J.M.; Schembri, M.A. Functional Analysis of Antigen 43 in Uropathogenic Escherichia coli Reveals a Role in Long-Term Persistence in the Urinary Tract. Infect. Immun. 2007, 75, 3233–3244. [Google Scholar] [CrossRef]
- Lüthje, P.; Brauner, A. Ag43 Promotes Persistence of Uropathogenic Escherichia coli Isolates in the Urinary Tract. J. Clin. Microbiol. 2010, 48, 2316–2317. [Google Scholar] [CrossRef]
- de Luna, M.G.; Scott-Tucker, A.; Desvaux, M.; Ferguson, P.; Morin, N.P.; Dudley, E.G.; Turner, S.; Nataro, J.P.; Owen, P.; Henderson, I.R. The Escherichia coli Biofilm-Promoting Protein Antigen 43 Does Not Contribute to Intestinal Colonization. FEMS Microbiol. Lett. 2008, 284, 237–246. [Google Scholar] [CrossRef]
- Fexby, S.; Bjarnsholt, T.; Jensen, P.O.; Roos, V.; Hoiby, N.; Givskov, M.; Klemm, P. Biological Trojan Horse: Antigen 43 Provides Specific Bacterial Uptake and Survival in Human Neutrophils. Infect. Immun. 2007, 75, 30–34. [Google Scholar] [CrossRef]
- Kitts, P.A.; Church, D.M.; Thibaud-Nissen, F.; Choi, J.; Hem, V.; Sapojnikov, V.; Smith, R.G.; Tatusova, T.; Xiang, C.; Zherikov, A.; et al. Assembly: A Resource for Assembled Genomes at NCBI. Nucleic Acids Res. 2016, 44, D73–D80. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and Applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Godzik, A. Cd-Hit: A Fast Program for Clustering and Comparing Large Sets of Protein or Nucleotide Sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Godzik, A. VISSA: A Program to Visualize Structural Features from Structure Sequence Alignment. Bioinformatics 2006, 22, 887–888. [Google Scholar] [CrossRef] [PubMed]
- Almagro Armenteros, J.J.; Tsirigos, K.D.; Sonderby, C.K.; Petersen, T.N.; Winther, O.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 5.0 Improves Signal Peptide Predictions Using Deep Neural Networks. Nat. Biotechnol. 2019, 37, 420–423. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Attwood, T.K.; Babbitt, P.C.; Bateman, A.; Bork, P.; Bridge, A.J.; Chang, H.Y.; Dosztanyi, Z.; El-Gebali, S.; Fraser, M.; et al. InterPro in 2017-beyond Protein Family and Domain Annotations. Nucleic Acids Res. 2017, 45, D190–D199. [Google Scholar] [CrossRef]
- Jones, P.; Binns, D.; Chang, H.Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-Scale Protein Function Classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef]
- Notredame, C.; Higgins, D.G.; Heringa, J. T-Coffee: A Novel Method for Fast and Accurate Multiple Sequence Alignment. J. Mol. Biol. 2000, 302, 205–217. [Google Scholar] [CrossRef]
- Di Tommaso, P.; Moretti, S.; Xenarios, I.; Orobitg, M.; Montanyola, A.; Chang, J.M.; Taly, J.F.; Notredame, C. T-Coffee: A Web Server for the Multiple Sequence Alignment of Protein and RNA Sequences Using Structural Information and Homology Extension. Nucleic Acids Res. 2011, 39, W13–W17. [Google Scholar] [CrossRef]
- Magis, C.; Taly, J.F.; Bussotti, G.; Chang, J.M.; Di Tommaso, P.; Erb, I.; Espinosa-Carrasco, J.; Notredame, C. T-Coffee: Tree-Based Consistency Objective Function for Alignment Evaluation. Methods Mol. Biol. 2014, 1079, 117–129. [Google Scholar] [CrossRef]
- Armougom, F.; Moretti, S.; Poirot, O.; Audic, S.; Dumas, P.; Schaeli, B.; Keduas, V.; Notredame, C. Expresso: Automatic Incorporation of Structural Information in Multiple Sequence Alignments Using 3D-Coffee. Nucleic Acids Res. 2006, 34, W604–W608. [Google Scholar] [CrossRef]
- O’Sullivan, O.; Suhre, K.; Abergel, C.; Higgins, D.G.; Notredame, C. 3DCoffee: Combining Protein Sequences and Structures within Multiple Sequence Alignments. J. Mol. Biol. 2004, 340, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Berman, H.M.; Bhat, T.N.; Bourne, P.E.; Feng, Z.; Gilliland, G.; Weissig, H.; Westbrook, J. The Protein Data Bank and the Challenge of Structural Genomics. Nat. Struct. Biol. 2000, 7, 957–959. [Google Scholar] [CrossRef] [PubMed]
- Barnard, T.J.; Gumbart, J.; Peterson, J.H.; Noinaj, N.; Easley, N.C.; Dautin, N.; Kuszak, A.J.; Tajkhorshid, E.; Bernstein, H.D.; Buchanan, S.K. Molecular Basis for the Activation of a Catalytic Asparagine Residue in a Self-Cleaving Bacterial Autotransporter. J. Mol. Biol. 2012, 415, 128–142. [Google Scholar] [CrossRef] [PubMed]
- van den Berg, B. Crystal Structure of a Full-Length Autotransporter. J. Mol. Biol. 2010, 396, 627–633. [Google Scholar] [CrossRef] [PubMed]
- Tajima, N.; Kawai, F.; Park, S.Y.; Tame, J.R. A Novel Intein-like Autoproteolytic Mechanism in Autotransporter Proteins. J. Mol. Biol. 2010, 402, 645–656. [Google Scholar] [CrossRef]
- Oomen, C.J.; van Ulsen, P.; van Gelder, P.; Feijen, M.; Tommassen, J.; Gros, P. Structure of the Translocator Domain of a Bacterial Autotransporter. EMBO J. 2004, 23, 1257–1266. [Google Scholar] [CrossRef]
- Kloepper, T.H.; Huson, D.H. Drawing Explicit Phylogenetic Networks and Their Integration into SplitsTree. BMC Evol. Biol. 2008, 8, 22. [Google Scholar] [CrossRef]
- Mangiafico, S.S. Summary and Analysis of Extension Program Evaluation in R. version 1.20.01. Rutgers Cooperative Extension, New Brunswick. 2016. Available online: https://rcompanion.org/documents/RHandbookProgramEvaluation.pdf (accessed on 5 March 2023).
- Mangiafico, S.S. An R Companion for the Handbook of Biological Statistics; version 1.3.4. Rutgers Cooperative Extension, New Brunswick. 2015. Available online: https://rcompanion.org/documents/RCompanionBioStatistics.pdf (accessed on 5 March 2023).
- Dray, S.; Dufour, A.-B. The Ade4 Package: Implementing the Duality Diagram for Ecologists. J. Stat. Softw. 2007, 22, 1–20. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ageorges, V.; Wawrzyniak, I.; Ruiz, P.; Bicep, C.; Zorgani, M.A.; Paxman, J.J.; Heras, B.; Henderson, I.R.; Leroy, S.; Bailly, X.; et al. Genome-Wide Analysis of Antigen 43 (Ag43) Variants: New Insights in Their Diversity, Distribution and Prevalence in Bacteria. Int. J. Mol. Sci. 2023, 24, 5500. https://doi.org/10.3390/ijms24065500
Ageorges V, Wawrzyniak I, Ruiz P, Bicep C, Zorgani MA, Paxman JJ, Heras B, Henderson IR, Leroy S, Bailly X, et al. Genome-Wide Analysis of Antigen 43 (Ag43) Variants: New Insights in Their Diversity, Distribution and Prevalence in Bacteria. International Journal of Molecular Sciences. 2023; 24(6):5500. https://doi.org/10.3390/ijms24065500
Chicago/Turabian StyleAgeorges, Valentin, Ivan Wawrzyniak, Philippe Ruiz, Cédric Bicep, Mohamed A. Zorgani, Jason J. Paxman, Begoña Heras, Ian R. Henderson, Sabine Leroy, Xavier Bailly, and et al. 2023. "Genome-Wide Analysis of Antigen 43 (Ag43) Variants: New Insights in Their Diversity, Distribution and Prevalence in Bacteria" International Journal of Molecular Sciences 24, no. 6: 5500. https://doi.org/10.3390/ijms24065500