
Transcriptome Analysis of Diffuse Large B-Cell Lymphoma Cells Inducibly Expressing MyD88 L265P Mutation Identifies Upregulated CD44, LGALS3, NFKBIZ, and BATF as Downstream Targets of Oncogenic NF-κB Signaling
 , , , , ,
, , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
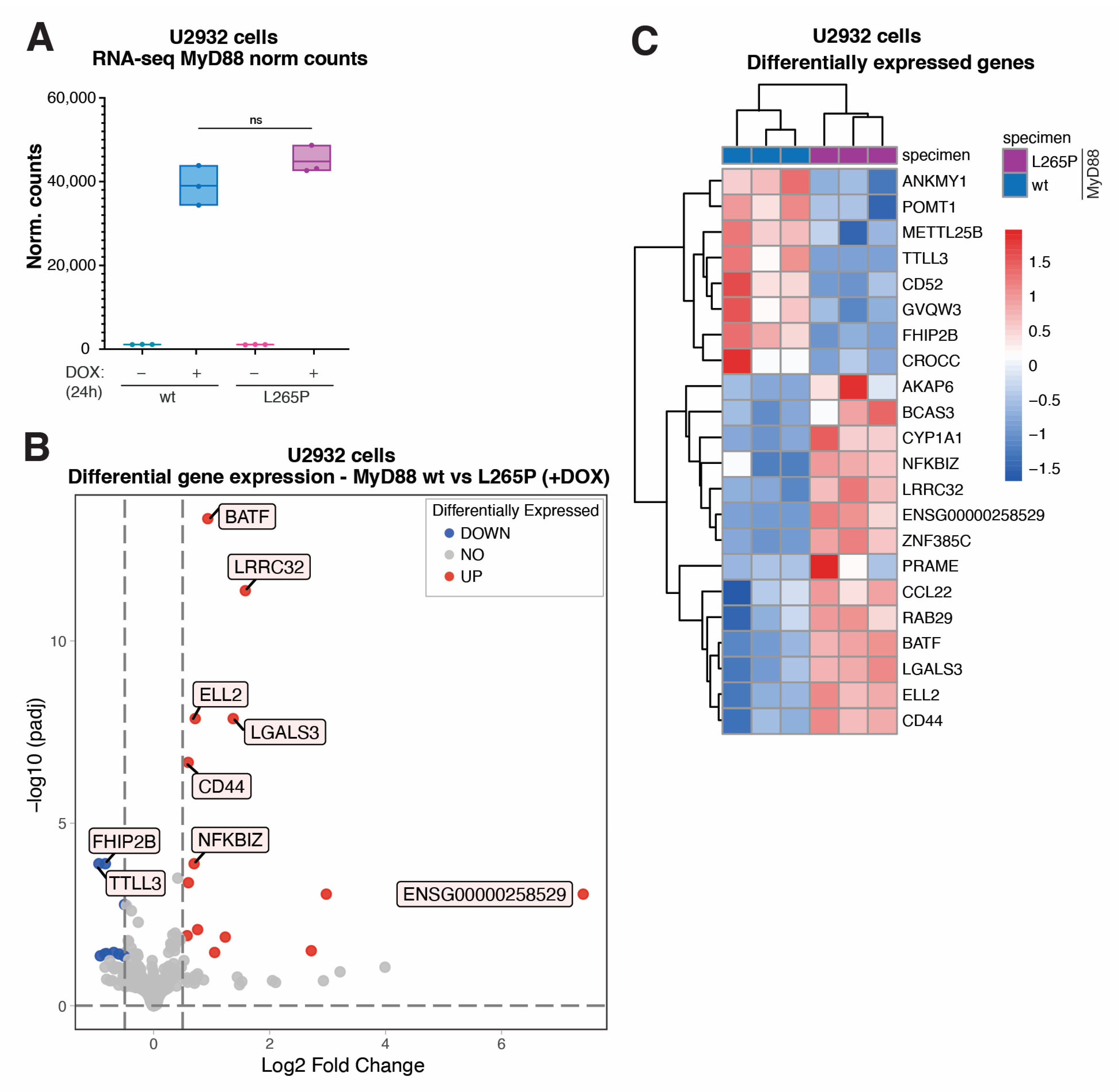
2.1. Lentiviral Inducible System Makes Acute MyD88L265P Expression Possible in Lymphoma Cells to Study Early Events of NF-κB-Mediated Cell Transformation
2.2. Transcriptome Analysis (RNA-Seq) of Genes Differentially Expressed in U2932 Lymphoma Cells Acutely Expressing MyD88L265P
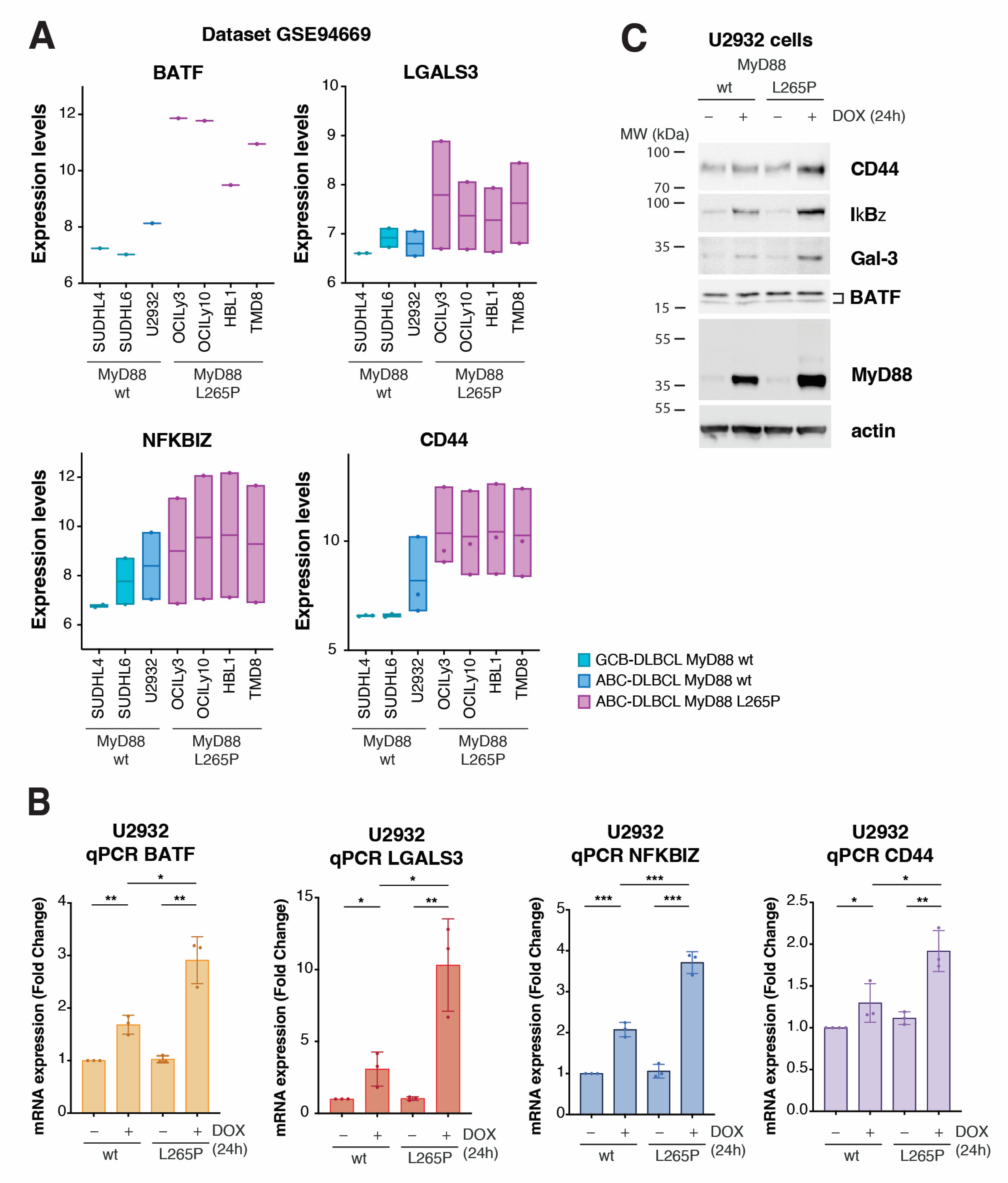
2.3. Validation of Top Upregulated Genes Identified with RNA-Seq Analysis Using Public Expression Datasets and with qPCR and Western Blotting
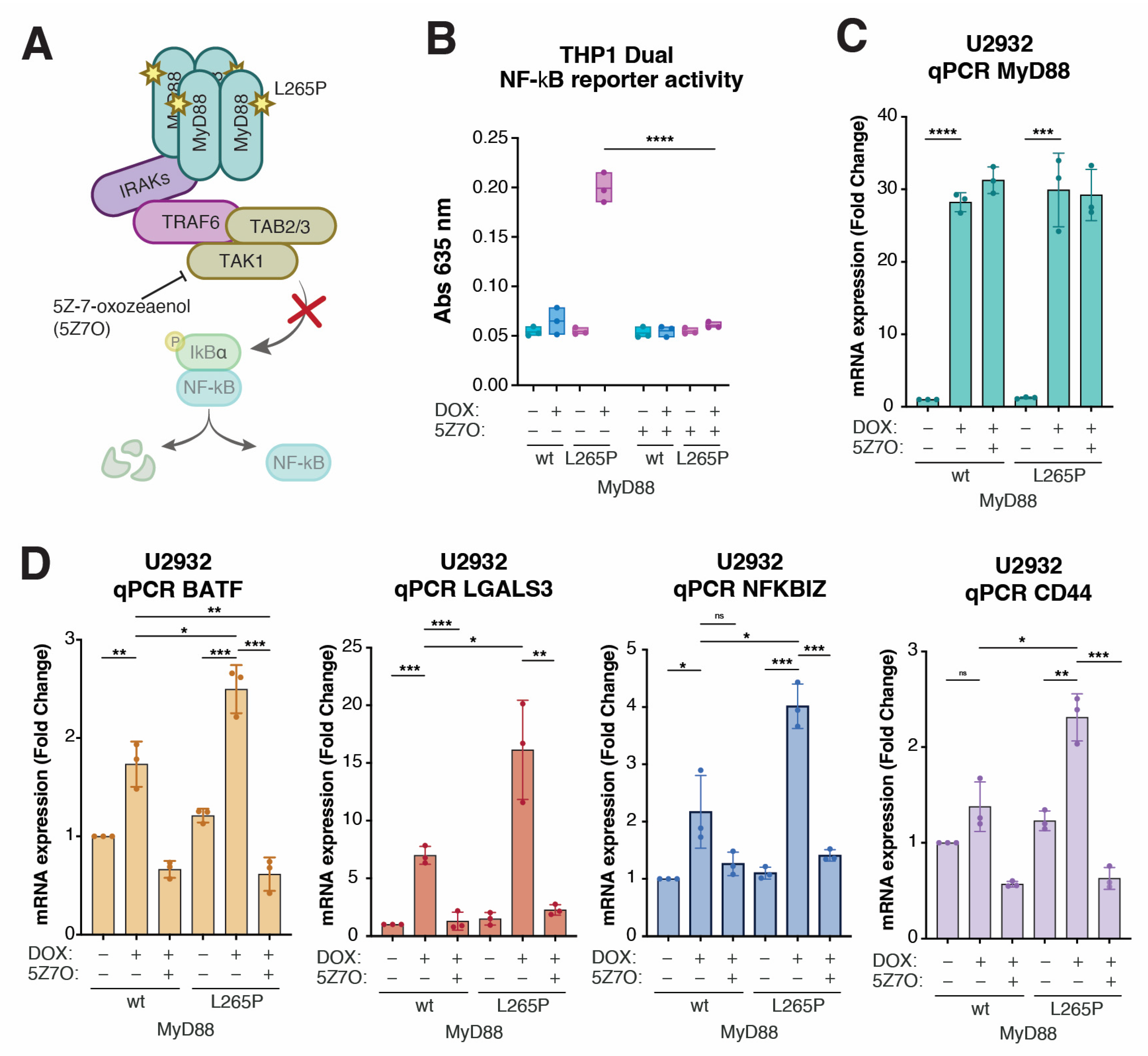
2.4. CD44 Is a Downstream Target of Oncogenic NF-κB Signaling in MyD88L265P-Expressing Lymphoma Cells
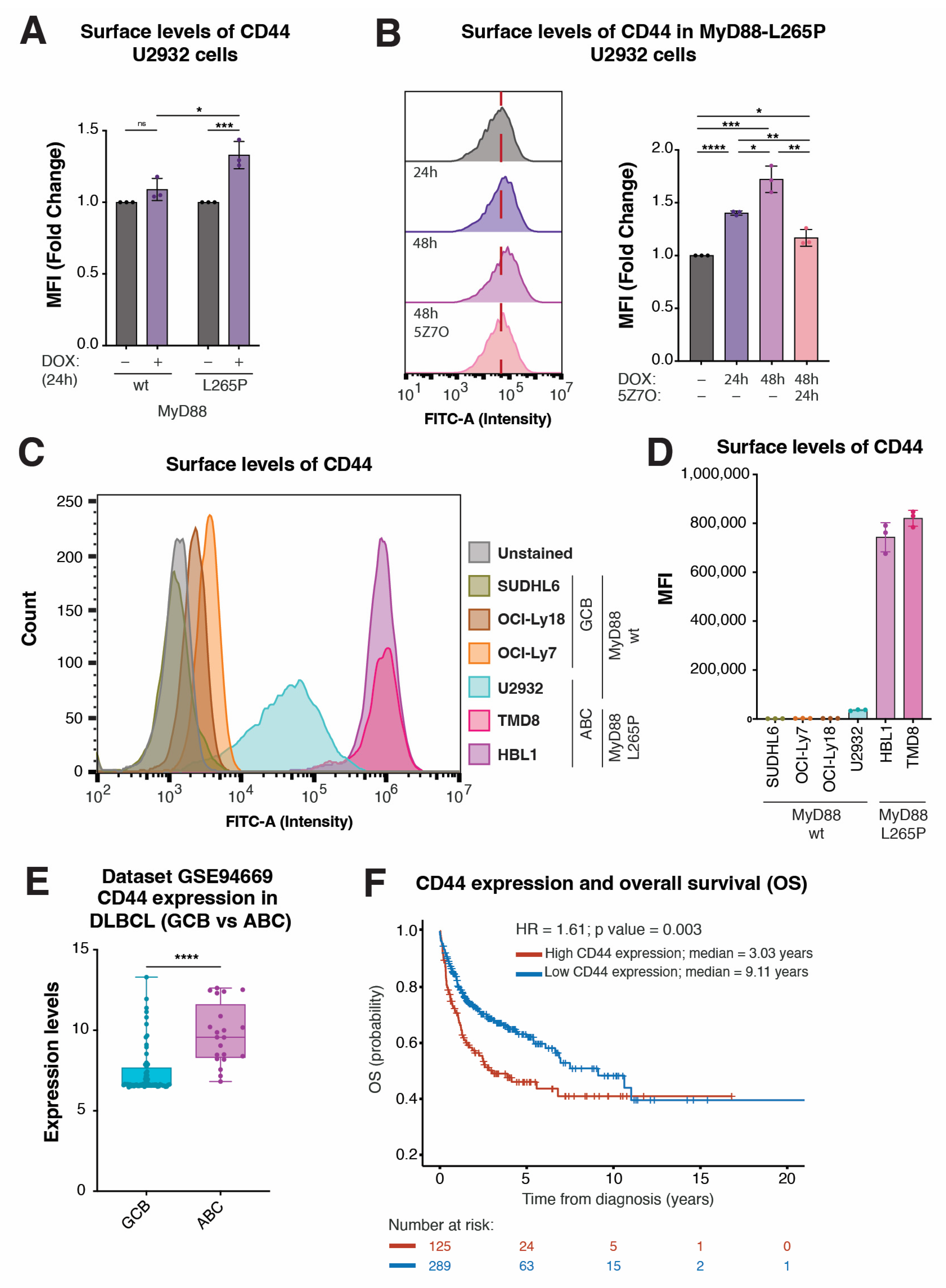
2.5. CD44 Surface Levels Are Correlated with NF-κB-Activating MyD88L265P Expression, and CD44 Expression Stratifies DLBCL Subsets and Predicts Overall Survival in DLBCL Patients
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Cloning
4.3. Lentiviral Particle Production and Cell Transduction
4.4. Western Blotting
4.5. THP1 Dual Reporter Assay
4.6. RNA Isolation
4.7. RNA Sequencing and Transcriptome Analysis
4.8. Quantitative Real-Time Polymerase Chain Reaction
4.9. Data Analysis for RNA-Seq Validation
4.10. CD44 Surface Phenotype
4.11. Survival Analysis
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Deguine, J.; Barton, G.M. MyD88: A central player in innate immune signaling. F1000Prime Rep. 2014, 6, 97. [Google Scholar] [CrossRef] [PubMed]
- Gay, N.J.; Symmons, M.F.; Gangloff, M.; Bryant, C.E. Assembly and localization of Toll-like receptor signalling complexes. Nat. Rev. Immunol. 2014, 14, 546–558. [Google Scholar] [CrossRef] [PubMed]
- Motshwene, P.G.; Moncrieffe, M.C.; Grossmann, J.G.; Kao, C.; Ayaluru, M.; Sandercock, A.M.; Robinson, C.V.; Latz, E.; Gay, N.J. An Oligomeric Signaling Platform Formed by the Toll-like Receptor Signal Transducers MyD88 and IRAK-4. J. Biol. Chem. 2009, 284, 25404–25411. [Google Scholar] [CrossRef] [PubMed]
- Ngo, V.N.; Young, R.M.; Schmitz, R.; Jhavar, S.; Xiao, W.; Lim, K.-H.; Kohlhammer, H.; Xu, W.; Yang, Y.; Zhao, H.; et al. Oncogenically active MYD88 mutations in human lymphoma. Nature 2011, 470, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Akira, S. TLR signaling pathways. Semin. Immunol. 2004, 16, 3–9. [Google Scholar] [CrossRef]
- Picard, C.; Casanova, J.L.; Puel, A. Infectious Diseases in Patients with IRAK-4, MyD88, NEMO, or IκBα Deficiency. Clin. Microbiol. Rev. 2011, 24, 490–497. [Google Scholar] [CrossRef] [PubMed]
- Yun, S.; Johnson, A.C.; Okolo, O.N.; Arnold, S.J.; McBride, A.; Zhang, L.; Baz, R.C.; Anwer, F. Waldenström Macroglobulinemia: Review of Pathogenesis and Management. Clin. Lymphoma Myeloma Leuk. 2017, 17, 252–262. [Google Scholar] [CrossRef]
- Yang, G.; Wang, J.; Tan, L.; Munshi, M.; Liu, X.; Kofides, A.; Chen, J.G.; Tsakmaklis, N.; Demos, M.G.; Guerrera, M.L.; et al. The HCK/BTK inhibitor KIN-8194 is active in MYD88-driven lymphomas and overcomes mutated BTKCys481 ibrutinib resistance. Blood 2021, 138, 1966–1979. [Google Scholar] [CrossRef]
- Cohen, P.; Kelsall, I.R.; Nanda, S.K.; Zhang, J. HOIL-1, an atypical E3 ligase that controls MyD88 signalling by forming ester bonds between ubiquitin and components of the Myddosome. Adv. Biol. Regul. 2020, 75, 100666. [Google Scholar] [CrossRef]
- Yang, Y.; Schmitz, R.; Mitala, J.; Whiting, A.; Xiao, W.; Ceribelli, M.; Wright, G.W.; Zhao, H.; Yang, Y.; Xu, W.; et al. Essential Role of the Linear Ubiquitin Chain Assembly Complex in Lymphoma Revealed by Rare Germline Polymorphisms. Cancer Discov. 2014, 4, 480–493. [Google Scholar] [CrossRef]
- Dobashi, A. Molecular Pathogenesis of Diffuse Large B-Cell Lymphoma. J. Clin. Exp. Hematop. 2016, 56, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Krappmann, D.; Vincendeau, M. Mechanisms of NF-κB deregulation in lymphoid malignancies. Semin. Cancer Biol. 2016, 39, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Li, W.; Deng, Q.; Li, L.; Hsi, E.D.; Young, K.H.; Zhang, M.; Li, Y. MYD88 L265P Mutation in Lymphoid Malignancies. Cancer Res 2018, 78, 2457–2462. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Q.; Jeelall, Y.S.; Ferguson, L.L.; Horikawa, K. Toll-Like Receptors and Cancer: MYD88 Mutation and Inflammation. Front. Immunol. 2014, 5, 367. [Google Scholar] [CrossRef]
- Weber, A.N.R.; Gloria, Y.C.; Çınar, Ö.; Reinhardt, H.C.; Pezzutto, A.; Wolz, O.O. Oncogenic MYD88 mutations in lymphoma: Novel insights and therapeutic possibilities. Cancer Immunol. Immunother. 2018, 67, 1797–1807. [Google Scholar] [CrossRef]
- Treon, S.P.; Xu, L.; Yang, G.; Zhou, Y.; Liu, X.; Cao, Y.; Sheehy, P.; Manning, R.J.; Patterson, C.J.; Tripsas, C.; et al. MYD88 L265P Somatic Mutation in Waldenström’s Macroglobulinemia. N. Engl. J. Med. 2012, 367, 826–833. [Google Scholar] [CrossRef]
- Varettoni, M.; Arcaini, L.; Zibellini, S.; Boveri, E.; Rattotti, S.; Riboni, R.; Corso, A.; Orlandi, E.; Bonfichi, M.; Gotti, M.; et al. Prevalence and clinical significance of the MYD88 (L265P) somatic mutation in Waldenström’s macroglobulinemia and related lymphoid neoplasms. Blood 2013, 121, 2522–2528. [Google Scholar] [CrossRef]
- de Groen, R.A.L.; Schrader, A.M.R.; Kersten, M.J.; Pals, S.T.; Vermaat, J.S.P. MYD88 in the driver’s seat of B-cell lymphomagenesis: From molecular mechanisms to clinical implications. Haematologica 2019, 104, 2337–2348. [Google Scholar] [CrossRef]
- Loiarro, M.; Volpe, E.; Ruggiero, V.; Gallo, G.; Furlan, R.; Maiorino, C.; Battistini, L.; Sette, C. Mutational Analysis Identifies Residues Crucial for Homodimerization of Myeloid Differentiation Factor 88 (MyD88) and for Its Function in Immune Cells. J. Biol. Chem. 2013, 288, 30210–30222. [Google Scholar] [CrossRef]
- Avbelj, M.; Wolz, O.O.; Fekonja, O.; Benčina, M.; Repič, M.; Mavri, J.; Krüger, J.; Schärfe, C.; Garcia, M.D.; Panter, G.; et al. Activation of lymphoma-associated MyD88 mutations via allostery-induced TIR-domain oligomerization. Blood 2014, 124, 3896. [Google Scholar] [CrossRef]
- Wang, J.Q.; Jeelall, Y.S.; Beutler, B.; Horikawa, K.; Goodnow, C.C. Consequences of the recurrent MYD88L265P somatic mutation for B cell tolerance. J. Exp. Med. 2014, 211, 413–426. [Google Scholar] [CrossRef]
- Knittel, G.; Liedgens, P.; Korovkina, D.; Seeger, J.M.; Al-Baldawi, Y.; Al-Maarri, M.; Fritz, C.; Vlantis, K.; Bezhanova, S.; Scheel, A.H.; et al. B-cell–specific conditional expression of Myd88p.L252P leads to the development of diffuse large B-cell lymphoma in mice. Blood 2016, 127, 2732–2741. [Google Scholar] [CrossRef]
- Sewastianik, T.; Guerrera, M.L.; Adler, K.; Dennis, P.S.; Wright, K.; Shanmugam, V.; Huang, Y.; Tanton, H.; Jiang, M.; Kofides, A.; et al. Human MYD88L265P is insufficient by itself to drive neoplastic transformation in mature mouse B cells. Blood Adv. 2019, 3, 3360–3374. [Google Scholar] [CrossRef]
- Rodriguez, S.; Celay, J.; Goicoechea, I.; Jimenez, C.; Botta, C.; Garcia-Barchino, M.J.; Garces, J.J.; Larrayoz, M.; Santos, S.; Alignani, D.; et al. Preneoplastic somatic mutations including MYD88 L265P in lymphoplasmacytic lymphoma. Sci. Adv. 2022, 8. [Google Scholar] [CrossRef]
- Flümann, R.; Rehkämper, T.; Nieper, P.; Pfeiffer, P.; Holzem, A.; Klein, S.; Bhatia, S.; Kochanek, M.; Kisis, I.; Pelzer, B.W.; et al. An Autochthonous Mouse Model of Myd88- and BCL2-Driven Diffuse Large B-cell Lymphoma Reveals Actionable Molecular Vulnerabilities. Blood Cancer Discov. 2021, 2, 70–91. [Google Scholar] [CrossRef]
- Phelan, J.D.; Young, R.M.; Webster, D.E.; Roulland, S.; Wright, G.W.; Kasbekar, M.; Shaffer, A.L.; Ceribelli, M.; Wang, J.Q.; Schmitz, R.; et al. A multiprotein supercomplex controlling oncogenic signalling in lymphoma. Nature 2018, 560, 387–391. [Google Scholar] [CrossRef]
- Yang, G.; Zhou, Y.; Liu, X.; Xu, L.; Cao, Y.; Manning, R.J.; Patterson, C.J.; Buhrlage, S.J.; Gray, N.; Tai, Y.T.; et al. A mutation in MYD88 (L265P) supports the survival of lymphoplasmacytic cells by activation of Bruton tyrosine kinase in Waldenström macroglobulinemia. Blood 2013, 122, 1222–1232. [Google Scholar] [CrossRef]
- An, B.; Zhu, S.; Li, T.; Wu, J.; Zang, G.; Lv, X.; Qiao, Y.; Huang, J.; Shao, Y.; Cui, J.; et al. A Dual TLR7/TLR9 Inhibitor HJ901 Inhibits ABC-DLBCL Expressing the MyD88 L265P Mutation. Front. Cell Dev. Biol. 2020, 8, 262. [Google Scholar] [CrossRef]
- Dubois, S.; Viailly, P.J.; Bohers, E.; Bertrand, P.; Ruminy, P.; Marchand, V.; Maingonnat, C.; Mareschal, S.; Picquenot, J.M.; Penther, D.; et al. Biological and Clinical Relevance of Associated Genomic Alterations in MYD88 L265P and non-L265P–Mutated Diffuse Large B-Cell Lymphoma: Analysis of 361 Cases. Clin. Cancer Res. 2017, 23, 2232–2244. [Google Scholar] [CrossRef]
- Radke, J.; Ishaque, N.; Koll, R.; Gu, Z.; Schumann, E.; Sieverling, L.; Uhrig, S.; Hübschmann, D.; Toprak, U.H.; López, C.; et al. The genomic and transcriptional landscape of primary central nervous system lymphoma. Nat. Commun. 2022, 13, 2558. [Google Scholar] [CrossRef]
- Schmitz, R.; Wright, G.W.; Huang, D.W.; Johnson, C.A.; Phelan, J.D.; Wang, J.Q.; Roulland, S.; Kasbekar, M.; Young, R.M.; Shaffer, A.L.; et al. Genetics and Pathogenesis of Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2018, 378, 1396–1407. [Google Scholar] [CrossRef] [PubMed]
- Tarantelli, C.; Gaudio, E.; Arribas, A.J.; Kwee, I.; Hillmann, P.; Rinaldi, A.; Cascione, L.; Spriano, F.; Bernasconi, E.; Guidetti, F.; et al. PQR309 Is a Novel Dual PI3K/mTOR Inhibitor with Preclinical Antitumor Activity in Lymphomas as a Single Agent and in Combination Therapy. Clin. Cancer Res. 2018, 24, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Alcoceba, M.; García-Álvarez, M.; Medina, A.; Maldonado, R.; González-Calle, V.; Chillón, M.C.; Sarasquete, M.E.; González, M.; García-Sanz, R.; Jiménez, C. MYD88 Mutations: Transforming the Landscape of IgM Monoclonal Gammopathies. Int. J. Mol. Sci. 2022, 23, 5570. [Google Scholar] [CrossRef] [PubMed]
- Axelrod, H.D.; Valkenburg, K.C.; Amend, S.R.; Hicks, J.L.; Parsana, P.; Torga, G.; De Marzo, A.M.; Pienta, K.J. AXL Is a Putative Tumor Suppressor and Dormancy Regulator in Prostate Cancer. Mol. Cancer Res. 2019, 17, 356–369. [Google Scholar] [CrossRef]
- Feng, Y.; Duan, T.; Du, Y.; Jin, S.; Wang, M.; Cui, J.; Wang, R.F. LRRC25 Functions as an Inhibitor of NF-κB Signaling Pathway by Promoting p65/RelA for Autophagic Degradation. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef]
- Chen, Y.; Cao, B.; Zheng, W.; Sun, Y.; Xu, T. eIF3k inhibits NF-κB signaling by targeting MyD88 for ATG5-mediated autophagic degradation in teleost fish. J. Biol. Chem. 2022, 298, 101730. [Google Scholar] [CrossRef]
- Tanimura, A.; Nakazato, A.; Tanaka, N. MYD88 signals induce tumour-initiating cell generation through the NF-κB-HIF-1α activation cascade. Sci. Rep. 2021, 11, 3991. [Google Scholar] [CrossRef]
- Boudesco, C.; Verhoeyen, E.; Martin, L.; Chassagne-Clement, C.; Salmi, L.; Mhaidly, R.; Pangault, C.; Fest, T.; Ramla, S.; Jardin, F.; et al. HSP110 sustains chronic NF-κB signaling in activated B-cell diffuse large B-cell lymphoma through MyD88 stabilization. Blood 2018, 132, 510–520. [Google Scholar] [CrossRef]
- Poulain, S.; Roumier, C.; Decambron, A.; Renneville, A.; Herbaux, C.; Bertrand, E.; Tricot, S.; Daudignon, A.; Galiègue-Zouitina, S.; Soenen, V.; et al. MYD88 L265P mutation in Waldenstrom macroglobulinemia. Blood 2013, 121, 4504–4511. [Google Scholar] [CrossRef]
- Schafer, A.R.M.; Smith, J.L.; Pryke, K.M.; DeFilippis, V.R.; Hirsch, A.J. The E3 Ubiquitin Ligase SIAH1 Targets MyD88 for Proteasomal Degradation During Dengue Virus Infection. Front. Microbiol. 2020, 11, 24. [Google Scholar] [CrossRef]
- Li, Q.; Wang, F.; Wang, Q.; Zhang, N.; Zheng, J.; Zheng, M.; Liu, R.; Cui, H.; Wen, J.; Zhao, G. SPOP promotes ubiquitination and degradation of MyD88 to suppress the innate immune response. PLoS Pathog. 2020, 16, e1008188. [Google Scholar] [CrossRef]
- Cohen, P.; Strickson, S. The role of hybrid ubiquitin chains in the MyD88 and other innate immune signalling pathways. Cell Death Differ. 2017, 24, 1153–1159. [Google Scholar] [CrossRef]
- Yu, X.; Li, W.; Deng, Q.; Liu, H.; Wang, X.; Hu, H.; Cao, Y.; Xu-Monette, Z.Y.; Li, L.; Zhang, M.; et al. MYD88 L265P elicits mutation-specific ubiquitination to drive NF-κB activation and lymphomagenesis. Blood 2021, 137, 1615–1627. [Google Scholar] [CrossRef]
- Morman, R.E.; Schweickert, P.G.; Konieczny, S.F.; Taparowsky, E.J. BATF regulates the expression of Nfil3, Wnt10a and miR155hg for efficient induction of antibody class switch recombination in mice. Eur. J. Immunol. 2018, 48, 1492. [Google Scholar] [CrossRef]
- Nogai, H.; Wenzel, S.S.; Hailfinger, S.; Grau, M.; Kaergel, E.; Seitz, V.; Wollert-Wulf, B.; Pfeifer, M.; Wolf, A.; Frick, M.; et al. IκB-ζ controls the constitutive NF-κB target gene network and survival of ABC DLBCL. Blood 2013, 122, 2242–2250. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 1–9. [Google Scholar] [CrossRef]
- Guan, S.; Lu, J.; Zhao, Y.; Woodfield, S.E.; Zhang, H.; Xu, X.; Yu, Y.; Zhao, J.; Bieerkehazhi, S.; Liang, H.; et al. TAK1 inhibitor 5Z-7-oxozeaenol sensitizes cervical cancer to doxorubicin-induced apoptosis. Oncotarget 2017, 8, 33666–33675. [Google Scholar] [CrossRef]
- Feng, S.; Wang, K.; Shao, Z.; Lin, Q.; Li, B.; Liu, P. MiR-373/miR-520s-CD44 Axis Significantly Inhibits the Growth and Invasion of Human Glioblastoma Cells. Arch. Med Res. 2022, 53, 550–561. [Google Scholar] [CrossRef] [PubMed]
- Markasz, L.; Savani, R.C.; Jonzon, A.; Sindelar, R. CD44 and RHAMM expression patterns in the human developing lung. Pediatr. Res. 2020, 89, 134–142. [Google Scholar] [CrossRef]
- Zhang, X.; Yang, L.; Lei, W.; Hou, Q.; Huang, M.; Zhou, R.; Enver, T.; Wu, S. Single-cell sequencing reveals CD133+CD44−-originating evolution and novel stemness related variants in human colorectal cancer. Ebiomedicine 2022, 82. [Google Scholar] [CrossRef]
- Zhang, H.; Brown, R.L.; Wei, Y.; Zhao, P.; Liu, S.; Liu, X.; Deng, Y.; Hu, X.; Zhang, J.; Gao, X.D.; et al. CD44 splice isoform switching determines breast cancer stem cell state. Genes Dev. 2019, 33, 166–179. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Xue, C.; Yu, Y.; Chen, J.; Chen, X.; Ren, F.; Ren, Z.; Cui, G.; Sun, R. CD44 is overexpressed and correlated with tumor progression in gallbladder cancer. Cancer Manag. Res. 2018, 10, 3857–3865. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Niu, M.; Yuan, X.; Wu, K.; Liu, A. CD44 as a tumor biomarker and therapeutic target. Exp. Hematol. Oncol. 2020, 9, 1–14. [Google Scholar] [CrossRef]
- Senbanjo, L.T.; Chellaiah, M.A. CD44: A Multifunctional Cell Surface Adhesion Receptor Is a Regulator of Progression and Metastasis of Cancer Cells. Front. Cell Dev. Biol. 2017, 5, 18. [Google Scholar] [CrossRef]
- Stauder, R.; Eisterer, W.; Thaler, J.; Gunthert, U. CD44 variant isoforms in non-Hodgkin’s lymphoma: A new independent prognostic factor. Blood 1995, 85, 2885–2899. [Google Scholar] [CrossRef]
- Higashi, M.; Sugaya, Y.; Soeta, S.; Yokota, A.; Ishii, G.; Harigaya, K. CD44 expression during tumor progression of follicular lymphoma. Oncol. Rep. 2009, 22, 1135–1140. [Google Scholar] [CrossRef]
- Espasa, A.; Tapia, G.; Vergara, S.; Raya, M.; Juncà, J.; Sorigue, M. Flow cytometric expression of CD71, CD81, CD44 and CD39 in B cell lymphoma. Scand. J. Clin. Lab. Investig. 2021, 81, 413–417. [Google Scholar] [CrossRef]
- Babst, N.; Isbell, L.K.; Rommel, F.; Tura, A.; Ranjbar, M.; Grisanti, S.; Tschuch, C.; Schueler, J.; Doostkam, S.; Reinacher, P.C.; et al. CXCR4, CXCR5 and CD44 May Be Involved in Homing of Lymphoma Cells into the Eye in a Patient Derived Xenograft Homing Mouse Model for Primary Vitreoretinal Lymphoma. Int. J. Mol. Sci. 2022, 23, 11757. [Google Scholar] [CrossRef]
- Eberth, S.; Schneider, B.; Rosenwald, A.; Hartmann, E.M.; Romani, J.; Zaborski, M.; Siebert, R.; Drexler, H.G.; Quentmeier, H. Epigenetic regulation of CD44in Hodgkin and non-Hodgkin lymphoma. BMC Cancer 2010, 10, 517. [Google Scholar] [CrossRef]
- Wallach-Dayan, S.B.; Grabovsky, V.; Moll, J.; Sleeman, J.; Herrlich, P.; Alon, R.; Naor, D. CD44-dependent lymphoma cell dissemination: A cell surface CD44 variant, rather than standard CD44, supports in vitro lymphoma cell rolling on hyaluronic acid substrate and its in vivo accumulation in the peripheral lymph nodes. J. Cell Sci. 2001, 114, 3463–3477. [Google Scholar] [CrossRef]
- Lenz, G.; Wright, G.; Dave, S.S.; Xiao, W.; Powell, J.; Zhao, H.; Xu, W.; Tan, B.; Goldschmidt, N.; Iqbal, J.; et al. Stromal Gene Signatures in Large-B-Cell Lymphomas. N. Eng. J. Med. 2008, 359, 2313–2323. [Google Scholar] [CrossRef] [PubMed]
- Cardesa-Salzmann, T.M.; Colomo, L.; Gutierrez, G.; Chan, W.C.; Weisenburger, D.; Climent, F.; González-Barca, E.; Mercadal, S.; Arenillas, L.; Serrano, S.; et al. High microvessel density determines a poor outcome in patients with diffuse large B-cell lymphoma treated with rituximab plus chemotherapy. Haematologica 2011, 96, 996–1001. [Google Scholar] [CrossRef] [PubMed]
- Prescott, J.A.; Mitchell, J.P.; Cook, S.J. Inhibitory feedback control of NF-κB signalling in health and disease. Biochem. J. 2021, 478, 2619. [Google Scholar] [CrossRef] [PubMed]
- Ruland, J. Return to Homeostasis: Downregulation of NF-ΚB Responses. Nat Immunol 2011, 12, 709–714. [Google Scholar] [CrossRef]
- Wenzl, K.; Manske, M.K.; Sarangi, V.; Asmann, Y.W.; Greipp, P.T.; Schoon, H.R.; Braggio, E.; Maurer, M.J.; Feldman, A.L.; Witzig, T.E.; et al. Loss of TNFAIP3 enhances MYD88L265P-driven signaling in non-Hodgkin lymphoma. Blood Cancer J. 2018, 8, 97. [Google Scholar] [CrossRef]
- Choi, J.W.; Kim, Y.; Lee, J.H.; Kim, Y.S. MYD88 expression and L265P mutation in diffuse large B-cell lymphoma. Hum. Pathol. 2013, 44, 1375–1381. [Google Scholar] [CrossRef]
- Lacy, S.E.; Barrans, S.L.; Beer, P.A.; Painter, D.; Smith, A.G.; Roman, E.; Cooke, S.L.; Ruiz, C.; Glover, P.; Van Hoppe, S.J.L.; et al. Targeted sequencing in DLBCL, molecular subtypes, and outcomes: A Haematological Malignancy Research Network report. Blood 2020, 135, 1759–1771. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, G.; Sui, Y.; Yang, Z.; Chu, Y.; Tang, H.; Guo, B.; Zhang, C.; Wu, C. CD52 Is a Prognostic Biomarker and Associated with Tumor Microenvironment in Breast Cancer. Front. Genet. 2020, 11. [Google Scholar] [CrossRef]
- Rodig, S.J.; Abramson, J.S.; Pinkus, G.S.; Treon, S.P.; Dorfman, D.M.; Dong, H.Y.; Shipp, M.A.; Kutok, J.L. Heterogeneous CD52 Expression among Hematologic Neoplasms: Implications for the Use of Alemtuzumab (CAMPATH-1H). Clin. Cancer Res. 2006, 12, 7174–7179. [Google Scholar] [CrossRef]
- Craig, J.W.; Mina, M.J.; Crombie, J.L.; LaCasce, A.S.; Weinstock, D.M.; Pinkus, G.S.; Pozdnyakova, O. Assessment of CD52 expression in "double-hit" and "double-expressor" lymphomas: Implications for clinical trial eligibility. PLoS ONE 2018, 13, e0199708. [Google Scholar] [CrossRef]
- Teo, E.C.Y.; Chew, Y.; Phipps, C. A review of monoclonal antibody therapies in lymphoma. Crit. Rev. Oncol. 2016, 97, 1–10. [Google Scholar] [CrossRef]
- Matsushita, M.; Yamazaki, R.; Ikeda, H.; Kawakami, Y. Preferentially Expressed Antigen of Melanoma (PRAME) in the Development of Diagnostic and Therapeutic Methods for Hematological Malignancies. Leuk. Lymphoma 2003, 44, 439–444. [Google Scholar] [CrossRef]
- Wadelin, F.; Fulton, J.; McEwan, P.A.; Spriggs, K.A.; Emsley, J.; Heery, D.M. Leucine-rich repeat protein PRAME: Expression, potential functions and clinical implications for leukaemia. Mol. Cancer 2010, 9, 226. [Google Scholar] [CrossRef]
- Kewitz, S.; Staege, M.S. Knock-Down of PRAME Increases Retinoic Acid Signaling and Cytotoxic Drug Sensitivity of Hodgkin Lymphoma Cells. PLoS ONE 2013, 8, e55897. [Google Scholar] [CrossRef]
- Weber, G.; Caruana, I.; Rouce, R.H.; Barrett, A.J.; Gerdemann, U.; Leen, A.M.; Rabin, K.R.; Bollard, C.M. Generation of Tumor Antigen-Specific T Cell Lines from Pediatric Patients with Acute Lymphoblastic Leukemia—Implications for Immunotherapy. Clin. Cancer Res. 2013, 19, 5079–5091. [Google Scholar] [CrossRef]
- Pujol, J.L.; De Pas, T.; Rittmeyer, A.; Vallières, E.; Kubisa, B.; Levchenko, E.; Wiesemann, S.; Masters, G.A.; Shen, R.; Tjulandin, S.A.; et al. Safety and Immunogenicity of the PRAME Cancer Immunotherapeutic in Patients with Resected Non–Small Cell Lung Cancer: A Phase I Dose Escalation Study. J. Thorac. Oncol. 2016, 11, 2208–2217. [Google Scholar] [CrossRef]
- Orlando, D.; Miele, E.; de Angelis, B.; Guercio, M.; Boffa, I.; Sinibaldi, M.; Po, A.; Caruana, I.; Abballe, L.; Carai, A.; et al. Adoptive Immunotherapy Using PRAME-Specific T Cells in Medulloblastoma. Cancer Res 2018, 78, 3337–3349. [Google Scholar] [CrossRef]
- Sun, Z.; Wu, Z.; Zhang, F.; Guo, Q.; Li, L.; Li, K.; Chen, H.; Zhao, J.; Song, D.; Huang, Q.; et al. Prame is critical for breast cancer growth and metastasis. Gene 2016, 594, 160–164. [Google Scholar] [CrossRef]
- Takata, K.; Chong, L.C.; Ennishi, D.; Aoki, T.; Li, M.Y.; Thakur, A.; Healy, S.; Viganò, E.; Dao, T.; Kwon, D.; et al. Tumor-associated antigen PRAME exhibits dualistic functions that are targetable in diffuse large B cell lymphoma. J. Clin. Investig. 2022, 132. [Google Scholar] [CrossRef]
- Martincic, K.; Alkan, S.A.; Cheatle, A.; Borghesi, L.; Milcarek, C. Transcription elongation factor ELL2 directs immunoglobulin secretion in plasma cells by stimulating altered RNA processing. Nat. Immunol. 2009, 10, 1102–1109. [Google Scholar] [CrossRef]
- Park, K.S.; Bayles, I.; Szlachta-McGinn, A.; Paul, J.; Boiko, J.; Santos, P.; Liu, J.; Wang, Z.; Borghesi, L.; Milcarek, C. Transcription Elongation Factor ELL2 Drives Ig Secretory-Specific mRNA Production and the Unfolded Protein Response. J. Immunol. 2014, 193, 4663–4674. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Vos, S.M.; Dienemann, C.; Ninov, M.; Urlaub, H.; Cramer, P. Allosteric transcription stimulation by RNA polymerase II super elongation complex. Mol. Cell 2021, 81, 3386–3399.e10. [Google Scholar] [CrossRef]
- Care, M.; Barrans, S.; Worrillow, L.; Jack, A.; Westhead, D.R.; Tooze, R.M. A Microarray Platform-Independent Classification Tool for Cell of Origin Class Allows Comparative Analysis of Gene Expression in Diffuse Large B-cell Lymphoma. PLoS ONE 2013, 8, e55895. [Google Scholar] [CrossRef] [PubMed]
- Tran, D.Q.; Andersson, J.; Wang, R.; Ramsey, H.; Unutmaz, D.; Shevach, E.M. GARP (LRRC32) is essential for the surface expression of latent TGF-β on platelets and activated FOXP3 + regulatory T cells. Proc. Natl. Acad. Sci. USA 2009, 106, 13445–13450. [Google Scholar] [CrossRef] [PubMed]
- Wallace, C.H.; Wu, B.X.; Salem, M.; Ansa-Addo, E.A.; Metelli, A.; Sun, S.; Gilkeson, G.; Shlomchik, M.J.; Liu, B.; Li, Z. B lymphocytes confer immune tolerance via cell surface GARP-TGF-β complex. J. Clin. Investig. 2018, 3, e99863. [Google Scholar] [CrossRef]
- Bouchard, A.; Collin, B.; Garrido, C.; Bellaye, P.S.; Kohli, E. GARP: A Key Target to Evaluate Tumor Immunosuppressive Microenvironment. Biology 2021, 10, 836. [Google Scholar] [CrossRef]
- Zimmer, N.; Trzeciak, E.R.; Graefen, B.; Satoh, K.; Tuettenberg, A. GARP as a Therapeutic Target for the Modulation of Regulatory T Cells in Cancer and Autoimmunity. Front. Immunol. 2022, 13, 928450. [Google Scholar] [CrossRef]
- Carrillo-Gálvez, A.B.; Quintero, J.E.; Rodríguez, R.; Menéndez, S.T.; González, M.V.; Blanco-Lorenzo, V.; Allonca, E.; Farias, V.D.A.; González-Correa, J.E.; Erill-Sagalés, N.; et al. GARP promotes the proliferation and therapeutic resistance of bone sarcoma cancer cells through the activation of TGF-β. Cell Death Dis. 2020, 11, 1–12. [Google Scholar] [CrossRef]
- Metelli, A.; Wu, B.X.; Fugle, C.W.; Rachidi, S.; Sun, S.; Zhang, Y.; Wu, J.; Tomlinson, S.; Howe, P.H.; Yang, Y.; et al. Surface Expression of TGFβ Docking Receptor GARP Promotes Oncogenesis and Immune Tolerance in Breast Cancer. Cancer Res 2016, 76, 7106–7117. [Google Scholar] [CrossRef]
- Li, A.; Chang, Y.; Song, N.J.; Wu, X.; Chung, D.; Riesenberg, B.P.; Velegraki, M.; Giuliani, G.D.; Das, K.; Okimoto, T.; et al. Selective targeting of GARP-LTGFβ axis in the tumor microenvironment augments PD-1 blockade via enhancing CD8+ T cell antitumor immunity. J. Immunother. Cancer 2022, 10, e005433. [Google Scholar] [CrossRef]
- Korbecki, J.; Kojder, K.; Simińska, D.; Bohatyrewicz, R.; Gutowska, I.; Chlubek, D.; Baranowska-Bosiacka, I. CC Chemokines in a Tumor: A Review of Pro-Cancer and Anti-Cancer Properties of the Ligands of Receptors CCR1, CCR2, CCR3, and CCR4. Int. J. Mol. Sci. 2020, 21, 8412. [Google Scholar] [CrossRef]
- Niens, M.; Visser, L.; Nolte, I.M.; van der Steege, G.; Diepstra, A.; Cordano, P.; Jarrett, R.F.; Meerman, G.J.T.; Poppema, S.; Van Der Berg, A. Serum chemokine levels in Hodgkin lymphoma patients: Highly increased levels of CCL17 and CCL22. Br. J. Haematol. 2008, 140, 527–536. [Google Scholar] [CrossRef]
- Kumai, T.; Nagato, T.; Kobayashi, H.; Komabayashi, Y.; Ueda, S.; Kishibe, K.; Ohkuri, T.; Takahara, M.; Celis, E.; Harabuchi, Y. CCL17 and CCL22/CCR4 signaling is a strong candidate for novel targeted therapy against nasal natural killer/T-cell lymphoma. Cancer Immunol. Immunother. 2015, 64, 697. [Google Scholar] [CrossRef]
- Döring, C.; Hansmann, M.L.; Agostinelli, C.; Piccaluga, P.P.; Facchetti, F.; Pileri, S.; Küppers, R.; Newrzela, S.; Hartmann, S. A novel immunohistochemical classifier to distinguish Hodgkin lymphoma from ALK anaplastic large cell lymphoma. Mod. Pathol. 2014, 27, 1345–1354. [Google Scholar] [CrossRef]
- Takegawa, S.; Jin, Z.; Nakayama, T.; Oyama, T.; Hieshima, K.; Nagakubo, D.; Shirakawa, A.K.; Tsuzuki, T.; Nakamura, S.; Yoshie, O. Expression of CCL17 and CCL22 by latent membrane protein 1-positive tumor cells in age-related Epstein–Barr virus-associated B-cell lymphoproliferative disorder. Cancer Sci. 2008, 99, 296–302. [Google Scholar] [CrossRef]
- Zeng, Q.; Gupta, A.; Xin, L.; Poon, M.; Schwarz, H. Plasma Factors for the Differentiation of Hodgkin’s Lymphoma and Diffused Large B Cell Lymphoma and for Monitoring Remission. J. Hematol. 2019, 8, 47–54. [Google Scholar] [CrossRef]
- Murphy, T.L.; Tussiwand, R.; Murphy, K.M. Specificity through cooperation: BATF–IRF interactions control immune-regulatory networks. Nat. Rev. Immunol. 2013, 13, 499–509. [Google Scholar] [CrossRef]
- Betz, B.C.; Jordan-Williams, K.L.; Wang, C.; Kang, S.G.; Liao, J.; Logan, M.R.; Kim, C.H.; Taparowsky, E.J. Batf coordinates multiple aspects of B and T cell function required for normal antibody responses. J. Exp. Med. 2010, 207, 933–942. [Google Scholar] [CrossRef]
- Kurachi, M.; Barnitz, R.A.; Yosef, N.; Odorizzi, P.M.; A DiIorio, M.; E Lemieux, M.; Yates, K.; Godec, J.; Klatt, M.G.; Regev, A.; et al. The transcription factor BATF operates as an essential differentiation checkpoint in early effector CD8+ T cells. Nat. Immunol. 2014, 15, 373. [Google Scholar] [CrossRef]
- Sahoo, A.; Alekseev, A.; Tanaka, K.; Obertas, L.; Lerman, B.; Haymaker, C.; Clise-Dwyer, K.; McMurray, J.S.; Nurieva, R. Batf is important for IL-4 expression in T follicular helper cells. Nat. Commun. 2015, 6, 1–10. [Google Scholar] [CrossRef]
- Schleussner, N.; Merkel, O.; Costanza, M.; Liang, H.C.; Hummel, F.; Romagnani, C.; Durek, P.; Anagnostopoulos, I.; Hummel, M.; Jöhrens, K.; et al. The AP-1-BATF and -BATF3 module is essential for growth, survival and TH17/ILC3 skewing of anaplastic large cell lymphoma. Leukemia 2018, 32, 1994. [Google Scholar] [CrossRef] [PubMed]
- Seo, H.; González-Avalos, E.; Zhang, W.; Ramchandani, P.; Yang, C.; Lio, C.-W.J.; Rao, A.; Hogan, P.G. BATF and IRF4 cooperate to counter exhaustion in tumor-infiltrating CAR T cells. Nat. Immunol. 2021, 22, 983–995. [Google Scholar] [CrossRef] [PubMed]
- Ise, W.; Kohyama, M.; Schraml, B.; Zhang, T.; Schwer, B.; Basu, U.; Alt, F.W.; Tang, J.; Oltz, E.M.; Murphy, T.L.; et al. The transcription factor BATF controls the global regulators of class-switch recombination in both B cells and T cells. Nat. Immunol. 2011, 12, 536–543. [Google Scholar] [CrossRef] [PubMed]
- Sopel, N.; Graser, A.; Mousset, S.; Finotto, S. The transcription factor BATF modulates cytokine-mediated responses in T cells. Cytokine Growth Factor Rev. 2016, 30, 39–45. [Google Scholar] [CrossRef]
- Liao, J.; Humphrey, S.E.; Poston, S.; Taparowsky, E.J. Batf Promotes Growth Arrest and Terminal Differentiation of Mouse Myeloid Leukemia Cells. Mol. Cancer Res. 2011, 9, 350. [Google Scholar] [CrossRef]
- Jia, C.; Ma, Y.; Wang, M.; Liu, W.; Tang, F.; Chen, J. Evidence of Omics, Immune Infiltration, and Pharmacogenomics for BATF in a Pan-Cancer Cohort. Front. Mol. Biosci. 2022, 9, 392. [Google Scholar] [CrossRef]
- Care, M.; Cocco, M.; Laye, J.P.; Barnes, N.; Huang, Y.; Wang, M.; Barrans, S.; Du, M.; Jack, A.; Westhead, D.; et al. SPIB and BATF provide alternate determinants of IRF4 occupancy in diffuse large B-cell lymphoma linked to disease heterogeneity. Nucleic Acids Res. 2014, 42, 7591–7610. [Google Scholar] [CrossRef]
- Rouillard, A.D.; Gundersen, G.W.; Fernandez, N.F.; Wang, Z.; Monteiro, C.D.; McDermott, M.G.; Ma’Ayan, A. The harmonizome: A collection of processed datasets gathered to serve and mine knowledge about genes and proteins. Database 2016, 2016, 100. [Google Scholar] [CrossRef]
- Schuster, M.; Annemann, M.; Plaza-Sirvent, C.; Schmitz, I. Atypical IκB Proteins - Nuclear Modulators of NF-ΚB Signaling. Cell Commun. Signal. 2013, 11, 1–11. [Google Scholar] [CrossRef]
- Willems, M.; Dubois, N.; Musumeci, L.; Bours, V.; Robe, P.A. IκBζ: An emerging player in cancer. Oncotarget 2016, 7, 66310. [Google Scholar] [CrossRef]
- Gautam, P.; Maenner, S.; Cailotto, F.; Reboul, P.; Labialle, S.; Jouzeau, J.; Bourgaud, F.; Moulin, D. Emerging role of IκBζ in inflammation: Emphasis on psoriasis. Clin. Transl. Med. 2022, 12, e1032. [Google Scholar] [CrossRef]
- Motoyama, M.; Yamazaki, S.; Eto-Kimura, A.; Takeshige, K.; Muta, T. Positive and Negative Regulation of Nuclear Factor-κB-mediated Transcription by IκB-ζ, an Inducible Nuclear Protein. J. Biol. Chem. 2005, 280, 7444–7451. [Google Scholar] [CrossRef]
- Xu, T.; Rao, T.; Yu, W.M.; Ning, J.Z.; Yu, X.; Zhu, S.M.; Yang, K.; Bai, T.; Cheng, F. Upregulation of NFKBIZ affects bladder cancer progression via the PTEN/PI3K/Akt signaling pathway. Int. J. Mol. Med. 2021, 47, 1–12. [Google Scholar] [CrossRef]
- Lenz, G.; Wright, G.W.; Emre, N.C.T.; Kohlhammer, H.; Dave, S.S.; Davis, R.E.; Carty, S.; Lam, L.T.; Shaffer, A.L.; Xiao, W.; et al. Molecular subtypes of diffuse large B-cell lymphoma arise by distinct genetic pathways. Proc. Natl. Acad. Sci. USA 2008, 105, 13520. [Google Scholar] [CrossRef]
- Arthur, S.E.; Thomas, N.; Rushton, C.; Tang, J.; Alcaide, M.; Healy, S.; Telenius, A.; Mottok, A.; Scott, D.W.; Steidl, C.; et al. Nfkbiz 3′ UTR Mutations Confer Selective Growth Advantage and Affect Drug Response in Diffuse Large B-Cell Lymphoma. Blood 2020, 136, 31. [Google Scholar] [CrossRef]
- Arthur, S.E.; Jiang, A.; Grande, B.M.; Alcaide, M.; Cojocaru, R.; Rushton, C.K.; Mottok, A.; Hilton, L.K.; Lat, P.K.; Zhao, E.Y.; et al. Genome-wide discovery of somatic regulatory variants in diffuse large B-cell lymphoma. Nat. Commun. 2018, 9, 4001. [Google Scholar] [CrossRef]
- Hanihara, F.; Takahashi, Y.; Okuma, A.; Ohba, T.; Muta, T. Transcriptional and post-transcriptional regulation of IκB-ζ upon engagement of the BCR, TLRs and FcγR. Int. Immunol. 2013, 25, 531–544. [Google Scholar] [CrossRef]
- Pricci, F.; Leto, G.; Amadio, L.; Iacobini, C.; Romeo, G.; Cordone, S.; Gradini, R.; Barsotti, P.; Liu, F.T.; Di Mario, U.; et al. Role of galectin-3 as a receptor for advanced glycosylation end products. Kidney Int. 2000, 77, S31–S39. [Google Scholar] [CrossRef]
- Sciacchitano, S.; Lavra, L.; Morgante, A.; Ulivieri, A.; Magi, F.; De Francesco, G.P.; Bellotti, C.; Salehi, L.B.; Ricci, A. Galectin-3: One Molecule for an Alphabet of Diseases, from A to Z. Int. J. Mol. Sci. 2018, 19, 379. [Google Scholar] [CrossRef]
- Díaz-Alvarez, L.; Ortega, E. The Many Roles of Galectin-3, a Multifaceted Molecule, in Innate Immune Responses against Pathogens. Mediat. Inflamm. 2017, 2017, 1–10. [Google Scholar] [CrossRef]
- Nangia-Makker, P.; Hogan, V.; Raz, A. Galectin-3 and cancer stemness. Glycobiology 2018, 28, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Ruvolo, P.P. Galectin 3 as a guardian of the tumor microenvironment. Biochim. et Biophys. Acta (BBA)-Mol. Cell Res. 2016, 1863, 427–437. [Google Scholar] [CrossRef] [PubMed]
- Dong, R.; Zhang, M.; Hu, Q.; Zheng, S.; Soh, A.; Zheng, Y.; Yuan, H. Galectin-3 as a novel biomarker for disease diagnosis and a target for therapy (Review). Int. J. Mol. Med. 2018, 41, 599–614. [Google Scholar] [CrossRef] [PubMed]
- Ruvolo, P.P.; Hu, C.W.; Qiu, Y.; Ruvolo, V.R.; Go, R.L.; Hubner, S.E.; Coombes, K.R.; Andreeff, M.; Qutub, A.A.; Kornblau, S.M. LGALS3 is connected to CD74 in a previously unknown protein network that is associated with poor survival in patients with AML. Ebiomedicine 2019, 44, 126–137. [Google Scholar] [CrossRef] [PubMed]
- Michalová, Z.; Čoma, M.; Kičová, M.; Gabzdilová, J.; Dedinská, K.; Guman, T.; Hájiková, M.; Veselinyová, D.; Giertlova, M.; Gál, P.; et al. Overexpression of Galectin-3 in Chronic Lymphocytic Leukemia Is Associated With 17p Deletion: A Short Report. Anticancer. Res. 2019, 39, 2805–2810. [Google Scholar] [CrossRef]
- Koh, Y.W.; Jung, S.J.; Park, C.S.; Yoon, D.H.; Suh, C.; Huh, J. LGALS3 as a prognostic factor for classical Hodgkin’s lymphoma. Mod. Pathol. 2014, 27, 1338–1344. [Google Scholar] [CrossRef]
- Shipp, M.A.; Ross, K.N.; Tamayo, P.; Weng, A.P.; Aguiar, R.C.T.; Gaasenbeek, M.; Angelo, M.; Reich, M.; Pinkus, G.S.; Ray, T.S.; et al. Diffuse large B-cell lymphoma outcome prediction by gene-expression profiling and supervised machine learning. Nat. Med. 2002, 8, 68–74. [Google Scholar] [CrossRef]
- Shi, Y.; Tang, D.; Li, X.; Xie, X.; Ye, Y.; Wang, L. Galectin Family Members: Emerging Novel Targets for Lymphoma Therapy? Front. Oncol 2022, 12, 2092. [Google Scholar] [CrossRef]
- Pena, C.; Mirandola, L.; Figueroa, J.A.; Hosiriluck, N.; Suvorava, N.; Trotter, K.; Reidy, A.; Rakhshanda, R.; Payne, D.; Jenkins, M.; et al. Galectins as therapeutic targets for hematological malignancies: A hopeful sweetness. Ann. Transl. Med. 2014, 2, 87. [Google Scholar] [CrossRef]
- Clark, M.C.; Pang, M.; Hsu, D.K.; Liu, F.T.; de Vos, S.; Gascoyne, R.D.; Said, J.; Baum, L.G. Galectin-3 binds to CD45 on diffuse large B-cell lymphoma cells to regulate susceptibility to cell death. Blood 2012, 120, 4635–4644. [Google Scholar] [CrossRef]
- Hoyer, K.K.; Pang, M.; Gui, D.; Shintaku, I.P.; Kuwabara, I.; Liu, F.T.; Said, J.W.; Baum, L.G.; Teitell, M.A. An Anti-Apoptotic Role for Galectin-3 in Diffuse Large B-Cell Lymphomas. Am. J. Pathol. 2004, 164, 893–902. [Google Scholar] [CrossRef]
- André, U.; Dictor, M.; Jerkeman, M.; Berglund, M.; Sundström, C.; Linderoth, J.; Rosenquist, R.; Borrebaeck, C.A.K.; Ek, S. Identification of molecular targets associated with transformed diffuse large B cell lymphoma using highly purified tumor cells. Am. J. Hematol. 2009, 84, 803–808. [Google Scholar] [CrossRef]
- Kim, S.J.; Lee, S.J.; Sung, H.J.; Choi, I.K.; Choi, C.W.; Kim, B.S.; Kim, J.S.; Yu, W.; Hwang, H.S.; Kim, I.S. Increased Serum 90K and Galectin-3 Expression Are Associated with Advanced Stage and a Worse Prognosis in Diffuse Large B-Cell Lymphomas. Acta Haematol. 2008, 120, 211–216. [Google Scholar] [CrossRef]
- FDA approves anti-LAG3 checkpoint. Nat. Biotechnol. 2022, 40, 625. [CrossRef]
- Bae, J.; Accardi, F.; Hideshima, T.; Tai, Y.T.; Prabhala, R.; Shambley, A.; Wen, K.; Rowell, S.; Richardson, P.G.; Munshi, N.C.; et al. Targeting LAG3/GAL-3 to overcome immunosuppression and enhance anti-tumor immune responses in multiple myeloma. Leukemia 2021, 36, 138–154. [Google Scholar] [CrossRef]
- Nakajima, K.; Balan, V.; Raz, A. Galectin-3: An immune checkpoint target for musculoskeletal tumor patients. Cancer Metastasis Rev. 2020, 40, 297–302. [Google Scholar] [CrossRef]
- Yan, Y.; Zuo, X.; Wei, D. Concise Review: Emerging Role of CD44 in Cancer Stem Cells: A Promising Biomarker and Therapeutic Target. STEM CELLS Transl. Med. 2015, 4, 1033–1043. [Google Scholar] [CrossRef]
- Thapa, R.; Wilson, G.D. The Importance of CD44 as a Stem Cell Biomarker and Therapeutic Target in Cancer. Stem Cells Int. 2016, 2016, 1–15. [Google Scholar] [CrossRef]
- Goodison, S.; Urquidi, V.; Tarin, D. CD44 Cell Adhesion Molecules. J. Clin. Pathol. Mol. Pathol. 1999, 52, 189–196. [Google Scholar] [CrossRef]
- Batsché, E.; Yi, J.; Mauger, O.; Kornobis, E.; Hopkins, B.; Hanmer-Lloyd, C.; Muchardt, C. CD44 alternative splicing senses intragenic DNA methylation in tumors via direct and indirect mechanisms. Nucleic Acids Res. 2021, 49, 6213–6237. [Google Scholar] [CrossRef]
- Prochazka, L.; Tesarik, R.; Turanek, J. Regulation of alternative splicing of CD44 in cancer. Cell. Signal. 2014, 26, 2234–2239. [Google Scholar] [CrossRef] [PubMed]
- Dingemans, K.P.; Ramkema, M.D.; Pals, S.T. CD44 Is Exposed to the Extracellular Matrix at Invasive Sites in Basal Cell Carcinomas. Lab. Investig. 2002, 82, 313–322. [Google Scholar] [CrossRef]
- Hertweck, M.K.; Erdfelder, F.; Kreuzer, K.A. CD44 in hematological neoplasias. Ann. Hematol. 2011, 90, 493–508. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.Y.; Lin, J.N.; Hsieh, J.T.; Chou, S.C.; Lai, C.H.; Yun, E.J.; Lo, U.G.; Pong, R.C.; Lin, J.H.; Lin, Y.H. Nanoparticle Targeting CD44-Positive Cancer Cells for Site-Specific Drug Delivery in Prostate Cancer Therapy. ACS Appl. Mater. Interfaces 2016, 8, 30722–30734. [Google Scholar] [CrossRef] [PubMed]
- Orian-Rousseau, V.; Ponta, H. Perspectives of CD44 targeting therapies. Arch. Toxicol. 2014, 89, 3–14. [Google Scholar] [CrossRef]
- Hsiao, Y.W.; Chi, J.Y.; Li, C.F.; Chen, L.Y.; Chen, Y.T.; Liang, H.Y.; Lo, Y.C.; Hong, J.Y.; Chuu, C.P.; Hung, L.Y.; et al. Disruption of the pentraxin 3/CD44 interaction as an efficient therapy for triple-negative breast cancers. Clin. Transl. Med. 2022, 12, e724. [Google Scholar] [CrossRef]
- Reimann, M.; Schrezenmeier, J.F.; Richter-Pechanska, P.; Dolnik, A.; Hick, T.P.; Schleich, K.; Cai, X.; Fan, D.N.Y.; Lohneis, P.; Masswig, S.; et al. Adaptive T-cell immunity controls senescence-prone MyD88- or CARD11-mutant B-cell lymphomas. Blood 2021, 137, 2785–2799. [Google Scholar] [CrossRef]
- Hardee, J.; Ouyang, Z.; Zhang, Y.; Kundaje, A.; Lacroute, P.; Snyder, M. STAT3 Targets Suggest Mechanisms of Aggressive Tumorigenesis in Diffuse Large B-Cell Lymphoma. G3 (Bethesda) 2013, 3, 2173–2185. [Google Scholar] [CrossRef]
- Nørgaard, C.H.; Jakobsen, L.H.; Gentles, A.J.; Dybkær, K.; El-Galaly, T.C.; Bødker, J.S.; Schmitz, A.; Johansen, P.; Herold, T.; Spiekermann, K.; et al. Subtype assignment of CLL based on B-cell subset associated gene signatures from normal bone marrow – A proof of concept study. PLoS ONE 2018, 13, e0193249. [Google Scholar] [CrossRef]
- Dybkær, K.; Bøgsted, M.; Falgreen, S.; Bødker, J.S.; Kjeldsen, M.K.; Schmitz, A.; Bilgrau, A.E.; Xu-Monette, Z.Y.; Li, L.; Bergkvist, K.S.; et al. Diffuse Large B-Cell Lymphoma Classification System That Associates Normal B-Cell Subset Phenotypes with Prognosis. J. Clin. Oncol. 2015, 33, 1379–1388. [Google Scholar] [CrossRef]
- Frei, E.; Visco, C.; Xu-Monette, Z.Y.; Dirnhofer, S.; Dybkær, K.; Orazi, A.; Bhagat, G.; Hsi, E.D.; van Krieken, J.H.; Ponzoni, M.; et al. Addition of rituximab to chemotherapy overcomes the negative prognostic impact of cyclin E expression in diffuse large B-cell lymphoma. J. Clin. Pathol. 2013, 66, 956–961. [Google Scholar] [CrossRef]
- NF-KB Target Genes NF-KB Transcription Factors Boston University. Available online: https://www.bu.edu/nf-kb/gene-resources/target-genes/ (accessed on 29 January 2023).
- Xue, X.; Zeng, N.; Gao, Z.; Du, M.Q. Diffuse large B-cell lymphoma: Sub-classification by massive parallel quantitative RT-PCR. Lab. Investig. 2015, 95, 113–120. [Google Scholar] [CrossRef]
- Mölder, F.; Jablonski, K.P.; Letcher, B.; Hall, M.B.; Tomkins-Tinch, C.H.; Sochat, V.; Forster, J.; Lee, S.; Twardziok, S.O.; Kanitz, A.; et al. Sustainable data analysis with Snakemake. F1000Research 2021, 10, 33. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10. [Google Scholar] [CrossRef]
- Kopylova, E.; Noé, L.; Touzet, H. SortMeRNA: Fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics 2012, 28, 3211–3217. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- García-Alcalde, F.; Okonechnikov, K.; Carbonell, J.; Cruz, L.M.; Götz, S.; Tarazona, S.; Dopazo, J.; Meyer, T.F.; Conesa, A. Qualimap: Evaluating next-generation sequencing alignment data. Bioinformatics 2012, 28, 2678–2679. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol 2014, 15, 1–21. [Google Scholar] [CrossRef]
- Wickham, H. Ggplot2. Wiley Interdiscip. Rev. Comput. Stat. 2016, 3, 180–185. [Google Scholar] [CrossRef]
- Budczies, J.; Klauschen, F.; Sinn, B.V.; Gyorffy, B.; Schmitt, W.D.; Darb-Esfahani, S.; Denkert, C. Cutoff Finder: A Comprehensive and Straightforward Web Application Enabling Rapid Biomarker Cutoff Optimization. PLoS ONE 2012, 7, e51862. [Google Scholar] [CrossRef]
- Leyva-Vega, M.; Gerfen, J.; Thiel, B.D.; Jurkiewicz, D.; Rand, E.B.; Pawlowska, J.; Kaminska, D.; Russo, P.; Gai, X.; Krantz, I.D.; et al. Genomic alterations in biliary atresia suggest region of potential disease susceptibility in 2q37.3. Am. J. Med Genet. Part A 2010, 152A, 886–895. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Qin, G.; Liu, J.; Wang, X.; Ye, B. Integrated Genome-Wide Methylation and Expression Analyses Reveal Key Regulators in Osteosarcoma. Comput. Math. Methods Med. 2020, 2020, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Ma, J.; Zhang, Q.; Xiong, K.; Zhang, Z.; Chen, C.; Xiao, H.; Wang, D. A CTL/M2 macrophage-related four-gene signature predicting metastasis-free survival in triple-negative breast cancer treated with adjuvant radiotherapy. Breast Cancer Res. Treat. 2021, 190, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Willer, T.; Prados, B.; Falcón-Pérez, J.M.; Renner-Müller, I.; Przemeck, G.K.H.; Lommel, M.; Coloma, A.; Valero, M.C.; de Angelis, M.H.; Tanner, W.; et al. Targeted disruption of the Walker–Warburg syndrome gene Pomt1 in mouse results in embryonic lethality. Proc. Natl. Acad. Sci. 2004, 101, 14126–14131. [Google Scholar] [CrossRef] [PubMed]
- Jaeken, J. Congenital disorders of glycosylation. Handb Clin Neurol 2013, 113, 1737–1743. [Google Scholar] [CrossRef]
- Alonso-Rangel, L.; Benítez-Guerrero, T.; Martínez-Vieyra, I.; Cisneros, B.; Martínez-Tovar, A.; Winder, S.J.; Cerecedo, D. A role for dystroglycan in the pathophysiology of acute leukemic cells. Life Sci. 2017, 182, 1–9. [Google Scholar] [CrossRef]
- Quereda, C.; Pastor, A.; Martín-Nieto, J. Involvement of abnormal dystroglycan expression and matriglycan levels in cancer pathogenesis. Cancer Cell Int. 2022, 22, 1–30. [Google Scholar] [CrossRef]
- Wloga, D.; Webster, D.M.; Rogowski, K.; Bré, M.H.; Levilliers, N.; Jerka-Dziadosz, M.; Janke, C.; Dougan, S.T.; Gaertig, J. TTLL3 Is a Tubulin Glycine Ligase that Regulates the Assembly of Cilia. Dev. Cell 2009, 16, 867–876. [Google Scholar] [CrossRef]
- Meár, L.; Sutantiwanichkul, T.; Östman, J.; Damdimopoulou, P.; Lindskog, C. Spatial Proteomics for Further Exploration of Missing Proteins: A Case Study of the Ovary. J. Proteome Res. 2022. [Google Scholar] [CrossRef]
- Yang, J.; Liu, X.; Yue, G.; Adamian, M.; Bulgakov, O.; Li, T. Rootletin, a novel coiled-coil protein, is a structural component of the ciliary rootlet. J. Cell Biol. 2002, 159, 431–440. [Google Scholar] [CrossRef]
- Ko, D.; Kim, J.; Rhee, K.; Choi, H.-J. Identification of a Structurally Dynamic Domain for Oligomer Formation in Rootletin. J. Mol. Biol. 2020, 432, 3915–3932. [Google Scholar] [CrossRef]
- Bahe, S.; Stierhof, Y.D.; Wilkinson, C.; Leiss, F.; Nigg, E.A. Rootletin forms centriole-associated filaments and functions in centrosome cohesion. J. Cell Biol. 2005, 171, 27–33. [Google Scholar] [CrossRef]
- Fabbri, L.; Bost, F.; Mazure, N.M. Primary Cilium in Cancer Hallmarks. Int. J. Mol. Sci. 2019, 20, 1336. [Google Scholar] [CrossRef]
- Higgins, M.; Obaidi, I.; McMorrow, T. Primary cilia and their role in cancer (Review). Oncol. Lett. 2019, 17, 3041–3047. [Google Scholar] [CrossRef]
- Blümel, L.; Qin, N.; Berlandi, J.; Paisana, E.; Cascão, R.; Custódia, C.; Pauck, D.; Picard, D.; Langini, M.; Stühler, K.; et al. Primary cilia contribute to the aggressiveness of atypical teratoid/rhabdoid tumors. Cell Death. Dis. 2022, 13, 1–13. [Google Scholar] [CrossRef]
- Wang, J.; Liu, Y.; Yang, Z.; Sui, Y.; Tian, J.; Tao, L.; Yao, J.; Wu, C. CD52 Is a Prognostic Biomarker and Correlated with Immune Features in Breast Cancer. Res. Sq. 2020. [Google Scholar] [CrossRef]
- Hochgreb-Hägele, T.; Koo, D.E.; Bronner, M.E. Znf385C mediates a novel p53-dependent transcriptional switch to control timing of facial bone formation. Dev. Biol. 2015, 400, 23–32. [Google Scholar] [CrossRef]
- Costessi, A.; Mahrour, N.; Tijchon, E.; Stunnenberg, R.; Stoel, M.A.; Jansen, P.W.; Sela, D.; Martin-Brown, S.; Washburn, M.; Florens, L.; et al. The tumour antigen PRAME is a subunit of a Cul2 ubiquitin ligase and associates with active NFY promoters. EMBO J. 2011, 30, 3786–3798. [Google Scholar] [CrossRef]
- Epping, M.T.; Wang, L.; Edel, M.J.; Carlée, L.; Hernandez, M.; Bernards, R. The Human Tumor Antigen PRAME Is a Dominant Repressor of Retinoic Acid Receptor Signaling. Cell 2005, 122, 835–847. [Google Scholar] [CrossRef]
- Wang, Z.; Pascal, L.E.; Chandran, U.R.; Chaparala, S.; Lv, S.; Ding, H.; Qi, L.; Wang, Z. ELL2 Is Required for the Growth and Survival of AR-Negative Prostate Cancer Cells. Cancer Manag. Res. 2020, 12, 4411–4427. [Google Scholar] [CrossRef]
- Zang, Y.; Pascal, L.E.; Zhou, Y.; Qiu, X.; Wei, L.; Ai, J.; Nelson, J.B.; Zhong, M.; Xue, B.; Wang, S.; et al. ELL2 regulates DNA non-homologous end joining (NHEJ) repair in prostate cancer cells. Cancer Lett. 2018, 415, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.; Ajore, R.; Wihlborg, A.K.; Niroula, A.; Swaminathan, B.; Johnsson, E.; Stephens, O.W.; Morgan, G.; Meissner, T.; Turesson, I.; et al. The multiple myeloma risk allele at 5q15 lowers ELL2 expression and increases ribosomal gene expression. Nat. Commun. 2018, 9, 1–9. [Google Scholar] [CrossRef]
- Li, N.; Johnson, D.C.; Weinhold, N.; Kimber, S.; Dobbins, S.E.; Mitchell, J.S.; Kinnersley, B.; Sud, A.; Law, P.J.; Orlando, G.; et al. Genetic Predisposition to Multiple Myeloma at 5q15 Is Mediated by an ELL2 Enhancer Polymorphism. Cell Rep. 2017, 20, 2556–2564. [Google Scholar] [CrossRef] [PubMed]
- Georgiadis, P.; Topinka, J.; Vlachodimitropoulos, D.; Stoikidou, M.; Gioka, M.; Stephanou, G.; Autrup, H.; Demopoulos, N.A.; Katsouyanni, K.; Sram, R.; et al. Interactions between CYP1A1 polymorphisms and exposure to environmental tobacco smoke in the modulation of lymphocyte bulky DNA adducts and chromosomal aberrations. Carcinog 2004, 26, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Al-Dayel, F.; Al-Rasheed, M.; Ibrahim, M.; Bu, R.; Bavi, P.; Abubaker, J.; Al-Jomah, N.; Mohamed, G.H.; Moorji, A.; Uddin, S.; et al. Polymorphisms of drug-metabolizing enzymes CYP1A1, GSTT and GSTP contribute to the development of diffuse large B-cell lymphoma risk in the Saudi Arabian population. Leuk. Lymphoma 2008, 49, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Onnis, A.; Finetti, F.; Patrussi, L.; Gottardo, M.; Cassioli, C.; Spanò, S.; Baldari, C.T. The small GTPase Rab29 is a common regulator of immune synapse assembly and ciliogenesis. Cell Death Differ. 2015, 22, 1687. [Google Scholar] [CrossRef]
- MacLeod, D.A.; Rhinn, H.; Kuwahara, T.; Zolin, A.; Di Paolo, G.; McCabe, B.D.; Marder, K.S.; Honig, L.S.; Clark, L.N.; Small, S.A.; et al. RAB7L1 Interacts with LRRK2 to Modify Intraneuronal Protein Sorting and Parkinson’s Disease Risk. Neuron 2013, 77, 425–439. [Google Scholar] [CrossRef]
- Tröger, J.; Moutty, M.C.; Skroblin, P.; Klussmann, E. A-kinase anchoring proteins as potential drug targets. Br. J. Pharmacol. 2012, 166, 420–433. [Google Scholar] [CrossRef]
- Vergarajauregui, S.; Becker, R.; Steffen, U.; Sharkova, M.; Esser, T.; Petzold, J.; Billing, F.; Kapiloff, M.S.; Schett, G.; Thievessen, I.; et al. AKAP6 orchestrates the nuclear envelope microtubule-organizing center by linking golgi and nucleus via AKAP9. Elife 2020, 9, 1–30. [Google Scholar] [CrossRef]
- Perino, A.; Ghigo, A.; Scott, J.D.; Hirsch, E. Anchoring Proteins as Regulators of Signaling Pathways. Circ. Res. 2012, 111, 482–492. [Google Scholar] [CrossRef]
- Zhou, Z.; Qiu, R.; Liu, W.; Yang, T.; Li, G.; Huang, W.; Teng, X.; Yang, Y.; Yu, H.; Yang, Y.; et al. BCAS3 exhibits oncogenic properties by promoting CRL4A-mediated ubiquitination of p53 in breast cancer. Cell Prolif. 2021, 54, e13088. [Google Scholar] [CrossRef]
- Bärlund, M.; Monni, O.; Weaver, J.D.; Kauraniemi, P.; Sauter, G.; Heiskanen, M.; Kallioniemi, O.-P.; Kallioniemi, A. Cloning of BCAS3 (17q23) and BCAS4 (20q13) genes that undergo amplification, overexpression, and fusion in breast cancer†. Genes, Chromosom. Cancer 2002, 35, 311–317. [Google Scholar] [CrossRef]
- Metelli, A.; Wu, B.X.; Riesenberg, B.; Guglietta, S.; Huck, J.D.; Mills, C.; Li, A.; Rachidi, S.; Krieg, C.; Rubinstein, M.P.; et al. Thrombin Contributes to Cancer Immune Evasion via Proteolysis of Platelet-Bound GARP to Activate LTGF-β. Sci. Transl. Med. 2020, 12, 4860. [Google Scholar] [CrossRef]
- Hahn, S.A.; Neuhoff, A.; Landsberg, J.; Schupp, J.; Eberts, D.; Leukel, P.; Bros, M.; Weilbaecher, M.; Schuppan, D.; Grabbe, S.; et al. A key role of GARP in the immune suppressive tumor microenvironment. Oncotarget 2016, 7, 42996–43009. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Sun, L.; Tang, L.; Yu, W.; Li, H. Expression of GARP Is Increased in Tumor-Infiltrating Regulatory T Cells and Is Correlated to Clinicopathology of Lung Cancer Patients. Front. Immunol. 2017, 8, 138. [Google Scholar] [CrossRef]
- Faget, J.; Biota, C.; Bachelot, T.; Gobert, M.; Treilleux, I.; Goutagny, N.; Durand, I.; Léon-Goddard, S.; Blay, J.Y.; Caux, C.; et al. Early Detection of Tumor Cells by Innate Immune Cells Leads to Treg Recruitment through CCL22 Production by Tumor Cells. Cancer Res 2011, 71, 6143–6152. [Google Scholar] [CrossRef]
- Tsujikawa, T.; Yaguchi, T.; Ohmura, G.; Ohta, S.; Kobayashi, A.; Kawamura, N.; Fujita, T.; Nakano, H.; Shimada, T.; Takahashi, T.; et al. Autocrine and paracrine loops between cancer cells and macrophages promote lymph node metastasis via CCR4/CCL22 in head and neck squamous cell carcinoma. Int. J. Cancer 2013, 132, 2755–2766. [Google Scholar] [CrossRef]
- Kimura, S.; Nanbu, U.; Noguchi, H.; Harada, Y.; Kumamoto, K.; Sasaguri, Y.; Nakayama, T. Macrophage CCL22 expression in the tumor microenvironment and implications for survival in patients with squamous cell carcinoma of the tongue. J. Oral Pathol. Med. 2019, 48, 677–685. [Google Scholar] [CrossRef]
- Thomas, J.K.; Mir, H.; Kapur, N.; Bae, S.; Singh, S. CC chemokines are differentially expressed in Breast Cancer and are associated with disparity in overall survival. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef]
- Nakanishi, T.; Imaizumi, K.; Hasegawa, Y.; Kawabe, T.; Hashimoto, N.; Okamoto, M.; Shimokata, K. Expression of macrophage-derived chemokine (MDC)/CCL22 in human lung cancer. Cancer Immunol. Immunother. 2006, 55, 1320–1329. [Google Scholar] [CrossRef]
- Wågsäter, D.; Dienus, O.; Löfgren, S.; Hugander, A.; Dimberg, J. Quantification of the chemokines CCL17 and CCL22 in human colorectal adenocarcinomas. Mol. Med. Rep. 2008, 1, 211–217. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Turi, M.; Anilkumar Sithara, A.; Hofmanová, L.; Žihala, D.; Radhakrishnan, D.; Vdovin, A.; Knápková, S.; Ševčíková, T.; Chyra, Z.; Jelínek, T.; et al. Transcriptome Analysis of Diffuse Large B-Cell Lymphoma Cells Inducibly Expressing MyD88 L265P Mutation Identifies Upregulated CD44, LGALS3, NFKBIZ, and BATF as Downstream Targets of Oncogenic NF-κB Signaling. Int. J. Mol. Sci. 2023, 24, 5623. https://doi.org/10.3390/ijms24065623
Turi M, Anilkumar Sithara A, Hofmanová L, Žihala D, Radhakrishnan D, Vdovin A, Knápková S, Ševčíková T, Chyra Z, Jelínek T, et al. Transcriptome Analysis of Diffuse Large B-Cell Lymphoma Cells Inducibly Expressing MyD88 L265P Mutation Identifies Upregulated CD44, LGALS3, NFKBIZ, and BATF as Downstream Targets of Oncogenic NF-κB Signaling. International Journal of Molecular Sciences. 2023; 24(6):5623. https://doi.org/10.3390/ijms24065623
Chicago/Turabian StyleTuri, Marcello, Anjana Anilkumar Sithara, Lucie Hofmanová, David Žihala, Dhwani Radhakrishnan, Alexander Vdovin, Sofija Knápková, Tereza Ševčíková, Zuzana Chyra, Tomáš Jelínek, and et al. 2023. "Transcriptome Analysis of Diffuse Large B-Cell Lymphoma Cells Inducibly Expressing MyD88 L265P Mutation Identifies Upregulated CD44, LGALS3, NFKBIZ, and BATF as Downstream Targets of Oncogenic NF-κB Signaling" International Journal of Molecular Sciences 24, no. 6: 5623. https://doi.org/10.3390/ijms24065623