
Antidiabetic Drugs Can Reduce the Harmful Impact of Chronic Smoking on Post-Traumatic Brain Injuries

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
2.1. Effect of MF and RSG on Body Weight and Fasting Blood Glucose Levels of Premorbid TS-Exposed and TBI-Induced Mice
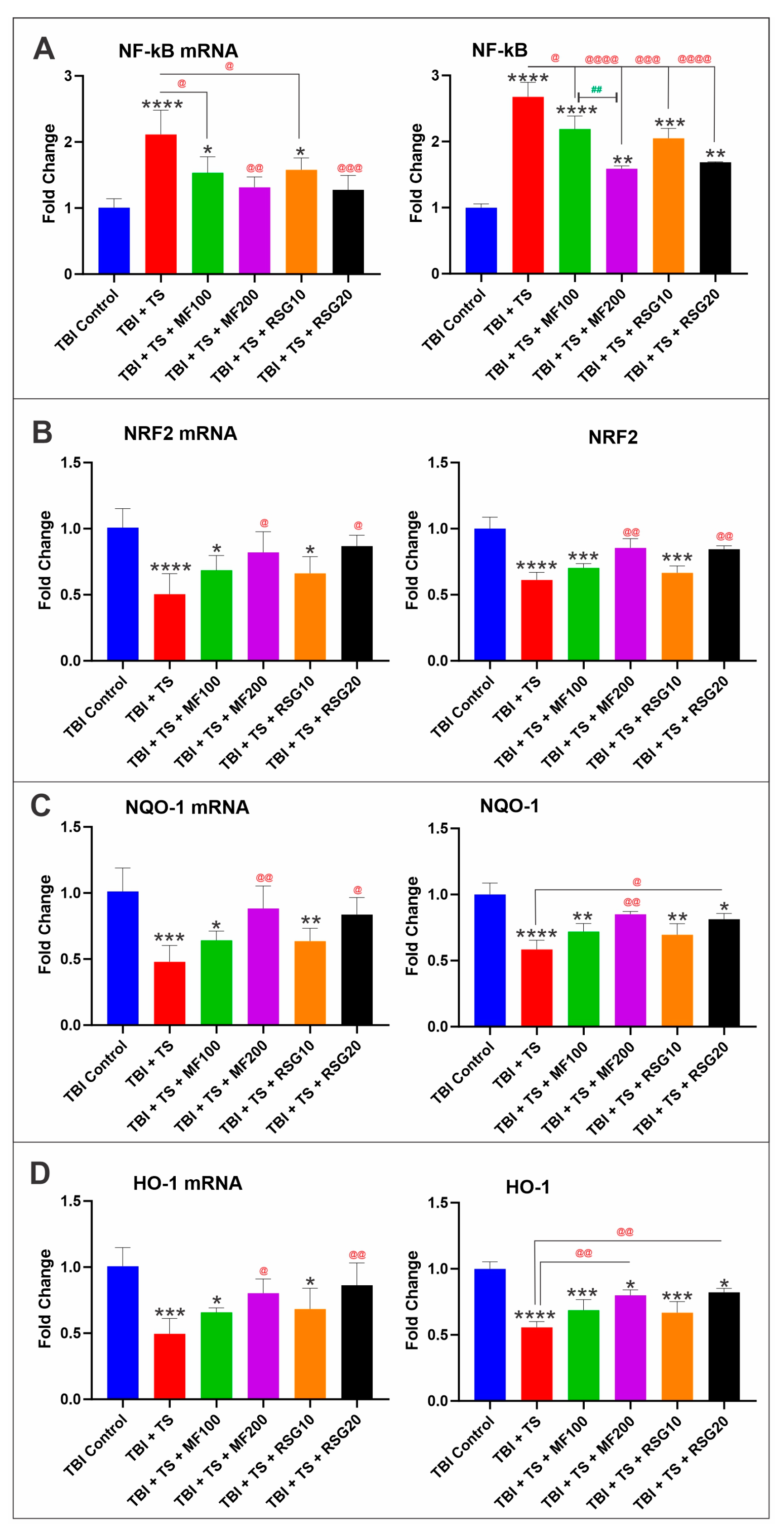
2.2. MF and RSG Upregulate NRF2 and Its Downstream Effector Molecules
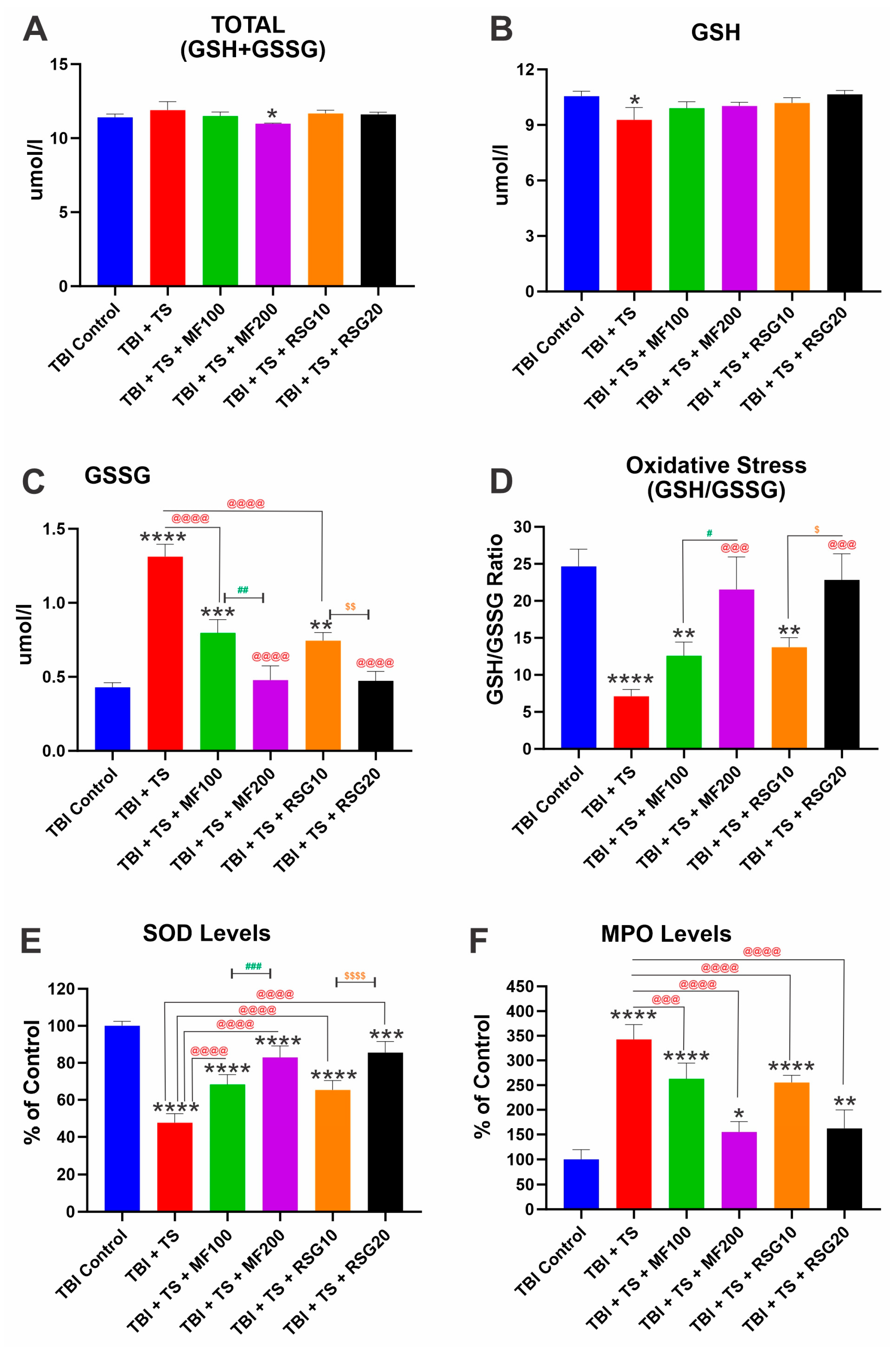
2.3. MF and RSG Reduce OS and Inflammatory Responses Enhanced by Premorbid TS Exposure
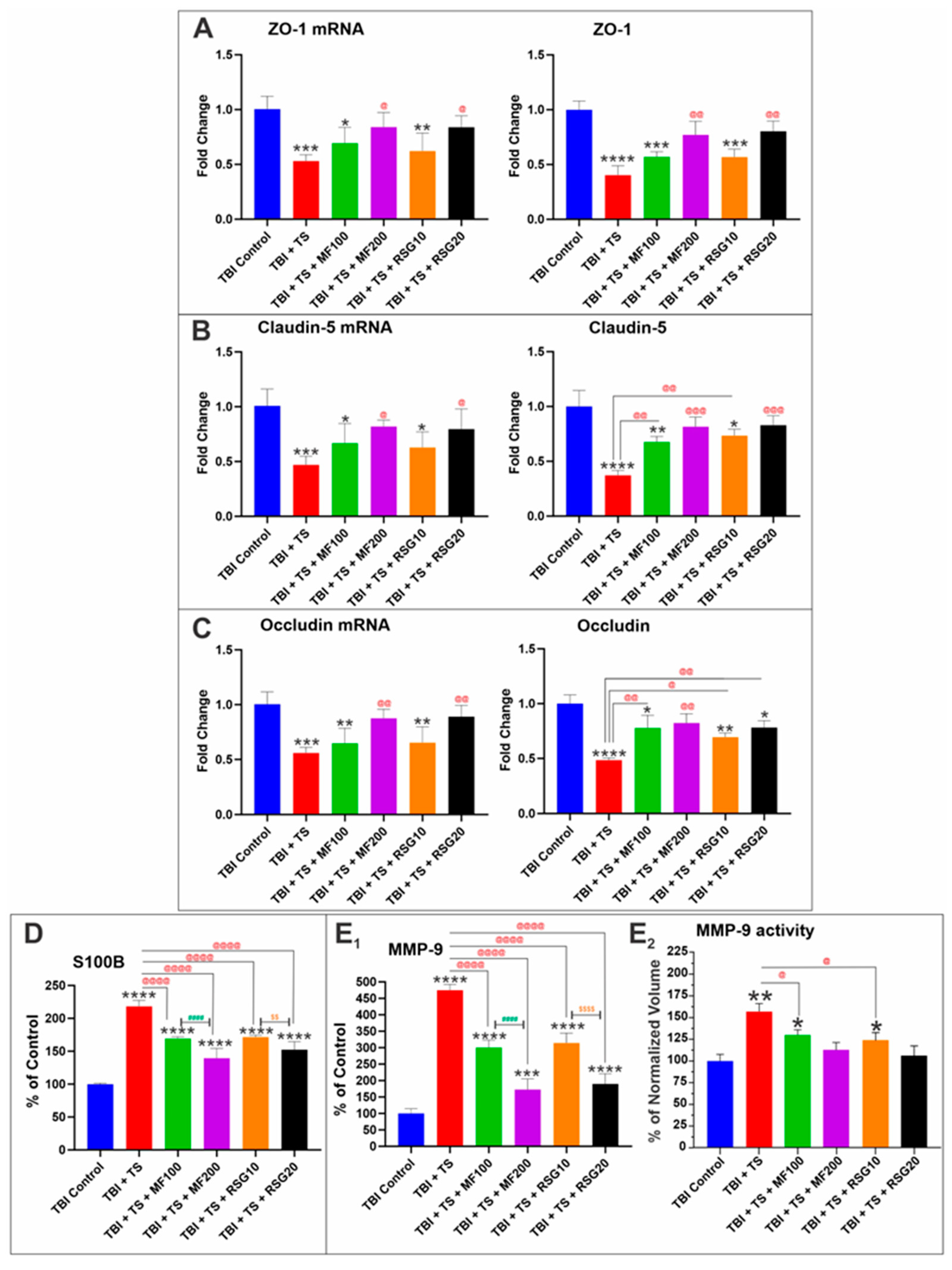
2.4. MF and RSG Reduced the Negative Impact of Chronic Premorbid TS Exposure on BBB Disruption by TBI
2.5. Downregulation of Thrombomodulin and UCH-L1 Prompted by Chronic TS Exposure in TBI-Induced Mice Is Reduced by MF and RSG
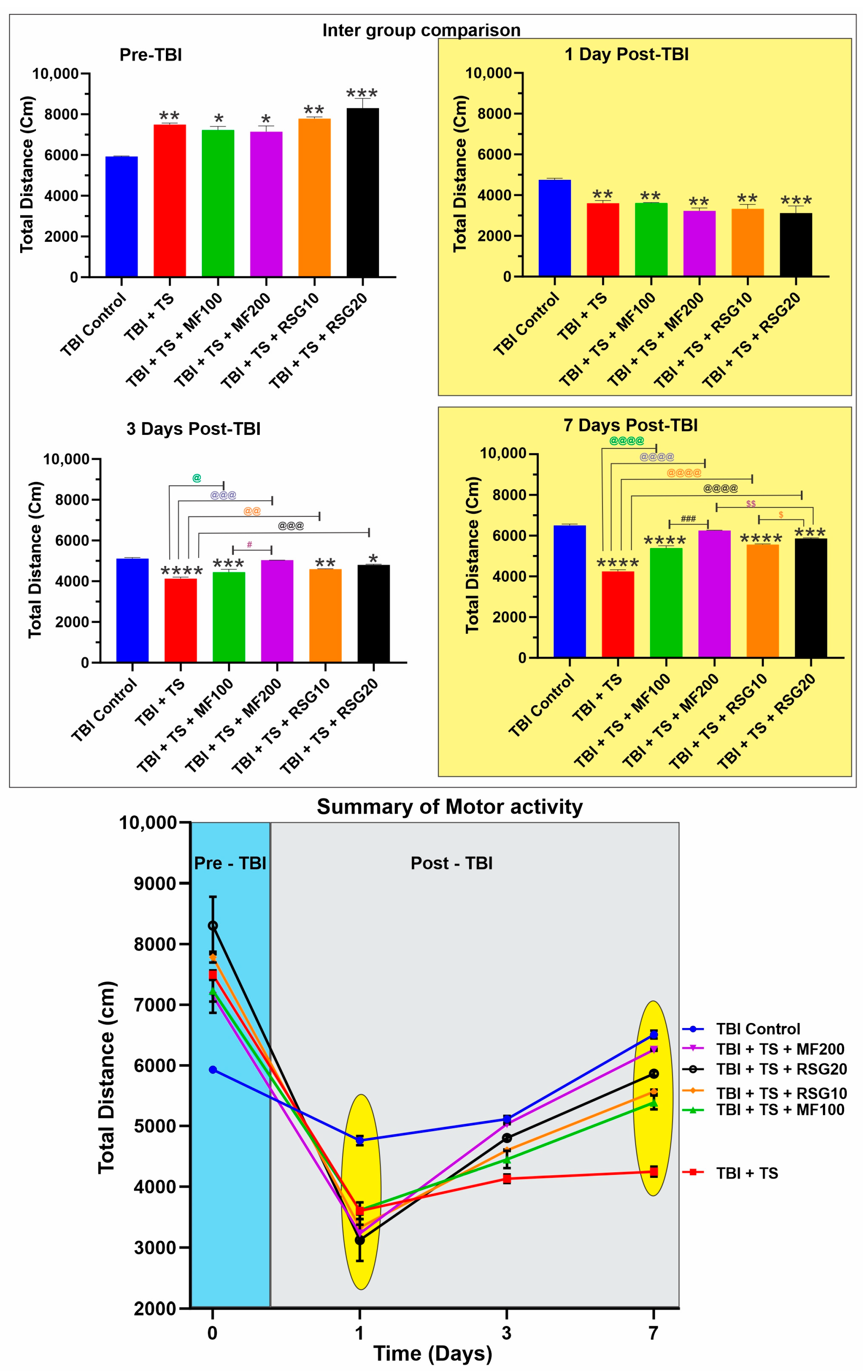
2.6. MF and RSG Decrease the Negative Effect of Chronic TS Exposure on the Loss of Motor Activity and Recovery Post-TBI
3. Discussion
4. Materials and Methods
4.1. Reagents and Materials
4.2. Experimental Design
4.3. Induction of Head Injury in Mice
4.4. Rosiglitazone and Metformin Treatment In Vivo
4.5. Fasting Blood Glucose Level Analysis
4.6. Open Field Test
4.7. Blood Collection and Brain Isolation
4.8. Hematoxylin and Eosin (H&E) and Nissl Staining
4.9. Preparation of Protein Extracts, ELISA, and Zymography
4.10. RNA Extraction and Quantitative Real-Time Polymerase Chain Reaction (RT-PCR)
4.11. Glutathione Levels Measurement
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ARE | Antioxidative Response Element |
| BBB | Blood–Brain Barrier |
| CNS | Central Nervous System |
| ELISA | Enzyme-Linked Immunosorbent Assay |
| HO-1 | Heme Oxygenase 1 |
| H&E | Hematoxylin and Eosin |
| ICAM-1 | Intercellular Adhesion Molecule-1 |
| KEAP1 | Kelch-Like ECH Associated Protein 1 |
| mBMEC | Mouse Brain Microvascular Endothelial Cells |
| MF | Metformin |
| MMP-9 | Matrix Metalloproteinase-9 |
| MPO | Myeloperoxidase |
| NF-κB | Nuclear Factor kappa B |
| NQO-1 | NAD(P)H: Quinone reductase I |
| NRF2 | Nuclear Factor erythroid-2 related Factor 2 |
| OS | Oxidative Stress |
| PECAM-1 | Platelet Endothelial Cell Adhesion Molecule 1 |
| PPARγ | Peroxisome Proliferator-Activated Receptor |
| ROS | Reactive Oxygen Species |
| RSG | Rosiglitazone |
| RT-PCR | Real-Time Polymerase Chain Reaction |
| SOD | Superoxide Dismutase |
| TBI | Traumatic Brain Injury |
| TJ | Tight Junction |
| TS | Tobacco Smoking |
| UCH-L1 | Ubiquitin C-terminal Hydrolase L1 |
| VCAM-1 | Vascular Cell Adhesion Protein 1 |
| ZO-1 | Zonulae Occludentes-1 |
References
- Mollayeva, T.; Mollayeva, S.; Colantonio, A. Traumatic brain injury: Sex, gender and intersecting vulnerabilities. Nat. Rev. Neurol. 2018, 14, 711–722. [Google Scholar] [CrossRef]
- Wu, A.-G.; Yong, Y.-Y.; Pan, Y.-R.; Zhang, L.; Wu, J.-M.; Zhang, Y.; Tang, Y.; Wei, J.; Yu, L.; Law, B.Y.-K.; et al. Targeting Nrf2-Mediated Oxidative Stress Response in Traumatic Brain Injury: Therapeutic Perspectives of Phytochemicals. Oxidative Med. Cell. Longev. 2022, 2022, 1015791. [Google Scholar] [CrossRef]
- Qin, D.; Wang, J.; Le, A.; Wang, T.J.; Chen, X.; Wang, J. Traumatic Brain Injury: Ultrastructural Features in Neuronal Ferroptosis, Glial Cell Activation and Polarization, and Blood–Brain Barrier Breakdown. Cells 2021, 10, 1009. [Google Scholar] [CrossRef] [PubMed]
- Janak, J.; Pugh, M.; Orman, J. Epidemiology of traumatic brain injury. Trauma. Brain Inj. Rehabil. Med. 2015, 6–35. [Google Scholar] [CrossRef]
- Tabet, M.; El-Kurdi, M.; Haidar, M.A.; Nasrallah, L.; Reslan, M.A.; Shear, D.; Pandya, J.D.; El-Yazbi, A.F.; Sabra, M.; Mondello, S.; et al. Mitoquinone supplementation alleviates oxidative stress and pathologic outcomes following repetitive mild traumatic brain injury at a chronic time point. Exp. Neurol. 2022, 351, 113987. [Google Scholar] [CrossRef]
- Ahmed, M.E.; Selvakumar, G.P.; Kempuraj, D.; Raikwar, S.P.; Thangavel, R.; Bazley, K.; Wu, K.; Khan, O.; Khan, A.; Zaheer, S.; et al. Glia maturation factor (GMF) regulates microglial expression phenotypes and the associated neurological deficits in a mouse model of traumatic brain injury. Mol. Neurobiol. 2020, 57, 4438–4450. [Google Scholar] [CrossRef] [PubMed]
- Akamatsu, Y.; Hanafy, K.A. Cell death and recovery in traumatic brain injury. Neurotherapeutics 2020, 17, 446–456. [Google Scholar] [CrossRef]
- Ahmed, M.E.; Selvakumar, G.P.; Kempuraj, D.; Raikwar, S.P.; Thangavel, R.; Bazley, K.; Wu, K.; Khan, O.; Kukulka, K.; Bussinger, B.; et al. Neuroinflammation mediated by glia maturation factor exacerbates neuronal injury in an in vitro model of traumatic brain injury. J. Neurotrauma 2020, 37, 1645–1655. [Google Scholar] [CrossRef] [PubMed]
- Charkviani, M.; Muradashvili, N.; Lominadze, D. Vascular and non-vascular contributors to memory reduction during traumatic brain injury. Eur. J. Neurosci. 2019, 50, 2860–2876. [Google Scholar] [CrossRef]
- Kempuraj, D.; Ahmed, M.E.; Selvakumar, G.P.; Thangavel, R.; Dhaliwal, A.S.; Dubova, I.; Mentor, S.; Premkumar, K.; Saeed, D.; Zahoor, H. Brain injury–mediated neuroinflammatory response and Alzheimer’s disease. Neuroscientist 2020, 26, 134–155. [Google Scholar] [CrossRef] [PubMed]
- Raikwar, S.P.; Thangavel, R.; Ahmed, M.E.; Selvakumar, G.P.; Kempuraj, D.; Wu, K.; Khan, O.; Bazley, K.; Bussinger, B.; Kukulka, K. Real-time noninvasive bioluminescence, ultrasound and photoacoustic imaging in NFκB-RE-Luc transgenic mice reveal glia maturation factor-mediated immediate and sustained spatio-temporal activation of NFκB signaling post-traumatic brain injury in a gender-specific manner. Cell. Mol. Neurobiol. 2021, 41, 1687–1706. [Google Scholar] [PubMed]
- Selvakumar, G.P.; Ahmed, M.E.; Iyer, S.S.; Thangavel, R.; Kempuraj, D.; Raikwar, S.P.; Bazley, K.; Wu, K.; Khan, A.; Kukulka, K. Absence of glia maturation factor protects from axonal injury and motor behavioral impairments after traumatic brain injury. Exp. Neurobiol. 2020, 29, 230. [Google Scholar] [CrossRef]
- Wang, M.; Luo, L. An effective NADPH oxidase 2 inhibitor provides neuroprotection and improves functional outcomes in animal model of traumatic brain injury. Neurochem. Res. 2020, 45, 1097–1106. [Google Scholar] [CrossRef] [PubMed]
- Chandran, R.; Kim, T.; Mehta, S.L.; Udho, E.; Chanana, V.; Cengiz, P.; Kim, H.; Kim, C.; Vemuganti, R. A combination antioxidant therapy to inhibit NOX2 and activate Nrf2 decreases secondary brain damage and improves functional recovery after traumatic brain injury. J. Cereb. Blood Flow Metab. 2018, 38, 1818–1827. [Google Scholar] [CrossRef]
- Galgano, M.; Toshkezi, G.; Qiu, X.; Russell, T.; Chin, L.; Zhao, L.-R. Traumatic brain injury: Current treatment strategies and future endeavors. Cell Transplant. 2017, 26, 1118–1130. [Google Scholar] [CrossRef] [Green Version]
- Dinet, V.; Petry, K.G.; Badaut, J. Brain–immune interactions and neuroinflammation after traumatic brain injury. Front. Neurosci. 2019, 13, 1178. [Google Scholar] [CrossRef] [Green Version]
- Crupi, R.; Cordaro, M.; Cuzzocrea, S.; Impellizzeri, D. Management of traumatic brain injury: From present to future. Antioxidants 2020, 9, 297. [Google Scholar] [CrossRef] [Green Version]
- Ladak, A.A.; Enam, S.A.; Ibrahim, M.T. A review of the molecular mechanisms of traumatic brain injury. World Neurosurg. 2019, 131, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.-Y.; Liou, T.-H.; Chang, K.-H.; Chi, W.-C.; Escorpizo, R.; Yen, C.-F.; Liao, H.-F.; Chiou, H.-Y.; Chiu, W.-T.; Tsai, J.-T. Functioning and disability analysis of patients with traumatic brain injury and spinal cord injury by using the world health organization disability assessment schedule 2.0. Int. J. Environ. Res. Public Health 2015, 12, 4116–4127. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Zhou, X.-M.; Wu, L.-Y.; Liu, G.-J.; Xu, W.-D.; Zhang, X.-S.; Gao, Y.-Y.; Tao, T.; Zhou, Y.; Lu, Y.; et al. Aucubin alleviates oxidative stress and inflammation via Nrf2-mediated signaling activity in experimental traumatic brain injury. J. Neuroinflamm. 2020, 17, 188. [Google Scholar] [CrossRef]
- Simon, D.W.; McGeachy, M.J.; Bayır, H.; Clark, R.S.; Loane, D.J.; Kochanek, P.M. The far-reaching scope of neuroinflammation after traumatic brain injury. Nat. Rev. Neurol. 2017, 13, 171–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capizzi, A.; Woo, J.; Verduzco-Gutierrez, M. Traumatic brain injury: An overview of epidemiology, pathophysiology, and medical management. Med. Clin. 2020, 104, 213–238. [Google Scholar]
- Shively, S.B.; Priemer, D.S.; Stein, M.B.; Perl, D.P. Pathophysiology of traumatic brain injury, chronic traumatic encephalopathy, and neuropsychiatric clinical expression. Psychiatr. Clin. 2021, 44, 443–458. [Google Scholar] [CrossRef]
- Loane, D.J.; Kumar, A. Microglia in the TBI brain: The good, the bad, and the dysregulated. Exp. Neurol. 2016, 275, 316–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donat, C.K.; Scott, G.; Gentleman, S.M.; Sastre, M. Microglial activation in traumatic brain injury. Front. Aging Neurosci. 2017, 9, 208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alam, A.; Thelin, E.P.; Tajsic, T.; Khan, D.Z.; Khellaf, A.; Patani, R.; Helmy, A. Cellular infiltration in traumatic brain injury. J. Neuroinflamm. 2020, 17, 328. [Google Scholar] [CrossRef]
- Hu, J.; Wang, X.; Chen, X.; Fang, Y.; Chen, K.; Peng, W.; Wang, Z.; Guo, K.; Tan, X.; Liang, F. Hydroxychloroquine attenuates neuroinflammation following traumatic brain injury by regulating the TLR4/NF-κB signaling pathway. J. Neuroinflamm. 2022, 19, 71. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Yin, D.; Ren, H.; Gao, W.; Li, F.; Sun, D.; Wu, Y.; Zhou, S.; Lyu, L.; Yang, M. Selective NLRP3 inflammasome inhibitor reduces neuroinflammation and improves long-term neurological outcomes in a murine model of traumatic brain injury. Neurobiol. Dis. 2018, 117, 15–27. [Google Scholar] [CrossRef]
- Gao, W.; Zhao, Z.; Yu, G.; Zhou, Z.; Zhou, Y.; Hu, T.; Jiang, R.; Zhang, J. VEGI attenuates the inflammatory injury and disruption of blood–brain barrier partly by suppressing the TLR4/NF-κB signaling pathway in experimental traumatic brain injury. Brain Res. 2015, 1622, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Muneer, P.; Chandra, N.; Haorah, J. Interactions of oxidative stress and neurovascular inflammation in the pathogenesis of traumatic brain injury. Mol. Neurobiol. 2015, 51, 966–979. [Google Scholar] [CrossRef]
- Lutton, E.M.; Farney, S.K.; Andrews, A.M.; Shuvaev, V.V.; Chuang, G.-Y.; Muzykantov, V.R.; Ramirez, S.H. Endothelial targeted strategies to combat oxidative stress: Improving outcomes in traumatic brain injury. Front. Neurol. 2019, 10, 582. [Google Scholar] [CrossRef] [Green Version]
- Archie, S.R.; Sharma, S.; Burks, E.; Abbruscato, T. Biological determinants impact the neurovascular toxicity of nicotine and tobacco smoke: A pharmacokinetic and pharmacodynamic perspective. NeuroToxicology 2022, 89, 140–160. [Google Scholar] [CrossRef] [PubMed]
- Bernard, A.; Ku, J.M.; Vlahos, R.; Miller, A.A. Cigarette smoke extract exacerbates hyperpermeability of cerebral endothelial cells after oxygen glucose deprivation and reoxygenation. Sci. Rep. 2019, 9, 15573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez-Rodriguez, A.; Jose Egea-Guerrero, J.; Murillo-Cabezas, F.; Carrillo-Vico, A. Oxidative stress in traumatic brain injury. Curr. Med. Chem. 2014, 21, 1201–1211. [Google Scholar] [CrossRef] [PubMed]
- Kaisar, M.A.; Villalba, H.; Prasad, S.; Liles, T.; Sifat, A.E.; Sajja, R.K.; Abbruscato, T.J.; Cucullo, L. Offsetting the impact of smoking and e-cigarette vaping on the cerebrovascular system and stroke injury: Is Metformin a viable countermeasure? Redox Biol. 2017, 13, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Bergold, P.J. Treatment of traumatic brain injury with anti-inflammatory drugs. Exp. Neurol. 2016, 275, 367–380. [Google Scholar] [CrossRef]
- Hellewell, S.; Semple, B.D.; Morganti-Kossmann, M.C. Therapies negating neuroinflammation after brain trauma. Brain Res. 2016, 1640, 36–56. [Google Scholar] [CrossRef]
- Wang, J.; Jiang, C.; Zhang, K.; Lan, X.; Chen, X.; Zang, W.; Wang, Z.; Guan, F.; Zhu, C.; Yang, X. Melatonin receptor activation provides cerebral protection after traumatic brain injury by mitigating oxidative stress and inflammation via the Nrf2 signaling pathway. Free Radic. Biol. Med. 2019, 131, 345–355. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Wang, H.; Zhou, J.; Dai, W.; Zhu, Y.; Zhou, Y.; Wang, X.; Zhou, M. Baicalin provides neuroprotection in traumatic brain injury mice model through Akt/Nrf2 pathway. Drug Des. Dev. Ther. 2018, 12, 2497. [Google Scholar] [CrossRef] [Green Version]
- Zeng, J.; Chen, Y.; Ding, R.; Feng, L.; Fu, Z.; Yang, S.; Deng, X.; Xie, Z.; Zheng, S. Isoliquiritigenin alleviates early brain injury after experimental intracerebral hemorrhage via suppressing ROS-and/or NF-κB-mediated NLRP3 inflammasome activation by promoting Nrf2 antioxidant pathway. J. Neuroinflamm. 2017, 14, 119. [Google Scholar] [CrossRef] [Green Version]
- Sajja, R.K.; Green, K.N.; Cucullo, L. Altered nrf2 signaling mediates hypoglycemia-induced blood–brain barrier endothelial dysfunction in vitro. PLoS ONE 2015, 10, e0122358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beyaz, S.; Yilmaz, Ö.H. Molecular pathways: Dietary regulation of stemness and tumor initiation by the ppar-δ pathway. Clin. Cancer Res. 2016, 22, 5636–5641. [Google Scholar] [CrossRef] [Green Version]
- Markowicz-Piasecka, M.; Sikora, J.; Szydłowska, A.; Skupień, A.; Mikiciuk-Olasik, E.; Huttunen, K.M. Metformin–a future therapy for neurodegenerative diseases. Pharm. Res. 2017, 34, 2614–2627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jimenez, R.; Toral, M.; Gómez-Guzmán, M.; Romero, M.; Sanchez, M.; Mahmoud, A.M.; Duarte, J. The role of Nrf2 signaling in PPARβ/δ-mediated vascular protection against hyperglycemia-induced oxidative stress. Oxidative Med. Cell. Longev. 2018, 2018, 5852706. [Google Scholar] [CrossRef] [Green Version]
- Ashabi, G.; Khalaj, L.; Khodagholi, F.; Goudarzvand, M.; Sarkaki, A. Pre-treatment with metformin activates Nrf2 antioxidant pathways and inhibits inflammatory responses through induction of AMPK after transient global cerebral ischemia. Metab. Brain Dis. 2015, 30, 747–754. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Nozohouri, S.; Vaidya, B.; Abbruscato, T. Repurposing metformin to treat age-related neurodegenerative disorders and ischemic stroke. Life Sci. 2021, 274, 119343. [Google Scholar] [CrossRef]
- Tao, L.; Li, D.; Liu, H.; Jiang, F.; Xu, Y.; Cao, Y.; Gao, R.; Chen, G. Neuroprotective effects of metformin on traumatic brain injury in rats associated with NF-κB and MAPK signaling pathway. Brain Res. Bull. 2018, 140, 154–161. [Google Scholar] [CrossRef]
- Sivandzade, F.; Cucullo, L. Anti-Diabetic Countermeasures Against Tobacco Smoke-Dependent Cerebrovascular Toxicity: Use and Effect of Rosiglitazone. Int. J. Mol. Sci. 2019, 20, 4225. [Google Scholar] [CrossRef] [Green Version]
- Prasad, S.; Sajja, R.K.; Kaisar, M.A.; Park, J.H.; Villalba, H.; Liles, T.; Abbruscato, T.; Cucullo, L. Role of Nrf2 and protective effects of Metformin against tobacco smoke-induced cerebrovascular toxicity. Redox Biol. 2017, 12, 58–69. [Google Scholar] [CrossRef]
- Sivandzade, F.; Cucullo, L. Assessing the protective effect of rosiglitazone against electronic cigarette/tobacco smoke-induced blood–brain barrier impairment. BMC Neurosci. 2019, 20, 15. [Google Scholar] [CrossRef] [Green Version]
- Prasad, S.; Sajja, R.K.; Park, J.H.; Naik, P.; Kaisar, M.A.; Cucullo, L. Impact of cigarette smoke extract and hyperglycemic conditions on blood–brain barrier endothelial cells. Fluids Barriers CNS 2015, 12, 18. [Google Scholar] [CrossRef] [Green Version]
- Oris, C.; Pereira, B.; Durif, J.; Simon-Pimmel, J.; Castellani, C.; Manzano, S.; Sapin, V.; Bouvier, D. The Biomarker S100B and Mild Traumatic Brain Injury: A Meta-analysis. Pediatrics 2018, 141, e20180037. [Google Scholar] [CrossRef] [Green Version]
- Goyal, A.; Failla, M.D.; Niyonkuru, C.; Amin, K.; Fabio, A.; Berger, R.P.; Wagner, A.K.; Ercole, A.; Thelin, E.P.; Holst, A.; et al. S100b as a prognostic biomarker in outcome prediction for patients with severe traumatic brain injury. J. Neurotrauma 2013, 30, 946–957. [Google Scholar] [CrossRef] [Green Version]
- Pijet, B.; Stefaniuk, M.; Kaczmarek, L. MMP-9 Contributes to Dendritic Spine Remodeling Following Traumatic Brain Injury. Neural Plast 2019, 2019, 3259295. [Google Scholar] [CrossRef] [Green Version]
- Pijet, B.; Stefaniuk, M.; Kostrzewska-Ksiezyk, A.; Tsilibary, P.E.; Tzinia, A.; Kaczmarek, L. Elevation of MMP-9 Levels Promotes Epileptogenesis After Traumatic Brain Injury. Mol. Neurobiol. 2018, 55, 9294–9306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guilfoyle, M.R.; Carpenter, K.L.; Helmy, A.; Pickard, J.D.; Menon, D.K.; Hutchinson, P.J. Matrix Metalloproteinase Expression in Contusional Traumatic Brain Injury: A Paired Microdialysis Study. J. Neurotrauma 2015, 32, 1553–1559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sivandzade, F.; Alqahtani, F.; Cucullo, L. Impact of chronic smoking on traumatic brain microvascular injury: An in vitro study. J. Cell. Mol. Med. 2021, 25, 7122–7134. [Google Scholar] [CrossRef] [PubMed]
- Sivandzade, F.; Alqahtani, F.; Sifat, A.; Cucullo, L. The cerebrovascular and neurological impact of chronic smoking on post-traumatic brain injury outcome and recovery: An in vivo study. J. Neuroinflamm. 2020, 17, 133. [Google Scholar] [CrossRef]
- Reinicke, A.T.; Laban, K.; Sachs, M.; Kraus, V.; Walden, M.; Damme, M.; Sachs, W.; Reichelt, J.; Schweizer, M.; Janiesch, P.C.; et al. Ubiquitin C-terminal hydrolase L1 (UCH-L1) loss causes neurodegeneration by altering protein turnover in the first postnatal weeks. Proc. Natl. Acad. Sci. USA 2019, 116, 7963–7972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Austin, V.; Crack, P.J.; Bozinovski, S.; Miller, A.A.; Vlahos, R. COPD and stroke: Are systemic inflammation and oxidative stress the missing links? Clin. Sci. 2016, 130, 1039–1050. [Google Scholar] [CrossRef] [Green Version]
- Naik, P.; Fofaria, N.; Prasad, S.; Sajja, R.K.; Weksler, B.; Couraud, P.O.; Romero, I.A.; Cucullo, L. Oxidative and pro-inflammatory impact of regular and denicotinized cigarettes on blood brain barrier endothelial cells: Is smoking reduced or nicotine-free products really safe? BMC Neurosci. 2014, 15, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.-H.; Cho, M.-H.; Choi, K.-C.; Lee, K.; Kim, K.-S.; Shim, S.-M. Oxidative stress induced by cigarette smoke extracts in human brain cells (T98G) and human brain microvascular endothelial cells (HBMEC) in mono-and co-culture. J. Toxicol. Environ. Health Part A 2015, 78, 1019–1027. [Google Scholar] [CrossRef]
- Naik, P.; Sajja, R.K.; Prasad, S.; Cucullo, L. Effect of full flavor and denicotinized cigarettes exposure on the brain microvascular endothelium: A microarray-based gene expression study using a human immortalized BBB endothelial cell line. BMC Neurosci. 2015, 16, 38. [Google Scholar] [CrossRef] [Green Version]
- Brandes, M.S.; Gray, N.E. NRF2 as a therapeutic target in neurodegenerative diseases. ASN Neuro 2020, 12, 1–23. [Google Scholar] [CrossRef]
- Scheen, A.; Esser, N.; Paquot, N. Antidiabetic agents: Potential anti-inflammatory activity beyond glucose control. Diabetes Metab. 2015, 41, 183–194. [Google Scholar] [CrossRef]
- Hammad, A.M.; Ibrahim, Y.A.; Khdair, S.I.; Hall, F.S.; Alfaraj, M.; Jarrar, Y.; Abed, A.F. Metformin reduces oxandrolone-induced depression-like behavior in rats via modulating the expression of IL-1β, IL-6, IL-10 and TNF-α. Behav. Brain Res. 2021, 414, 113475. [Google Scholar] [CrossRef]
- Cameron, A.R.; Morrison, V.L.; Levin, D.; Mohan, M.; Forteath, C.; Beall, C.; McNeilly, A.D.; Balfour, D.J.; Savinko, T.; Wong, A.K. Anti-inflammatory effects of metformin irrespective of diabetes status. Circ. Res. 2016, 119, 652–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasanpour Dehkordi, A.; Abbaszadeh, A.; Mir, S.; Hasanvand, A. Metformin and its anti-inflammatory and anti-oxidative effects; new concepts. J. Ren. Inj. Prev. 2019, 8, 54–61. [Google Scholar] [CrossRef]
- Tu, W.; Wang, H.; Li, S.; Liu, Q.; Sha, H. The anti-inflammatory and anti-oxidant mechanisms of the Keap1/Nrf2/ARE signaling pathway in chronic diseases. Aging Dis. 2019, 10, 637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadam, L.; Gomez-Lopez, N.; Mial, T.N.; Kohan-Ghadr, H.-R.; Drewlo, S. Rosiglitazone regulates TLR4 and rescues HO-1 and NRF2 expression in myometrial and decidual macrophages in inflammation-induced preterm birth. Reprod. Sci. 2017, 24, 1590–1599. [Google Scholar] [CrossRef]
- Kohan-Ghadr, H.-R.; Kilburn, B.A.; Kadam, L.; Johnson, E.; Kolb, B.L.; Rodriguez-Kovacs, J.; Hertz, M.; Armant, D.R.; Drewlo, S. Rosiglitazone augments antioxidant response in the human trophoblast and prevents apoptosis. Biol. Reprod. 2019, 100, 479–494. [Google Scholar] [CrossRef]
- Khalin, I.; Jamari, N.L.; Razak, N.B.; Hasain, Z.B.; Nor, M.A.; Zainudin, M.H.; Omar, A.B.; Alyautdin, R. A mouse model of weight-drop closed head injury: Emphasis on cognitive and neurological deficiency. Neural Regen Res. 2016, 11, 630–635. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.-M.; Chen, W.; Liu, R.-N.; Zhao, Y. Notch inhibitor can attenuate apparent diffusion coefficient and improve neurological function through downregulating NOX2-ROS in severe traumatic brain injury. Drug Des. Dev. Ther. 2018, 12, 3847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarrahi, A.; Braun, M.; Ahluwalia, M.; Gupta, R.; Wilson, M.; Munie, S.; Ahluwalia, P.; Vender, J.R.; Vale, F.L.; Dhandapani, K.M.; et al. Revisiting traumatic brain injury: From molecular mechanisms to therapeutic interventions. Biomedicines 2020, 8, 389. [Google Scholar] [CrossRef]
- Angeloni, C.; Prata, C.; Sega, F.V.D.; Piperno, R.; Hrelia, S. Traumatic brain injury and NADPH oxidase: A deep relationship. Oxidative Med. Cell. Longev. 2015, 2015, 370312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, J.; Liu, H.; Zhong, J.; Guo, Z.; Wu, J.; Zhang, H.; Huang, Z.; Jiang, L.; Li, H.; Zhang, Z.; et al. Bexarotene protects against neurotoxicity partially through a PPARgamma-dependent mechanism in mice following traumatic brain injury. Neurobiol. Dis. 2018, 117, 114–124. [Google Scholar] [CrossRef]
- Peng, J.; Wang, K.; Xiang, W.; Li, Y.; Hao, Y.; Guan, Y. Rosiglitazone polarizes microglia and protects against pilocarpine-induced status epilepticus. CNS Neurosci. 2019, 25, 1363–1372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Zhang, Q.; Yang, X.; Wang, L. PPAR-gamma agonist rosiglitazone reduces autophagy and promotes functional recovery in experimental traumaticspinal cord injury. Neurosci. Lett. 2017, 650, 89–96. [Google Scholar] [CrossRef]
- Kumar, A.P.; Kumar, B.R.P.; Jeyarani, V.; Dhanabal, S.P.; Justin, A. Glitazones, PPAR-gamma and Neuroprotection. Mini. Rev. Med. Chem. 2021, 21, 1457–1464. [Google Scholar] [CrossRef]
- Banks, W.A. From blood–brain barrier to blood–brain interface: New opportunities for CNS drug delivery. Nat. Rev. Drug Discov. 2016, 15, 275–292. [Google Scholar] [CrossRef]
- Chow, B.W.; Gu, C. The molecular constituents of the blood–brain barrier. Trends Neurosci. 2015, 38, 598–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pimentel, E.; Sivalingam, K.; Doke, M.; Samikkannu, T. Effects of drugs of abuse on the blood-brain barrier: A brief overview. Front. Neurosci. 2020, 14, 513. [Google Scholar] [CrossRef]
- Price, L.; Wilson, C.; Grant, G. Blood–Brain Barrier Pathophysiology Following Traumatic Brain Injury; Laskowitz, D., Grant, G., Eds.; Translational Research in Traumatic Brain Injury; CRC Press/Taylor and Francis Group: Boca Raton, FL, USA, 2016; Chapter 4. PMID: 26583184. [Google Scholar]
- Kanner, A.A.; Marchi, N.; Fazio, V.; Mayberg, M.R.; Koltz, M.T.; Siomin, V.; Stevens, G.H.; Masaryk, T.; Aumayr, B.; Vogelbaum, M.A.; et al. Serum S100beta: A noninvasive marker of blood-brain barrier function and brain lesions. Cancer 2003, 97, 2806–2813. [Google Scholar] [CrossRef] [Green Version]
- Marchi, N.; Rasmussen, P.; Kapural, M.; Fazio, V.; Kight, K.; Mayberg, M.R.; Kanner, A.; Ayumar, B.; Albensi, B.; Cavaglia, M.; et al. Peripheral markers of brain damage and blood-brain barrier dysfunction. Restor. Neurol. Neurosci. 2003, 21, 109–121. [Google Scholar] [PubMed]
- Suzuki, Y.; Nagai, N.; Umemura, K. A review of the mechanisms of blood-brain barrier permeability by tissue-type plasminogen activator treatment for cerebral ischemia. Front. Cell. Neurosci. 2016, 10, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, G.; Liu, C.; Li, X.; Chen, W.; He, R.; Wang, X.; Feng, P.; Lan, W. Induction of matrix metalloproteinase-1 by tumor necrosis factor-α is mediated by interleukin-6 in cultured fibroblasts of keratoconus. Exp. Biol. Med. 2016, 241, 2033–2041. [Google Scholar] [CrossRef] [Green Version]
- Sivandzade, F.; Prasad, S.; Bhalerao, A.; Cucullo, L. NRF2 and NF-қB interplay in cerebrovascular and neurodegenerative disorders: Molecular mechanisms and possible therapeutic approaches. Redox Biol. 2019, 21, 101059. [Google Scholar] [CrossRef]
- Corrigan, F.; Mander, K.A.; Leonard, A.V.; Vink, R. Neurogenic inflammation after traumatic brain injury and its potentiation of classical inflammation. J. Neuroinflamm. 2016, 13, 264. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-kappaB signaling in inflammation. Signal Transduct. Target. 2017, 2, 17023. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Wu, S.; Chen, C.; Xie, B.; Fang, Z.; Hu, W.; Chen, J.; Fu, H.; He, H. Omega-3 polyunsaturated fatty acid supplementation attenuates microglial-induced inflammation by inhibiting the HMGB1/TLR4/NF-κB pathway following experimental traumatic brain injury. J. Neuroinflamm. 2017, 14, 143. [Google Scholar] [CrossRef]
- Kumar, A.; Stoica, B.A.; Loane, D.J.; Yang, M.; Abulwerdi, G.; Khan, N.; Kumar, A.; Thom, S.R.; Faden, A.I. Microglial-derived microparticles mediate neuroinflammation after traumatic brain injury. J. Neuroinflamm. 2017, 14, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.K.W.; Kobeissy, F.H.; Shakkour, Z.; Tyndall, J.A. Thorough overview of ubiquitin C-terminal hydrolase-L1 and glial fibrillary acidic protein as tandem biomarkers recently cleared by US Food and Drug Administration for the evaluation of intracranial injuries among patients with traumatic brain injury. Acute Med. Surg. 2021, 8, e622. [Google Scholar] [CrossRef]
- Mi, Z.; Liu, H.; Rose, M.E.; Ma, X.; Reay, D.P.; Ma, J.; Henchir, J.; Dixon, C.E.; Graham, S.H. Abolishing UCHL1′s hydrolase activity exacerbates TBI-induced axonal injury and neuronal death in mice. Exp. Neurol. 2021, 336, 113524. [Google Scholar] [CrossRef]
- Choi, J.; Levey, A.I.; Weintraub, S.T.; Rees, H.D.; Gearing, M.; Chin, L.S.; Li, L. Oxidative modifications and down-regulation of ubiquitin carboxyl-terminal hydrolase L1 associated with idiopathic Parkinson’s and Alzheimer’s diseases. J. Biol. Chem. 2004, 279, 13256–13264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Namani, A.; Matiur Rahaman, M.; Chen, M.; Tang, X. Gene-expression signature regulated by the KEAP1-NRF2-CUL3 axis is associated with a poor prognosis in head and neck squamous cell cancer. BMC Cancer 2018, 18, 46. [Google Scholar] [CrossRef] [PubMed]
- Ivachtchenko, A.V.; Lavrovsky, Y.; Okun, I. AVN-101: A multi-target drug candidate for the treatment of CNS disorders. J. Alzheimer’s Dis. 2016, 53, 583–620. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Shen, J.K.; Choy, E.; Zhang, Z.; Mankin, H.J.; Hornicek, F.J.; Duan, Z. Pharmacokinetics and tolerability of NSC23925b, a novel P-glycoprotein inhibitor: Preclinical study in mice and rats. Sci. Rep. 2016, 6, 25659. [Google Scholar] [CrossRef] [PubMed]
- Kaisar, M.A.; Sivandzade, F.; Bhalerao, A.; Cucullo, L. Conventional and electronic cigarettes dysregulate the expression of iron transporters and detoxifying enzymes at the brain vascular endothelium: In vivo evidence of a gender-specific cellular response to chronic cigarette smoke exposure. Neurosci. Lett. 2018, 682, 1–9. [Google Scholar] [CrossRef]
- Prasad, S.; Sajja, R.K.; Naik, P.; Cucullo, L. Diabetes Mellitus and Blood-Brain Barrier Dysfunction: An Overview. J. Pharm. 2014, 2, 125. [Google Scholar] [CrossRef]
- Sajja, R.K.; Kaisar, M.A.; Vijay, V.; Desai, V.G.; Prasad, S.; Cucullo, L. In Vitro Modulation of Redox and Metabolism Interplay at the Brain Vascular Endothelium: Genomic and Proteomic Profiles of Sulforaphane Activity. Sci. Rep. 2018, 8, 12708. [Google Scholar] [CrossRef] [Green Version]
- Sajja, R.K.; Prasad, S.; Tang, S.; Kaisar, M.A.; Cucullo, L. Blood-brain barrier disruption in diabetic mice is linked to Nrf2 signaling deficits: Role of ABCB10? Neurosci. Lett. 2017, 653, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Benady, A.; Freidin, D.; Pick, C.G.; Rubovitch, V. GM1 ganglioside prevents axonal regeneration inhibition and cognitive deficits in a mouse model of traumatic brain injury. Sci. Rep. 2018, 8, 13340. [Google Scholar] [CrossRef] [Green Version]
- Kempuraj, D.; Ahmed, M.E.; Selvakumar, G.P.; Thangavel, R.; Raikwar, S.P.; Zaheer, S.A.; Iyer, S.S.; Govindarajan, R.; Nattanmai Chandrasekaran, P.; Burton, C. Acute traumatic brain injury-induced neuroinflammatory response and neurovascular disorders in the brain. Neurotox. Res. 2021, 39, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Kostich, W.; Hamman, B.D.; Li, Y.-W.; Naidu, S.; Dandapani, K.; Feng, J.; Easton, A.; Bourin, C.; Baker, K.; Allen, J.; et al. Inhibition of AAK1 kinase as a novel therapeutic approach to treat neuropathic pain. J. Pharmacol. Exp. Ther. 2016, 358, 371–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sajja, R.K.; Cucullo, L. Altered glycaemia differentially modulates efflux transporter expression and activity in hCMEC/D3 cell line. Neurosci. Lett. 2015, 598, 59–65. [Google Scholar] [CrossRef] [Green Version]
- Sivandzade, F.; Bhalerao, A.; Cucullo, L. Cerebrovascular and neurological disorders: Protective role of NRF2. Int. J. Mol. Sci. 2019, 20, 3433. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TBI Control | TBI + TS | TBI + TS + MF100 | TBI + TS + MF200 | TBI + TS + RSG10 | TBI + TS + RSG20 | |
|---|---|---|---|---|---|---|
| TBI | √ | √ | √ | √ | √ | √ |
| TS Exposure | - | √ | √ | √ | √ | √ |
| MF 100 mg/kg | - | - | √ | - | - | - |
| MF 200 mg/kg | - | - | - | √ | - | - |
| RSG 10 mg/kg | - | - | - | - | √ | - |
| RSG 20 mg/kg | - | - | - | - | - | √ |
| Target Gene | Forward | Reverse |
|---|---|---|
| NRF2 | 5′- GGC TCA GCA CCT TGT ATC TT -3′ | 5′- CAC ATT GCC ATC TCT GGT TTG -3′ |
| NQO-1 | 5′- GAG AAG AGC CCT GAT TGT ACT G -3′ | 5′- ACC TCC CAT CCT CTC TTC TT -3′ |
| HO-1 | 5′- CTC CCT GTG TTT CCT TTC TCT C -3′ | 5′- GCT GCT GGT TTC AAA GTT CAG -3′ |
| NF-kB | 5′- AGA CAT CCT TCC GCA AAC TC -3′ | 5′- TAG GTC CTT CCT GCC CAT AA -3′ |
| Claudin-5 | 5′- GGT GAA GTA GGC ACC AAA CT -3′ | 5′- TTT CTC CAG CTG CCC TTT C -3′ |
| Occludin | 5′- CAG CAG CAA TGG TAA CCT AGA G -3′ | 5′- CAC CTG TCG TGT AGT CTG TTT C -3′ |
| VCAM-1 | 5′- GAG GGA GAC ACC GTC ATT ATC -3′ | 5′- CGA GCC ATC CAC AGA CTT TA -3′ |
| PECAM-1 | 5′- CAA CAG AGC CAG CAG TAT GA -3′ | 5′- TGA CAA CCA CCG CAA TGA -3′ |
| ZO-1 | 5′- CAT TAC GAC CCT GAA GAG GAT G -3′ | 5′- AGC AGG AAG ATG TGC AGA AG -3′ |
| Β-Actin | 5′- GAG GTA TCC TGA CCC TGA AGT A -3′ | 5′- CAC ACG CAG CTC ATT GTA GA -3′ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sivandzade, F.; Alqahtani, F.; Dhaibar, H.; Cruz-Topete, D.; Cucullo, L. Antidiabetic Drugs Can Reduce the Harmful Impact of Chronic Smoking on Post-Traumatic Brain Injuries. Int. J. Mol. Sci. 2023, 24, 6219. https://doi.org/10.3390/ijms24076219
Sivandzade F, Alqahtani F, Dhaibar H, Cruz-Topete D, Cucullo L. Antidiabetic Drugs Can Reduce the Harmful Impact of Chronic Smoking on Post-Traumatic Brain Injuries. International Journal of Molecular Sciences. 2023; 24(7):6219. https://doi.org/10.3390/ijms24076219
Chicago/Turabian StyleSivandzade, Farzane, Faleh Alqahtani, Hemangini Dhaibar, Diana Cruz-Topete, and Luca Cucullo. 2023. "Antidiabetic Drugs Can Reduce the Harmful Impact of Chronic Smoking on Post-Traumatic Brain Injuries" International Journal of Molecular Sciences 24, no. 7: 6219. https://doi.org/10.3390/ijms24076219