
Gene Polymorphisms and Biological Effects of Vitamin D Receptor on Nonalcoholic Fatty Liver Disease Development and Progression


Abstract
:1. Introduction
2. Methodology
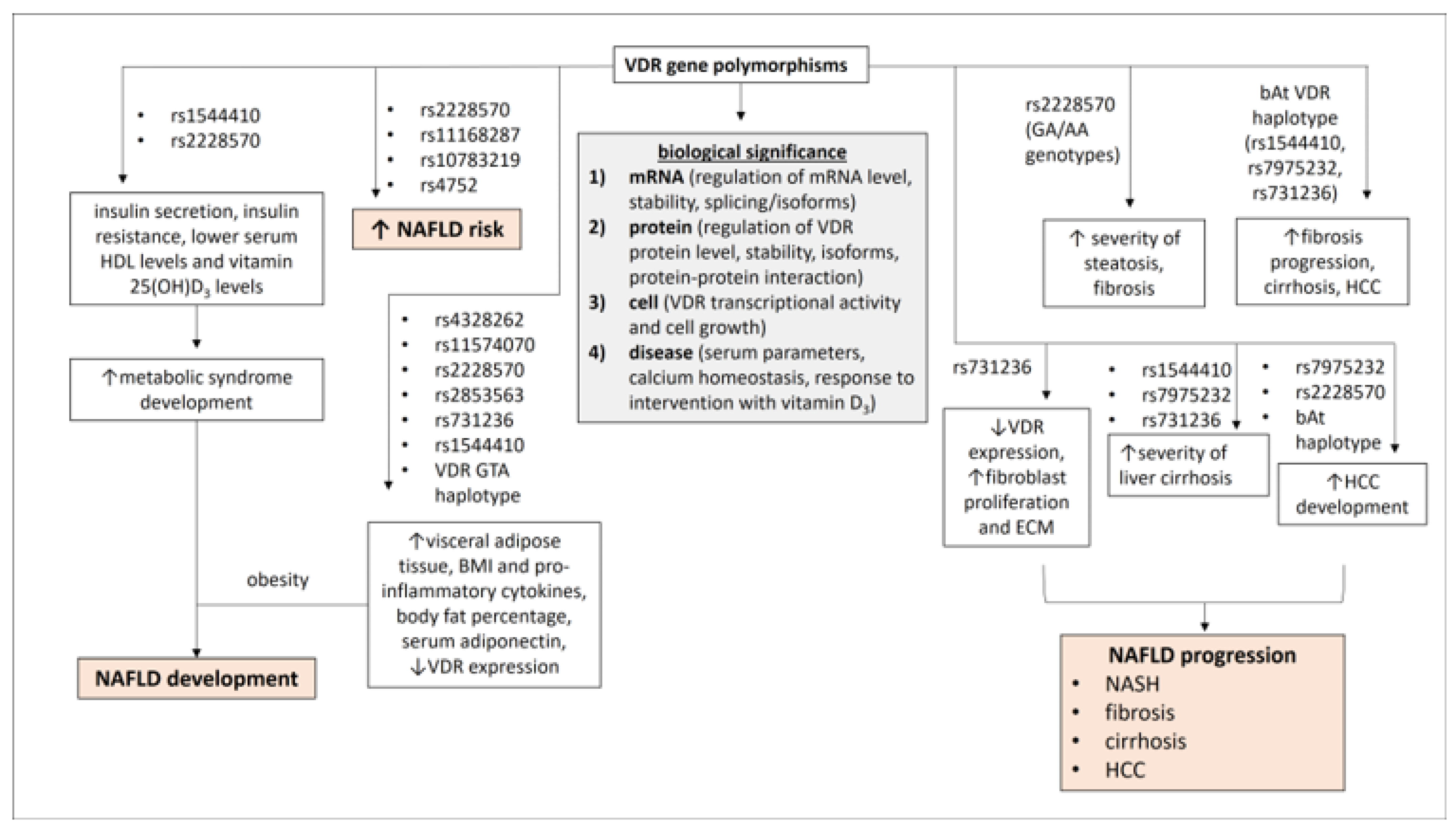
3. Genetics and Biology of VDR Gene Polymorphisms
3.1. Polymorphisms of the VDR Gene
3.2. Biological Significance of VDR Gene Polymorphisms
4. VDR Gene Polymorphisms in the Development of NAFLD
5. VDR Gene Polymorphisms in NAFLD Disease Progression
6. Vitamin D-VDR Signaling in Cells
7. The Role of Vitamin D-VDR Signaling in the Development of NAFLD
7.1. Direct Effects of VDR on the Development of NAFLD
7.2. Indirect Effects of VDR on the Development of NAFLD
8. VDR and Insulin Resistance
9. VDR and Gut Microbiota
10. The Role of Vitamin D-VDR Signaling in NAFLD Disease Progression
11. Vitamin D-VDR and NASH
11.1. VDR and Lipotoxicity in NASH
11.2. VDR and Immune Modulation in NASH
12. Vitamin D-VDR and Fibrosis-Liver Cirrhosis
12.1. VDR and HSCs Activation
12.2. VDR and MMPs/TIMPs
12.3. VDR and Fibrosis-Related Signal Transduction Pathways
13. VDR and HCC
13.1. VDR and CAFs
13.2. VDR and CAFs-Immune Cell Crosstalk

{kind=link}
{kind=link}
| VDR Mediated-Mechanism | Effects on NAFLD Disease Progression | Refs. |
|---|---|---|
| VDR and lipotoxicity in NASH | ||
|
| [83,84,85,86,87,88,89] |
| VDR and immune modulation in NASH | ||
|
| [14,72,85,90,91,92,93,94,95,96,97] |
| VDR and fibrosis-liver cirrhosis | ||
|
| [98,100,101,102,103] |
|
| [105,106,107,108] |
|
| [111,112,113,114,115,116] |
|
| [117,118] |
|
| [119,120,121,122,123] |
| VDR and hepatocellular carcinoma (HCC) | ||
|
| [113,126,127,128,129,130,131,132,133] |
|
| [142,143,144,145] |
|
| [142,146,147,148,149,150,151,152,153,154] |
14. Conclusions and Future Directions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Estes, C.; Razavi, H.; Loomba, R.; Younossi, Z.; Sanyal, A.J. Modeling the epidemic of nonalcoholic fatty liver disease demonstrates an exponential increase in burden of disease. Hepatology 2018, 67, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Powell, E.E.; Wong, V.W.S.; Rinella, M. Non-alcoholic fatty liver disease. Lancet 2021, 397, 2212–2224. [Google Scholar] [CrossRef] [PubMed]
- Mundi, M.S.; Velapati, S.; Patel, J.; Kellogg, T.A.; Abu Dayyeh, B.K.; Hurt, R.T. Evolution of NAFLD and Its Management. Nutr. Clin. Pract. 2020, 35, 72–84. [Google Scholar] [CrossRef]
- Kawagoe, F.; Mendoza, A.; Hayata, Y.; Asano, L.; Kotake, K.; Mototani, S.; Kawamura, S.; Kurosaki, S.; Akagi, Y.; Takemoto, Y.; et al. Discovery of a Vitamin D Receptor-Silent Vitamin D Derivative That Impairs Sterol Regulatory Element-Binding Protein In Vivo. J. Med. Chem. 2021, 64, 5689–5709. [Google Scholar] [CrossRef]
- Cimini, F.A.; Barchetta, I.; Carotti, S.; Morini, S.; Cavallo, M.G. Overview of studies of the vitamin D/vitamin D receptor system in the development of non-alcoholic fatty liver disease. World J. Gastrointest. Pathophysiol. 2019, 10, 11–16. [Google Scholar] [CrossRef]
- Barchetta, I.; Cimini, F.A.; Cavallo, M.G. Vitamin D and Metabolic Dysfunction-Associated Fatty Liver Disease (MAFLD): An Update. Nutrients 2020, 12, 3302. [Google Scholar] [CrossRef]
- Arai, T.; Atsukawa, M.; Tsubota, A.; Koeda, M.; Yoshida, Y.; Okubo, T.; Nakagawa, A.; Itokawa, N.; Kondo, C.; Nakatsuka, K.; et al. Association of vitamin D levels and vitamin D-related gene polymorphisms with liver fibrosis in patients with biopsy-proven nonalcoholic fatty liver disease. Dig. Liver Dis. 2019, 51, 1036–1042. [Google Scholar] [CrossRef]
- Yaghooti, H.; Ghanavati, F.; Seyedian, S.S.; Cheraghian, B.; Mohammadtaghvaei, N. The efficacy of calcitriol treatment in non-alcoholic fatty liver patients with different genotypes of vitamin D receptor FokI polymorphism. BMC Pharmacol. Toxicol. 2021, 22, 18. [Google Scholar] [CrossRef]
- Khan, R.J.; Riestra, P.; Gebreab, S.Y.; Wilson, J.G.; Gaye, A.; Xu, R.; Sharon, D.K. Vitamin D Receptor Gene Polymorphisms Are Associated with Abdominal Visceral Adipose Tissue Volume and Serum Adipokine Concentrations but Not with Body Mass Index or Waist Circumference in African Americans: The Jackson Heart Study123. J. Nutr. 2016, 146, 1476–1482. [Google Scholar] [CrossRef]
- Triantos, C.; Aggeletopoulou, I.; Kalafateli, M.; Spantidea, P.I.; Vourli, G.; Diamantopoulou, G.; Tapratzi, D.; Michalaki, M.; Manolakopoulos, S.; Gogos, C.; et al. Prognostic significance of vitamin D receptor (VDR) gene polymorphisms in liver cirrhosis. Sci. Rep. 2018, 8, 14065. [Google Scholar] [CrossRef]
- Mosaad, H.; Emam, E.A.; Hamed, E.F.; El Demerdash, E.A.; Hussein, S. Vitamin D receptor gene polymorphism and hepatocellular carcinoma in chronic hepatitis C patients. Egypt. Liver J. 2020, 10, 55. [Google Scholar] [CrossRef]
- Nurminen, V.; Seuter, S.; Carlberg, C. Primary Vitamin D Target Genes of Human Monocytes. Front. Physiol. 2019, 10, 194. [Google Scholar] [CrossRef]
- Kongsbak, M.; Levring, T.B.; Geisler, C.; Rode von Essen, M. The Vitamin D Receptor and T Cell Function. Front. Immunol. 2013, 4, 148. [Google Scholar] [CrossRef]
- Arai, H.; Miyamoto, K.I.; Yoshida, M.; Yamamoto, H.; Taketani, Y.; Morita, K.; Kubota, M.; Yoshida, S.; Ikeda, M.; Watabe, F.; et al. The polymorphism in the caudal-related homeodomain protein Cdx-2 binding element in the human vitamin D receptor gene. J. Bone Miner. Res. 2001, 16, 1256–1264. [Google Scholar] [CrossRef]
- Fang, Y.; van Meurs, J.B.J.; d’Alesio, A.; Jhamai, M.; Zhao, H.; Rivadeneira, F.; Hofman, A.; van Leeuwen, J.P.T.; Jehan, F.; Pols, H.A.P.; et al. Promoter and 3’-untranslated-region haplotypes in the vitamin d receptor gene predispose to osteoporotic fracture: The rotterdam study. Am. J. Hum. Genet. 2005, 77, 807–823. [Google Scholar] [CrossRef]
- Arai, H.; Miyamoto, K.; Taketani, Y.; Yamamoto, H.; Iemori, Y.; Morita, K.; Tonai, T.; Nishisho, T.; Mori, S.; Takeda, E. A vitamin D receptor gene polymorphism in the translation initiation codon: Effect on protein activity and relation to bone mineral density in Japanese women. J. Bone Miner. Res. 1997, 12, 915–921. [Google Scholar] [CrossRef]
- Saccone, D.; Asani, F.; Bornman, L. Regulation of the vitamin D receptor gene by environment, genetics and epigenetics. Gene 2015, 561, 171–180. [Google Scholar] [CrossRef]
- Morrison, N.A.; Qi, J.C.; Tokita, A.; Kelly, P.J.; Crofts, L.; Nguyen, T.V.; Sambrook, P.N.; Eisman, J.A. Prediction of bone density from vitamin D receptor alleles. Nature 1994, 367, 284–287. [Google Scholar] [CrossRef]
- Ye, W.Z.; Reis, A.F.; Velho, G. Identification of a novel Tru9 I polymorphism in the human vitamin D receptor gene. J. Hum. Genet. 2000, 45, 56–57. [Google Scholar] [CrossRef]
- Selvaraj, P.; Prabhu Anand, S.; Harishankar, M.; Alagarasu, K. Plasma 1,25 dihydroxy vitamin D3 level and expression of vitamin d receptor and cathelicidin in pulmonary tuberculosis. J. Clin. Immunol. 2009, 29, 470–478. [Google Scholar] [CrossRef] [PubMed]
- Andraos, C.; Koorsen, G.; Knight, J.C.; Bornman, L. Vitamin D receptor gene methylation is associated with ethnicity, tuberculosis, and TaqI polymorphism. Hum. Immunol. 2011, 72, 262–268. [Google Scholar] [CrossRef] [PubMed]
- Uitterlinden, A.G.; Fang, Y.; Van Meurs, J.B.J.; Pols, H.A.P.; Van Leeuwen, J.P.T.M. Genetics and biology of vitamin D receptor polymorphisms. Gene 2004, 338, 143–156. [Google Scholar] [CrossRef] [PubMed]
- Angulo, P. GI epidemiology: Nonalcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2007, 25, 883–889. [Google Scholar] [CrossRef] [PubMed]
- Jaroenlapnopparat, A.; Suppakitjanusant, P.; Ponvilawan, B.; Charoenngam, N. Vitamin D-Related Genetic Variations and Nonalcoholic Fatty Liver Disease: A Systematic Review. Int. J. Mol. Sci. 2022, 23, 9122. [Google Scholar] [CrossRef]
- Almeda-Valdés, P.; Cuevas-Ramos, D.; Aguilar-Salinas, C.A. Metabolic syndrome and non-alcoholic fatty liver disease. Ann. Hepatol. 2009, 8, S18–S24. [Google Scholar] [CrossRef]
- Schuch, N.J.; Garcia, V.C.; Gouvea Ferreiro Vivolo, S.R.; Martini, L.A. Relationship between Vitamin D Receptor gene polymorphisms and the components of metabolic syndrome. Nutr. J. 2013, 12, 96. [Google Scholar] [CrossRef]
- Fabbrini, E.; Sullivan, S.; Klein, S. Obesity and Nonalcoholic Fatty Liver Disease: Biochemical, Metabolic and Clinical Implications. Hepatology 2010, 51, 679–689. [Google Scholar] [CrossRef]
- Al-Daghri, N.M.; Guerini, F.R.; Al-Attas, O.S.; Alokail, M.S.; Alkharfy, K.M.; Draz, H.M.; Agliardi, C.; Costa, A.S.; Saulle, I.; Mohammed, A.K.; et al. Vitamin D receptor gene polymorphisms are associated with obesity and inflammosome activity. PLoS ONE 2014, 9, e102141. [Google Scholar] [CrossRef]
- Chiu, K.W.; Goto, S.; Nakano, T.; Hu, T.H.; Chen, D.W.; Huang, K.T.; Hsu, L.W.; Chen, C.L. Genetic polymorphisms of the hepatic pathways of fatty liver disease after living donor liver transplantation. Liver Int. 2018, 38, 2287–2293. [Google Scholar] [CrossRef]
- Petta, S.; Cammà, C.; Scazzone, C.; Tripodo, C.; Di Marco, V.; Bono, A.; Cabibi, D.; Licata, G.; Porcasi, R.; Marchesini, G.; et al. Low vitamin D serum level is related to severe fibrosis and low responsiveness to interferon-based therapy in genotype 1 chronic hepatitis C. Hepatology 2010, 51, 1158–1167. [Google Scholar] [CrossRef]
- Jablonski, K.L.; Jovanovich, A.; Holmen, J.; Targher, G.; McFann, K.; Kendrick, J.; Chonchol, M. Low 25-hydroxyvitamin D level is independently associated with non-alcoholic fatty liver disease. Nutr. Metab. Cardiovasc. Dis. 2013, 23, 792–798. [Google Scholar] [CrossRef]
- Kneeman, J.M.; Misdraji, J.; Corey, K.E. Secondary causes of nonalcoholic fatty liver disease. Ther. Adv. Gastroenterol. 2012, 5, 199–207. [Google Scholar] [CrossRef]
- Vernon, G.; Baranova, A.; Younossi, Z.M. Systematic review: The epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment. Pharmacol. Ther. 2011, 34, 274–285. [Google Scholar] [CrossRef]
- Farrell, G.C.; Larter, C.Z. Nonalcoholic fatty liver disease: From steatosis to cirrhosis. Hepatology 2006, 43, S99–S112. [Google Scholar] [CrossRef]
- Ascha, M.S.; Hanouneh, I.A.; Lopez, R.; Tamimi, T.A.R.; Feldstein, A.F.; Zein, N.N. The incidence and risk factors of hepatocellular carcinoma in patients with nonalcoholic steatohepatitis. Hepatology 2010, 51, 1972–1978. [Google Scholar] [CrossRef]
- Falleti, E.; Bitetto, D.; Fabris, C.; Cussigh, A.; Fontanini, E.; Fornasiere, E.; Fumolo, E.; Bignulin, S.; Cmet, S.; Minisini, R. Vitamin D receptor gene polymorphisms and hepatocellular carcinoma in alcoholic cirrhosis. World J. Gastroenterol. 2010, 16, 3016–3024. [Google Scholar] [CrossRef]
- Baur, K.; Mertens, J.C.; Schmitt, J.; Iwata, R.; Stieger, B.; Eloranta, J.J.; Frei, P.; Stickel, F.; Dill, M.T.; Seifert, B.; et al. Combined effect of 25-OH vitamin D plasma levels and genetic vitamin D receptor (NR 1I1) variants on fibrosis progression rate in HCV patients. Liver Int. 2012, 32, 635–643. [Google Scholar] [CrossRef]
- Hung, C.H.; Chiu, Y.C.; Hu, T.H.; Chen, C.H.; Lu, S.N.; Huang, C.M.; Wang, J.H.; Lee, C.M. Significance of vitamin d receptor gene polymorphisms for risk of hepatocellular carcinoma in chronic hepatitis C. Transl. Oncol. 2014, 7, 503–507. [Google Scholar] [CrossRef]
- Pontoriero, A.C.; Trinks, J.; Hulaniuk, M.L.; Caputo, M.; Fortuny, L.; Pratx, L.B.; Frias, A.; Torres, O.; Nunez, F.; Gadano, A.; et al. Influence of ethnicity on the distribution of genetic polymorphisms associated with risk of chronic liver disease in South American populations. BMC Genet. 2015, 16, 93. [Google Scholar] [CrossRef]
- Gibson, P.S.; Quaglia, A.; Dhawan, A.; Wu, H.; Lanham-New, S.; Hart, K.H.; Fitzpatrick, E.; Moore, J.B. Vitamin D status and associated genetic polymorphisms in a cohort of UK children with non-alcoholic fatty liver disease. Pediatr. Obes. 2018, 13, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Heyens, L.J.M.; Busschots, D.; Koek, G.H.; Robaeys, G.; Francque, S. Liver Fibrosis in Non-alcoholic Fatty Liver Disease: From Liver Biopsy to Non-invasive Biomarkers in Diagnosis and Treatment. Front. Med. 2021, 8, 615978. [Google Scholar] [CrossRef] [PubMed]
- Thanapirom, K.; Suksawatamnuay, S.; Sukeepaisarnjaroen, W.; Tangkijvanich, P.; Thaimai, P.; Wasitthankasem, R.; Poovorawan, Y.; Komolmit, P. Genetic associations of vitamin D receptor polymorphisms with advanced liver fibrosis and response to pegylated interferon-based therapy in chronic hepatitis C. PeerJ 2019, 7, e7666. [Google Scholar] [CrossRef] [PubMed]
- Gisbert-Ferrándiz, L.; Cosin-Roger, J.; Hernández, C.; Macias-Ceja, D.C.; Ortiz-Masiá, D.; Salvador, P.; Wildenberg, M.E.; Esplugues, J.V.; Alos, R.; Navarro, F.; et al. The vitamin D receptor Taq I polymorphism is associated with reduced VDR and increased PDIA3 protein levels in human intestinal fibroblasts. J. Steroid Biochem. Mol. Biol. 2020, 202, 105720. [Google Scholar] [CrossRef]
- Li, Y.J.; Tang, Y.W.; Shi, Y.Q.; Han, S.; Wang, J.B.; Zhou, X.M.; Chen, Y.; Wu, Z.D.; Han, Z.Y.; Han, Y.; et al. Polymorphisms in the vitamin D receptor gene and risk of primary biliary cirrhosis: A meta-analysis. J. Gastroenterol. Hepatol. 2014, 29, 706–715. [Google Scholar] [CrossRef]
- Fang, F.; Wang, J.; Pan, J.; Su, G.H.; Xu, L.X.; Li, G. Relationship between vitamin D (1,25-dihydroxyvitamin D3) receptor gene polymorphisms and primary biliary cirrhosis risk: A meta-analysis. Genet. Mol. Res. 2015, 14, 981–988. [Google Scholar] [CrossRef]
- Luis, C.B.; Adams, L.A. The Natural Course of Non-Alcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2016, 17, 774. [Google Scholar] [CrossRef]
- Quan, Y.; Yang, J.; Qin, T.; Hu, Y. Associations between twelve common gene polymorphisms and susceptibility to hepatocellular carcinoma: Evidence from a meta-analysis. World J. Surg. Oncol. 2019, 17, 216. [Google Scholar] [CrossRef]
- Barooah, P.; Saikia, S.; Bharadwaj, R.; Sarmah, P.; Bhattacharyya, M.; Goswami, B.; Medhi, S. Role of VDR, GC, and CYP2R1 Polymorphisms in the Development of Hepatocellular Carcinoma in Hepatitis C Virus-Infected Patients. Genet. Test. Mol. Biomark. 2019, 23, 325–331. [Google Scholar] [CrossRef]
- Stokes, C.S.; Volmer, D.A.; Grünhage, F.; Lammert, F. Vitamin D in chronic liver disease. Liver Int. 2013, 33, 338–352. [Google Scholar] [CrossRef]
- Gascon-Barré, M.; Demers, C.; Mirshahi, A.; Néron, S.; Zalzal, S.; Nanci, A. The normal liver harbors the vitamin D nuclear receptor in nonparenchymal and biliary epithelial cells. Hepatology 2003, 37, 1034–1042. [Google Scholar] [CrossRef]
- Zúñiga, S.; Firrincieli, D.; Housset, C.; Chignard, N. Vitamin D and the vitamin D receptor in liver pathophysiology. Clin. Res. Hepatol. Gastroenterol. 2011, 35, 295–302. [Google Scholar] [CrossRef]
- Bikle, D.D.; Feingold, K.R.; Anawalt, B.; Blackman, M.R.; Boyce, A.; Chrousos, G.; Corpas, E.; de Herder, W.W.; Dhatariya, K.; Dungan, K.; et al. Vitamin D: Production, Metabolism and Mechanisms of Action. Available online: http://www.ncbi.nlm.nih.gov/books/NBK278935/ (accessed on 20 February 2023).
- Scott, M.J. The upside-downside nature of Vitamin D signaling in liver. J. Leukoc. Biol. 2019, 106, 783–785. [Google Scholar] [CrossRef]
- Bagur, R.; Hajnóczky, G. Intracellular Ca2+ sensing: Role in calcium homeostasis and signaling. Mol. Cell 2017, 66, 780–788. [Google Scholar] [CrossRef]
- Mota, M.; Banini, B.A.; Cazanave, S.C.; Sanyal, A.J. Molecular mechanisms of lipotoxicity and glucotoxicity in nonalcoholic fatty liver disease. Metabolism 2016, 65, 1049–1061. [Google Scholar] [CrossRef]
- Geng, Y.; Faber, K.N.; de Meijer, V.E.; Blokzijl, H.; Moshage, H. How does hepatic lipid accumulation lead to lipotoxicity in non-alcoholic fatty liver disease? Hepatol. Int. 2021, 15, 21–35. [Google Scholar] [CrossRef]
- Bugianesi, E.; McCullough, A.J.; Marchesini, G. Insulin resistance: A metabolic pathway to chronic liver disease. Hepatology 2005, 42, 987–1000. [Google Scholar] [CrossRef]
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Invest. 2005, 115, 1343–1351. [Google Scholar] [CrossRef]
- Tao, T.; Kobelski, M.M.; Saini, V.; Demay, M.B. Adipose-specific VDR Deletion Leads to Hepatic Steatosis in Female Mice Fed a Low-Fat Diet. Endocrinology 2022, 163, bqab249. [Google Scholar] [CrossRef]
- Bozic, M.; Guzman, C.; Benet, M.; Sanchez-Campos, S.; Garcia-Monzon, C.; Gari, E.; Gatius, S.; Valdivielso, J.M.; Jover, R. Hepatocyte vitamin D receptor regulates lipid metabolism and mediates experimental diet-induced steatosis. J. Hepatol. 2016, 65, 748–757. [Google Scholar] [CrossRef]
- Jahn, D.; Dorbath, D.; Schilling, A.K.; Gildein, L.; Meier, C.; Vuille-Dit-Bille, R.N.; Schmitt, J.; Kraus, D.; Fleet, J.C.; Hermanns, H.M.; et al. Intestinal vitamin D receptor modulates lipid metabolism, adipose tissue inflammation and liver steatosis in obese mice. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1567–1578. [Google Scholar] [CrossRef] [PubMed]
- Zechner, R.; Strauss, J.; Frank, S.; Wagner, E.; Hofmann, W.; Kratky, D.; Hiden, M.; Levak-Frank, S. The role of lipoprotein lipase in adipose tissue development and metabolism. Int. J. Obes. Relat. Metab. Disord. 2000, 24, S53–S56. [Google Scholar] [CrossRef] [PubMed]
- Barchetta, I.; Cimini, F.A.; Chiappetta, C.; Bertoccini, L.; Ceccarelli, V.; Capoccia, D.; Gaggini, M.; Cristofano, C.D.; Rocca, C.D.; Silecchia, G.; et al. Relationship between hepatic and systemic angiopoietin-like 3, hepatic Vitamin D receptor expression and NAFLD in obesity. Liver Int. 2020, 40, 2139–2147. [Google Scholar] [CrossRef] [PubMed]
- García-Monzón, C.; Petrov, P.D.; Rey, E.; Marañón, P.; Del Pozo-Maroto, E.; Guzmán, C.; Rodriguez de Cia, J.; Casado-Collado, A.J.; Vargas-Castrillon, J.; Saez, A.; et al. Angiopoietin-Like Protein 8 Is a Novel Vitamin D Receptor Target Gene Involved in Nonalcoholic Fatty Liver Pathogenesis. Am. J. Pathol. 2018, 188, 2800–2810. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Shen, Z.; Lin, Y.; Zhang, J.; Zhang, Y.; Liu, P.; Zeng, H.; Yu, M.; Chen, X.; Ning, L.; et al. Vitamin D receptor targets hepatocyte nuclear factor 4α and mediates protective effects of vitamin D in nonalcoholic fatty liver disease. J. Biol. Chem. 2020, 295, 3891–3905. [Google Scholar] [CrossRef]
- Xu, Y.; Zalzala, M.; Xu, J.; Li, Y.; Yin, L.; Zhang, Y. A metabolic stress-inducible miR-34a-HNF4α pathway regulates lipid and lipoprotein metabolism. Nat. Commun. 2015, 6, 7466. [Google Scholar] [CrossRef]
- Italian Association for the Study of the Liver (AISF). AISF position paper on nonalcoholic fatty liver disease (NAFLD): Updates and future directions. Dig. Liver Dis. 2017, 49, 471–483. [Google Scholar] [CrossRef]
- Bashir, A.; Duseja, A.; De, A.; Mehta, M.; Tiwari, P. Non-alcoholic fatty liver disease development: A multifactorial pathogenic phenomena. Liver Res. 2022, 6, 72–83. [Google Scholar] [CrossRef]
- Utzschneider, K.M.; Kahn, S.E. The Role of Insulin Resistance in Nonalcoholic Fatty Liver Disease. J. Clin. Endocrinol. Metab. 2006, 91, 4753–4761. [Google Scholar] [CrossRef]
- Elhafiz, M.; Zhao, G.; Ismail, M.; Xu, D.; Das, D.; Fan, S.; Cheng, N.; Yousef, B.A.; Jiang, Z.; Zhang, L. Imbalanced insulin substrate-1 and insulin substrate-2 signaling trigger hepatic steatosis in vitamin D deficient rats: 8-methoxypsoralen, a vitamin D receptor ligand with a promising anti-steatotic action. Biochim. Biophys. Acta Mol. Cell. Biol. Lipids 2020, 1865, 158657. [Google Scholar] [CrossRef]
- Dong, B.; Zhou, Y.; Wang, W.; Scott, J.; Kim, K.; Sun, Z.; Guo, Q.; Lu, Y.; Gonzales, N.M.; Wu, H.; et al. Vitamin D Receptor Activation in Liver Macrophages Ameliorates Hepatic Inflammation, Steatosis, and Insulin Resistance in Mice. Hepatology 2020, 71, 1559–1574. [Google Scholar] [CrossRef]
- Oh, J.; Riek, A.E.; Darwech, I.; Funai, K.; Shao, J.; Chin, K.; Sierra, O.L.; Carmeliet, G.; Ostlund Jr, R.E.; Mizrachi, C.B. Deletion of Macrophage Vitamin D Receptor Promotes Insulin Resistance and Monocyte Cholesterol Transport to Accelerate Atherosclerosis in Mice. Cell. Rep. 2015, 10, 1872–1886. [Google Scholar] [CrossRef]
- Ogunkolade, B.W.; Boucher, B.J.; Prahl, J.M.; Bustin, S.A.; Burrin, J.M.; Noonan, K.; North, B.V.; Mannan, N.; McDermott, M.F.; DeLuca, H.F.; et al. Vitamin D receptor (VDR) mRNA and VDR protein levels in relation to vitamin D status, insulin secretory capacity, and VDR genotype in Bangladeshi Asians. Diabetes 2002, 51, 2294–2300. [Google Scholar] [CrossRef]
- Su, D.; Nie, Y.; Zhu, A.; Chen, Z.; Wu, P.; Zhang, L.; Luo, M.; Sun, Q.; Cai, Y.; Xiao, Z.; et al. Vitamin D Signaling through Induction of Paneth Cell Defensins Maintains Gut Microbiota and Improves Metabolic Disorders and Hepatic Steatosis in Animal Models. Front. Physiol. 2016, 7, 498. [Google Scholar] [CrossRef]
- Pendyala, S.; Walker, J.M.; Holt, P.R. A high-fat diet is associated with endotoxemia that originates from the gut. Gastroenterology 2012, 142, 1100–1101.e2. [Google Scholar] [CrossRef]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef]
- Kolodziejczyk, A.A.; Zheng, D.; Shibolet, O.; Elinav, E. The role of the microbiome in NAFLD and NASH. EMBO Mol. Med. 2019, 11, e9302. [Google Scholar] [CrossRef]
- Jin, D.; Wu, S.; Zhang, Y.G.; Lu, R.; Xia, Y.; Dong, H.; Sun, J. Lack of Vitamin D Receptor Causes Dysbiosis and Changes the Functions of the Murine Intestinal Microbiome. Clin. Ther. 2015, 37, 996–1009.e7. [Google Scholar] [CrossRef]
- Wu, S.; Zhang, Y.G.; Lu, R.; Xia, Y.; Zhou, D.; Petrof, E.O.; Claud, E.C.; Chen, D.; Chang, E.B.; Carmeliet, G.; et al. Intestinal epithelial vitamin D receptor deletion leads to defective autophagy in colitis. Gut 2015, 64, 1082–1094. [Google Scholar] [CrossRef]
- Akimbekov, N.S.; Digel, I.; Sherelkhan, D.K.; Lutfor, A.B.; Razzaque, M.S. Vitamin D and the Host-Gut Microbiome: A Brief Overview. Acta Histochem. Cytochem. 2020, 53, 33–42. [Google Scholar] [CrossRef]
- Chatterjee, I.; Lu, R.; Zhang, Y.; Zhang, J.; Dai, Y.; Xia, Y.; Sun, J. Vitamin D receptor promotes healthy microbial metabolites and microbiome. Sci. Rep. 2020, 10, 7340. [Google Scholar] [CrossRef] [PubMed]
- Musso, G.; Cassader, M.; Paschetta, E.; Gambino, R. Bioactive Lipid Species and Metabolic Pathways in Progression and Resolution of Nonalcoholic Steatohepatitis. Gastroenterology 2018, 155, 282–302.e8. [Google Scholar] [CrossRef] [PubMed]
- Neuschwander-Tetri, B.A. Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: The central role of nontriglyceride fatty acid metabolites. Hepatology 2010, 52, 774–788. [Google Scholar] [CrossRef] [PubMed]
- Ricca, C.; Aillon, A.; Bergandi, L.; Alotto, D.; Castagnoli, C.; Silvagno, F. Vitamin D Receptor Is Necessary for Mitochondrial Function and Cell Health. Int. J. Mol. Sci. 2018, 19, 1672. [Google Scholar] [CrossRef] [PubMed]
- Leamy, A.K.; Egnatchik, R.A.; Young, J.D. Molecular mechanisms and the role of saturated fatty acids in the progression of non-alcoholic fatty liver disease. Prog. Lipid Res. 2013, 52, 165–174. [Google Scholar] [CrossRef]
- Malhi, H.; Kaufman, R.J. Endoplasmic reticulum stress in liver disease. J. Hepatol. 2011, 54, 795–809. [Google Scholar] [CrossRef]
- Zhou, Y.; Dong, B.; Kim, K.H.; Choi, S.; Sun, Z.; Wu, N.; Wu, Y.; Scott, J.; Moore, D.D. Vitamin D Receptor Activation in Liver Macrophages Protects Against Hepatic Endoplasmic Reticulum Stress in Mice. Hepatology 2020, 71, 1453–1466. [Google Scholar] [CrossRef]
- Wen, G.; Eder, K.; Ringseis, R. 1,25-hydroxyvitamin D3 decreases endoplasmic reticulum stress-induced inflammatory response in mammary epithelial cells. PLoS ONE 2020, 15, e0228945. [Google Scholar] [CrossRef]
- Roh, Y.S.; Seki, E. Toll-like receptors in alcoholic liver disease, non-alcoholic steatohepatitis and carcinogenesis. J. Gastroenterol. Hepatol. 2013, 28, 38–42. [Google Scholar] [CrossRef]
- Tomita, K.; Tamiya, G.; Ando, S.; Ohsumi, K.; Chiyo, T.; Mizutani, A.; Kitamura, N.; Toda, K.; Kaneko, T.; Horie, Y.; et al. Tumour necrosis factor alpha signalling through activation of Kupffer cells plays an essential role in liver fibrosis of non-alcoholic steatohepatitis in mice. Gut 2006, 55, 415–424. [Google Scholar] [CrossRef]
- Fritsche, J.; Mondal, K.; Ehrnsperger, A.; Andreesen, R.; Kreutz, M. Regulation of 25-hydroxyvitamin D3-1 alpha-hydroxylase and production of 1 alpha,25-dihydroxyvitamin D3 by human dendritic cells. Blood 2003, 102, 3314–3316. [Google Scholar] [CrossRef]
- Arora, J.; Wang, J.; Weaver, V.; Zhang, Y.; Cantorna, M.T. Novel insight into the role of the vitamin D receptor in the development and function of the immune system. J. Steroid Biochem. Mol. Biol. 2022, 219, 106084. [Google Scholar] [CrossRef]
- Spittler, A.; Willheim, M.; Leutmezer, F.; Ohler, R.; Krugluger, W.; Reissner, C.; Luca, T.; Brodowicz, T.; Roth, E.; Boltz-Nitulescu, G. Effects of 1 alpha,25-dihydroxyvitamin D3 and cytokines on the expression of MHC antigens, complement receptors and other antigens on human blood monocytes and U937 cells: Role in cell differentiation, activation and phagocytosis. Immunology 1997, 90, 286–293. [Google Scholar] [CrossRef]
- Rendra, E.; Riabov, V.; Mossel, D.M.; Sevastyanova, T.; Harmsen, M.C.; Kzhyshkowska, J. Reactive oxygen species (ROS) in macrophage activation and function in diabetes. Immunobiology 2019, 224, 242–253. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, W.; Sun, T.; Huang, Y.; Wang, Y.; Deb, D.K.; Yoon, D.; Kong, J.; Thadhani, R.; Chun Li, Y. 1,25-Dihydroxyvitamin D promotes negative feedback regulation of TLR signaling via targeting microRNA-155-SOCS1 in macrophages. J. Immunol. 2013, 190, 3687–3695. [Google Scholar] [CrossRef]
- Ellergezen, P.; Alp, A.; Çavun, S. Vitamin D, VDR, and VDBP Levels Correlate with Anti-inflammatory Cytokine Profile in FMS Patients. Med. Rec. 2023, 5, 24–28. [Google Scholar] [CrossRef]
- Affo, S.; Yu, L.X.; Schwabe, R.F. The Role of Cancer-Associated Fibroblasts and Fibrosis in Liver Cancer. Annu. Rev. Pathol. 2017, 12, 153–186. [Google Scholar] [CrossRef]
- Mihm, S. Danger-Associated Molecular Patterns (DAMPs): Molecular Triggers for Sterile Inflammation in the Liver. Int. J. Mol. Sci. 2018, 19, 3104. [Google Scholar] [CrossRef]
- Watanabe, A.; Hashmi, A.; Gomes, D.A.; Town, T.; Badou, A.; Flavell, R.A.; Mehal, W.Z. Apoptotic hepatocyte DNA inhibits hepatic stellate cell chemotaxis via toll-like receptor 9. Hepatology 2007, 46, 1509–1518. [Google Scholar] [CrossRef]
- Duran, A.; Hernandez, E.D.; Reina-Campos, M.; Castilla, E.A.; Subramaniam, S.; Raghunandan, S.; Roberts, L.R.; Kisseleva, T.; Karin, M.; Diaz-Meco, M.T.; et al. p62/SQSTM1 by Binding to Vitamin D Receptor Inhibits Hepatic Stellate Cell Activity, Fibrosis, and Liver Cancer. Cancer Cell 2016, 30, 595–609. [Google Scholar] [CrossRef]
- Abramovitch, S.; Dahan-Bachar, L.; Sharvit, E.; Weisman, Y.; Tov, B.A.; Brazowski, E.; Reif, S. Vitamin D inhibits proliferation and profibrotic marker expression in hepatic stellate cells and decreases thioacetamide-induced liver fibrosis in rats. Gut 2011, 60, 1728–1737. [Google Scholar] [CrossRef]
- Neeman, R.; Abramovitch, S.; Sharvit, E.; Elad-Sfadia, G.; Haklai, R.; Kloog, Y.; Reif, S. Vitamin D and S-farnesylthiosalicylic acid have a synergistic effect on hepatic stellate cells proliferation. Dig. Dis. Sci. 2014, 59, 2462–2469. [Google Scholar] [CrossRef] [PubMed]
- Roderfeld, M. Matrix metalloproteinase functions in hepatic injury and fibrosis. Matrix Biol. 2018, 69, 452–462. [Google Scholar] [CrossRef] [PubMed]
- Veidal, S.S.; Vassiliadis, E.; Barascuk, N.; Zhang, C.; Segovia-Silvestre, T.; Klickstein, L.; Larsen, M.R.; Qvist, P.; Christiansen, C.; Vainer, B.; et al. Matrix metalloproteinase-9-mediated type III collagen degradation as a novel serological biochemical marker for liver fibrogenesis. Liver Int. 2010, 30, 1293–1304. [Google Scholar] [CrossRef]
- Schuppan, D.; Surabattula, R.; Wang, X.Y. Determinants of fibrosis progression and regression in NASH. J. Hepatol. 2018, 68, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Halder, S.K.; Osteen, K.G.; Al-Hendy, A. Vitamin D3 inhibits expression and activities of matrix metalloproteinase-2 and -9 in human uterine fibroid cells. Hum. Reprod. 2013, 28, 2407–2416. [Google Scholar] [CrossRef]
- Rahman, A.; Hershey, S.; Ahmed, S.; Nibbelink, K.; Simpson, R.U. Heart extracellular matrix gene expression profile in the vitamin D receptor knockout mice. J. Steroid Biochem. Mol. Biol. 2007, 103, 416–419. [Google Scholar] [CrossRef]
- Roehlen, N.; Crouchet, E.; Baumert, T.F. Liver Fibrosis: Mechanistic Concepts and Therapeutic Perspectives. Cells 2020, 9, 875. [Google Scholar] [CrossRef]
- Saffioti, F.; Pinzani, M. Development and Regression of Cirrhosis. Dig. Dis. 2016, 34, 374–381. [Google Scholar] [CrossRef]
- Abramovitch, S.; Sharvit, E.; Weisman, Y.; Bentov, A.; Brazowski, E.; Cohen, G.; Volovelsky, O.; Reif, S. Vitamin D inhibits development of liver fibrosis in an animal model but cannot ameliorate established cirrhosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 308, G112–G120. [Google Scholar] [CrossRef]
- Ying, H.Z.; Chen, Q.; Zhang, W.Y.; Zhang, H.H.; Ma, Y.; Zhang, S.Z.; Fang, J.; Yu, C.H. PDGF signaling pathway in hepatic fibrosis pathogenesis and therapeutics. Mol. Med. Rep. 2017, 16, 7879–7889. [Google Scholar] [CrossRef]
- Pedigo, N.; Zhang, H.; Koszewski, N.J.; Kaetzel, D.M. A 5’-distal element mediates vitamin D-inducibility of PDGF-A gene transcription. Growth Factors 2003, 21, 151–160. [Google Scholar] [CrossRef]
- Walton, K.L.; Johnson, K.E.; Harrison, C.A. Targeting TGF-β Mediated SMAD Signaling for the Prevention of Fibrosis. Front. Pharmacol. 2017, 8, 461. [Google Scholar] [CrossRef]
- Ding, N.; Yu, R.T.; Subramaniam, N.; Sherman, M.H.; Wilson, C.; Rao, R.; Leblanc, M.; Coulter, S.; He, M.; Scott, C.; et al. A vitamin D receptor/SMAD genomic circuit gates hepatic fibrotic response. Cell 2013, 153, 601–613. [Google Scholar] [CrossRef]
- Beilfuss, A.; Sowa, J.P.; Sydor, S.; Beste, M.; Bechmann, L.P.; Schlattjan, M.; Syn, W.K.; Wedemeyer, I.; Mathe, Z.; Jochum, C.; et al. Vitamin D counteracts fibrogenic TGF-β signalling in human hepatic stellate cells both receptor-dependently and independently. Gut 2015, 64, 791–799. [Google Scholar] [CrossRef]
- Wang, X.; Wang, G.; Qu, J.; Yuan, Z.; Pan, R.; Li, K. Calcipotriol Inhibits NLRP3 Signal Through YAP1 Activation to Alleviate Cholestatic Liver Injury and Fibrosis. Front. Pharmacol. 2020, 11, 200. [Google Scholar] [CrossRef]
- Rao, Z.; Chen, X.; Wu, J.; Xiao, M.; Zhang, J.; Wang, B.; Fang, L.; Zhang, H.; Wang, X.; Yang, S.; et al. Vitamin D Receptor Inhibits NLRP3 Activation by Impeding Its BRCC3-Mediated Deubiquitination. Front. Immunol. 2019, 10, 2783. [Google Scholar] [CrossRef]
- Harini, K.S.; Ezhilarasan, D. Wnt/beta-catenin signaling and its modulators in nonalcoholic fatty liver diseases. Hepatobiliary Pancreat. Dis. Int. 2022; in press. [Google Scholar] [CrossRef]
- Lecarpentier, Y.; Schussler, O.; Hébert, J.L.; Vallée, A. Multiple Targets of the Canonical WNT/β-Catenin Signaling in Cancers. Front. Oncol. 2019, 9, 1248. [Google Scholar] [CrossRef]
- Guo, Y.; Xiao, L.; Sun, L.; Liu, F. Wnt/beta-catenin signaling: A promising new target for fibrosis diseases. Physiol. Res. 2012, 61, 337–346. [Google Scholar] [CrossRef]
- Egan, J.B.; Thompson, P.A.; Vitanov, M.V.; Bartik, L.; Jacobs, E.T.; Haussler, M.R.; Gerner, E.W.; Jurutka, P.W. Vitamin D receptor ligands, adenomatous polyposis coli, and the vitamin D receptor FokI polymorphism collectively modulate beta-catenin activity in colon cancer cells. Mol. Carcinog. 2010, 49, 337–352. [Google Scholar] [CrossRef] [PubMed]
- Sari, E.; Oztay, F.; Tasci, A.E. Vitamin D modulates E-cadherin turnover by regulating TGF-β and Wnt signalings during EMT-mediated myofibroblast differentiation in A459 cells. J. Steroid Biochem. Mol. Biol. 2020, 202, 105723. [Google Scholar] [CrossRef]
- Khan, F.Z.; Perumpail, R.B.; Wong, R.J.; Ahmed, A. Advances in hepatocellular carcinoma: Nonalcoholic steatohepatitis-related hepatocellular carcinoma. World J. Hepatol. 2015, 7, 2155–2161. [Google Scholar] [CrossRef] [PubMed]
- Dhar, D.; Baglieri, J.; Kisseleva, T.; Brenner, D.A. Mechanisms of liver fibrosis and its role in liver cancer. Exp. Biol. Med. (Maywood) 2020, 245, 96–108. [Google Scholar] [CrossRef] [PubMed]
- Baglieri, J.; Brenner, D.A.; Kisseleva, T. The Role of Fibrosis and Liver-Associated Fibroblasts in the Pathogenesis of Hepatocellular Carcinoma. Int. J. Mol. Sci. 2019, 20, 1723. [Google Scholar] [CrossRef]
- Kong, F.; Li, L.; Wang, G.; Deng, X.; Li, Z.; Kong, X. VDR signaling inhibits cancer-associated-fibroblasts’ release of exosomal miR-10a-5p and limits their supportive effects on pancreatic cancer cells. Gut 2019, 68, 950–951. [Google Scholar] [CrossRef]
- Ferrer-Mayorga, G.; Gómez-López, G.; Barbáchano, A.; Fernández-Barral, A.; Peña, C.; Pisano, D.G.; Camtero, R.; Rojo, F.; Munoz, A.; Larriba, M.J. Vitamin D receptor expression and associated gene signature in tumour stromal fibroblasts predict clinical outcome in colorectal cancer. Gut 2017, 66, 1449–1462. [Google Scholar] [CrossRef]
- Campos, L.T.; Brentani, H.; Roela, R.A.; Katayama, M.L.H.; Lima, L.; Rolim, C.F.; Milani, C.; Azevedo Koike Folgueira, M.A.; Brentani, M.M. Differences in transcriptional effects of 1α,25 dihydroxyvitamin D3 on fibroblasts associated to breast carcinomas and from paired normal breast tissues. J. Steroid Biochem. Mol. Biol. 2013, 133, 12–24. [Google Scholar] [CrossRef]
- Zhao, Z.X.; Zhang, Y.Q.; Sun, H.; Chen, Z.Q.; Chang, J.J.; Wang, X.; Wang, X.; Tan, C.; Ni, S.J.; Weng, W.W.; et al. Calcipotriol abrogates cancer-associated fibroblast-derived IL-8-mediated oxaliplatin resistance in gastric cancer cells via blocking PI3K/Akt signaling. Acta Pharmacol. Sin. 2023, 44, 178–188. [Google Scholar] [CrossRef]
- Giulianelli, S.; Cerliani, J.P.; Lamb, C.A.; Fabris, V.T.; Bottino, M.C.; Gorostiaga, M.A.; Novaro, V.; Gongora, A.; Baldi, A.; Molinolo, A.; et al. Carcinoma-associated fibroblasts activate progesterone receptors and induce hormone independent mammary tumor growth: A role for the FGF-2/FGFR-2 axis. Int. J. Cancer 2008, 123, 2518–2531. [Google Scholar] [CrossRef]
- Peña, C.; Céspedes, M.V.; Lindh, M.B.; Kiflemariam, S.; Mezheyeuski, A.; Edqvist, P.H.; Hagglof, C.; Birgisson, H.; Bojmar, L.; Jirstrom, K.; et al. STC1 expression by cancer-associated fibroblasts drives metastasis of colorectal cancer. Cancer Res. 2013, 73, 1287–1297. [Google Scholar] [CrossRef]
- Sewell-Loftin, M.K.; Bayer, S.V.H.; Crist, E.; Hughes, T.; Joison, S.M.; Longmore, G.D.; George, S.C. Cancer-associated fibroblasts support vascular growth through mechanical force. Sci. Rep. 2017, 7, 12574. [Google Scholar] [CrossRef]
- Zheng, Q.; Martin, R.C.; Shi, X.; Pandit, H.; Yu, Y.; Liu, X.; Guo, W.; Tan, M.; Bai, Q.; Meng, X.; et al. Lack of FGF21 promotes NASH-HCC transition via hepatocyte-TLR4-IL-17A signaling. Theranostics 2020, 10, 9923–9936. [Google Scholar] [CrossRef]
- Gomes, A.L.; Teijeiro, A.; Burén, S.; Tummala, K.S.; Yilmaz, M.; Waisman, A.; Theurillat, J.P.; Perma, C.; Djouder, N. Metabolic Inflammation-Associated IL-17A Causes Non-alcoholic Steatohepatitis and Hepatocellular Carcinoma. Cancer Cell 2016, 30, 161–175. [Google Scholar] [CrossRef]
- Tezze, C.; Romanello, V.; Sandri, M. FGF21 as Modulator of Metabolism in Health and Disease. Front Physiol. 2019, 10, 419. [Google Scholar] [CrossRef]
- Wang, D.; Huang, H.J.; Kazlauskas, A.; Cavenee, W.K. Induction of vascular endothelial growth factor expression in endothelial cells by platelet-derived growth factor through the activation of phosphatidylinositol 3-kinase. Cancer Res. 1999, 59, 1464–1472. [Google Scholar] [PubMed]
- Jamali, N.; Song, Y.S.; Sorenson, C.M.; Sheibani, N. 1,25(OH)2D3 regulates the proangiogenic activity of pericyte through VDR-mediated modulation of VEGF production and signaling of VEGF and PDGF receptors. FASEB Bioadv 2019, 1, 415–434. [Google Scholar] [CrossRef]
- Kim, R.; Emi, M.; Tanabe, K. Cancer immunoediting from immune surveillance to immune escape. Immunology 2007, 121, 1–14. [Google Scholar] [CrossRef]
- Mougiakakos, D.; Choudhury, A.; Lladser, A.; Kiessling, R.; Johansson, C.C. Regulatory T cells in cancer. Adv. Cancer Res. 2010, 107, 57–117. [Google Scholar] [CrossRef]
- Ostrand-Rosenberg, S. Myeloid-derived suppressor cells: More mechanisms for inhibiting antitumor immunity. Cancer Immunol. Immunother. 2010, 59, 1593–1600. [Google Scholar] [CrossRef]
- Monteran, L.; Erez, N. The Dark Side of Fibroblasts: Cancer-Associated Fibroblasts as Mediators of Immunosuppression in the Tumor Microenvironment. Front. Immunol. 2019, 10, 1835. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Zhu, X.; Cao, G.; Wu, R.; Li, K.; Yuan, W.; Chen, B.; Sun, G.; Xia, X.; Zhang, H.; et al. 1α,25(OH)2D3 reverses exhaustion and enhances antitumor immunity of human cytotoxic T cells. J. Immunother. Cancer 2022, 10, e003477. [Google Scholar] [CrossRef] [PubMed]
- Bochen, F.; Balensiefer, B.; Körner, S.; Bittenbring, J.T.; Neumann, F.; Koch, A.; Bumm, K.; Marx, A.; Wemmert, S.; Papaspyrou, G.; et al. Vitamin D deficiency in head and neck cancer patients–prevalence, prognostic value and impact on immune function. Oncoimmunology 2018, 7, e1476817. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, P.E.; Pringle, J.H. Consideration of possible effects of vitamin D on established cancer, with reference to malignant melanoma. Pigment. Cell. Melanoma Res. 2022, 35, 408–424. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, J.; Ge, X.; Du, J.; Deb, D.K.; Li, Y.C. Vitamin D receptor inhibits nuclear factor κB activation by interacting with IκB kinase β protein. J. Biol. Chem. 2013, 288, 19450–19458. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-κB signaling in inflammation. Signal. Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef]
- Guo, J.; Ma, Z.; Ma, Q.; Wu, Z.; Fan, P.; Zhou, X.; Chen, L.; Zhou, S.; Goltzman, D.; Miao, D.; et al. 1, 25(OH)2D3 Inhibits Hepatocellular Carcinoma Development Through Reducing Secretion of Inflammatory Cytokines from Immunocytes. Curr. Med. Chem. 2013, 20, 4131–4141. [Google Scholar] [CrossRef]
- Bishop, L.E.; Ismailova, A.; Dimeloe, S.; Hewison, M.; White, J.H. Vitamin D and Immune Regulation: Antibacterial, Antiviral, Anti-Inflammatory. JBMR Plus 2021, 5, e10405. [Google Scholar] [CrossRef]
- von Essen, M.R.; Kongsbak, M.; Schjerling, P.; Olgaard, K.; Odum, N.; Geisler, C. Vitamin D controls T cell antigen receptor signaling and activation of human T cells. Nat. Immunol. 2010, 11, 344–349. [Google Scholar] [CrossRef]
- Lemire, J.M. Immunomodulatory role of 1,25-dihydroxyvitamin D3. J. Cell. Biochem. 1992, 49, 26–31. [Google Scholar] [CrossRef]
- Scolletta, S.; Colletti, M.; Luigi, L.D.; Crescioli, C. Vitamin D receptor agonists target CXCL10, New therapeutic tools for resolution of inflammation. Mediat. Inflamm. 2013, 2013, 876319. [Google Scholar] [CrossRef]
- Boonstra, A.; Barrat, F.J.; Crain, C.; Heath, V.L.; Savelkoul, H.F.; O’Garra, A. 1alpha,25-Dihydroxyvitamin d3 has a direct effect on naive CD4(+) T cells to enhance the development of Th2 cells. J. Immunol. 2001, 167, 4974–4980. [Google Scholar] [CrossRef]
- Penna, G.; Roncari, A.; Amuchastegui, S.; Daniel, K.C.; Berti, E.; Colonna, M.; Adorini, L. Expression of the inhibitory receptor ILT3 on dendritic cells is dispensable for induction of CD4+Foxp3+ regulatory T cells by 1,25-dihydroxyvitamin D3. Blood 2005, 106, 3490–3497. [Google Scholar] [CrossRef]
- Kim, Y.; Chang, Y.; Ryu, S.; Cho, I.Y.; Kwon, M.J.; Sohn, W.; Kim, M.K.; Wild, S.H.; Byrne, C.D. Resolution of, and Risk of Incident Non-alcoholic Fatty Liver Disease With Changes in Serum 25-hydroxy Vitamin D Status. J. Clin. Endocrinol. Metab. 2022, 107, e3437–e3447. [Google Scholar] [CrossRef]
- Chen, Y.; Feng, S.; Chang, Z.; Zhao, Y.; Liu, Y.; Fu, J.; Liu, Y.; Tang, S.; Han, Y.; Zhang, S.; et al. Higher Serum 25-Hydroxyvitamin D Is Associated with Lower All-Cause and Cardiovascular Mortality among US Adults with Nonalcoholic Fatty Liver Disease. Nutrients 2022, 14, 4013. [Google Scholar] [CrossRef]
- Gong, J.; Gong, H.; Liu, Y.; Tao, X.; Zhang, H. Calcipotriol attenuates liver fibrosis through the inhibition of vitamin D receptor-mediated NF-κB signaling pathway. Bioengineered 2022, 13, 2658–2672. [Google Scholar] [CrossRef]
- Dalhoff, K.; Dancey, J.; Astrup, L.; Skovsgaard, T.; Hamberg, K.J.; Lofts, F.J.; Rosmorduc, O.; Erlinger, S.; Bach Hansen, J.; Steward, W.P.; et al. A phase II study of the vitamin D analogue Seocalcitol in patients with inoperable hepatocellular carcinoma. Br. J. Cancer 2003, 89, 252–257. [Google Scholar] [CrossRef]


| VDR Mediated-Mechanism | Effects on NAFLD Development | Refs. |
|---|---|---|
| Direct effects of VDR | ||
|
| [60] |
|
| [61,65] |
|
| [62,63,64,65] |
|
| [66,67] |
| Indirect effects of VDR | ||
|
| [71] |
|
| [72] |
|
| [71,73,74] |
|
| [62,76,77,78,79,80,81,82] |
| ↑: increase, ↓: decrease | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tourkochristou, E.; Mouzaki, A.; Triantos, C. Gene Polymorphisms and Biological Effects of Vitamin D Receptor on Nonalcoholic Fatty Liver Disease Development and Progression. Int. J. Mol. Sci. 2023, 24, 8288. https://doi.org/10.3390/ijms24098288
Tourkochristou E, Mouzaki A, Triantos C. Gene Polymorphisms and Biological Effects of Vitamin D Receptor on Nonalcoholic Fatty Liver Disease Development and Progression. International Journal of Molecular Sciences. 2023; 24(9):8288. https://doi.org/10.3390/ijms24098288
Chicago/Turabian StyleTourkochristou, Evanthia, Athanasia Mouzaki, and Christos Triantos. 2023. "Gene Polymorphisms and Biological Effects of Vitamin D Receptor on Nonalcoholic Fatty Liver Disease Development and Progression" International Journal of Molecular Sciences 24, no. 9: 8288. https://doi.org/10.3390/ijms24098288