
Pushing the Resolution Limit of Stimulated Emission Depletion Optical Nanoscopy
 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
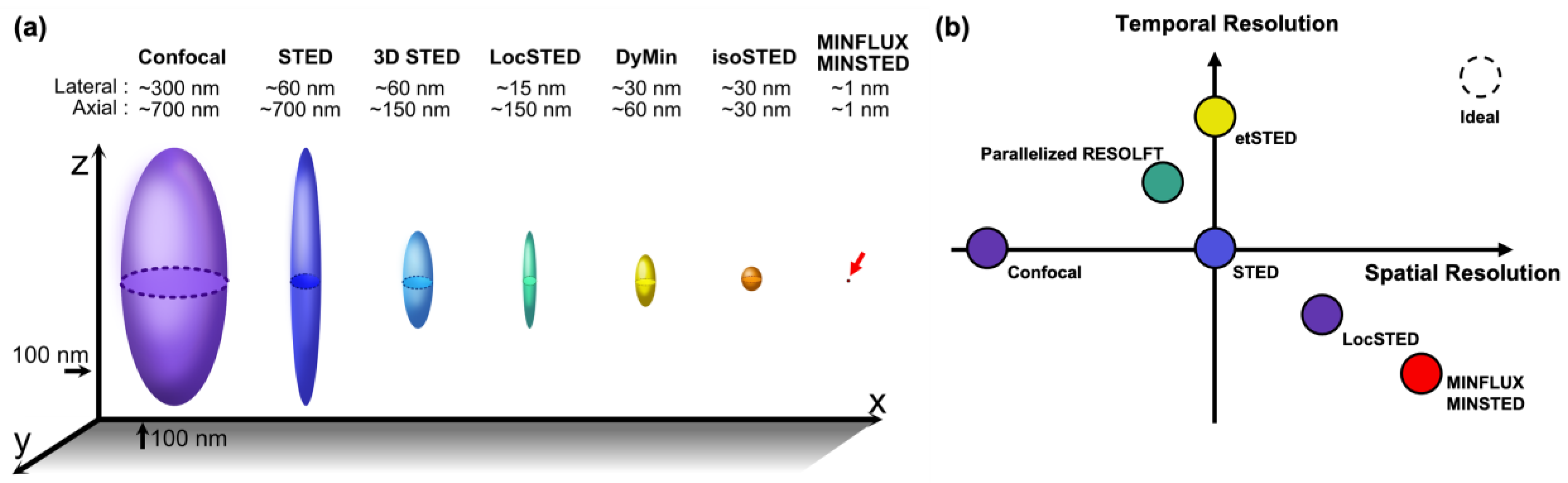
:1. Introduction
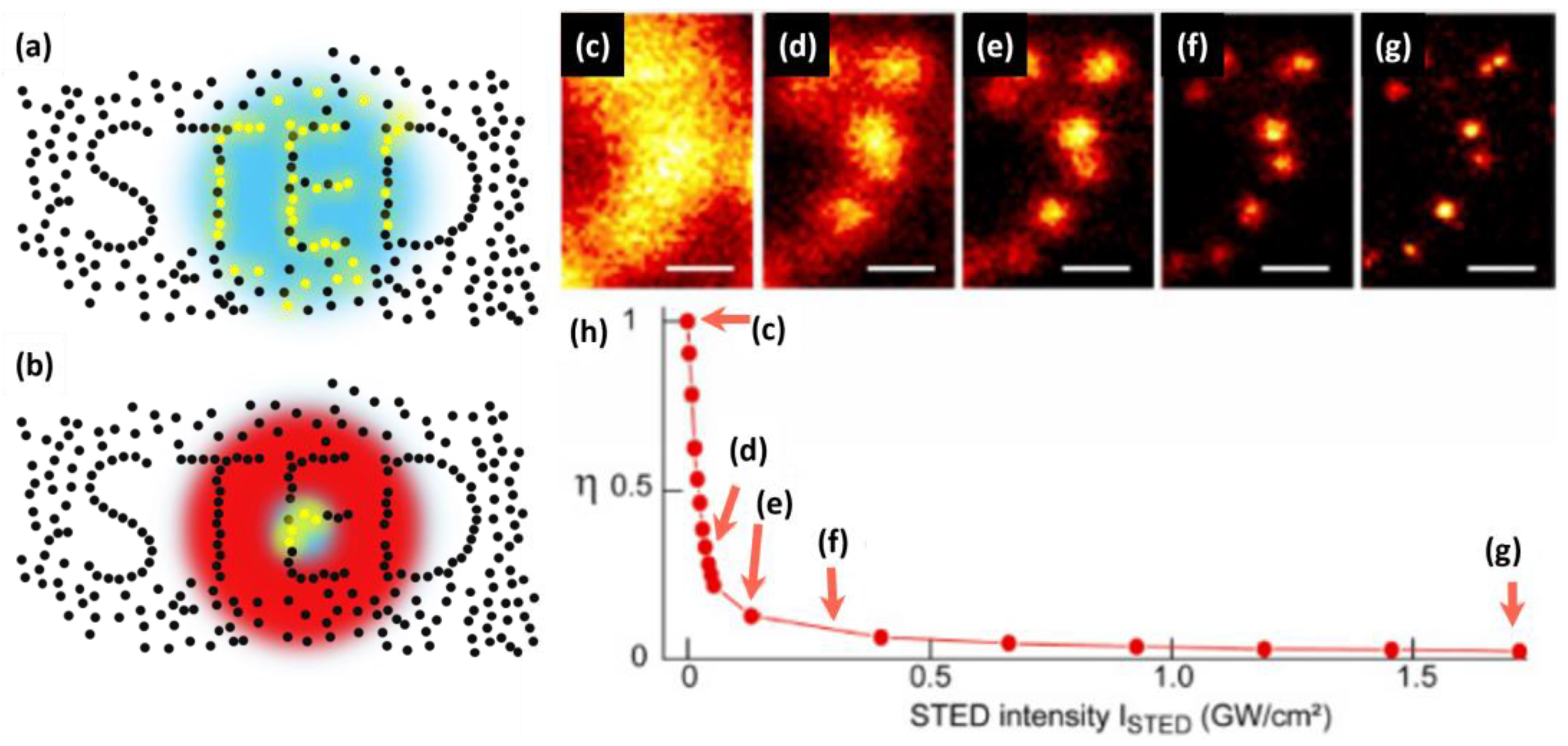
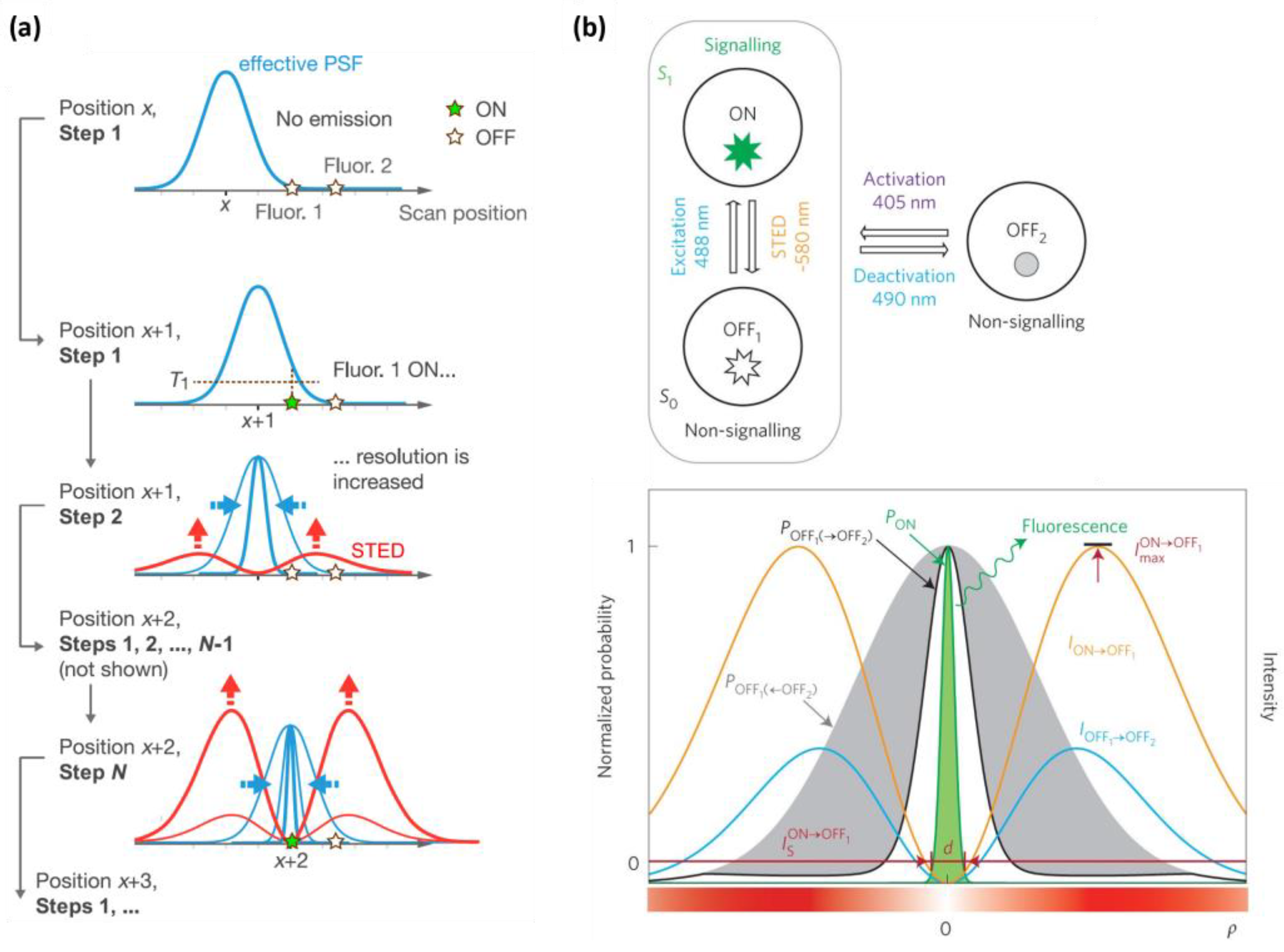
2. Lateral Resolution Improvement
2.1. Fluorophore Localization
2.1.1. LocSTED
2.1.2. MINSTED
2.2. Adaptive Illumination
2.2.1. RESCue
2.2.2. MINFIELD
2.2.3. DyMin
2.2.4. Protected STED
2.2.5. Differential STED
2.3. Fluorescence Lifetime Detection
2.3.1. g-STED
2.3.2. SPLIT STED
2.3.3. STED-FLIM with Phasor Plot
3. Axial Resolution Improvement
3.1. 3D-STED
3.2. Isotropic STED
4. Temporal Resolution Improvement
4.1. Labeling Methods for Fast, Live Cell STED Nanoscopy
4.2. Parallelized Illumination
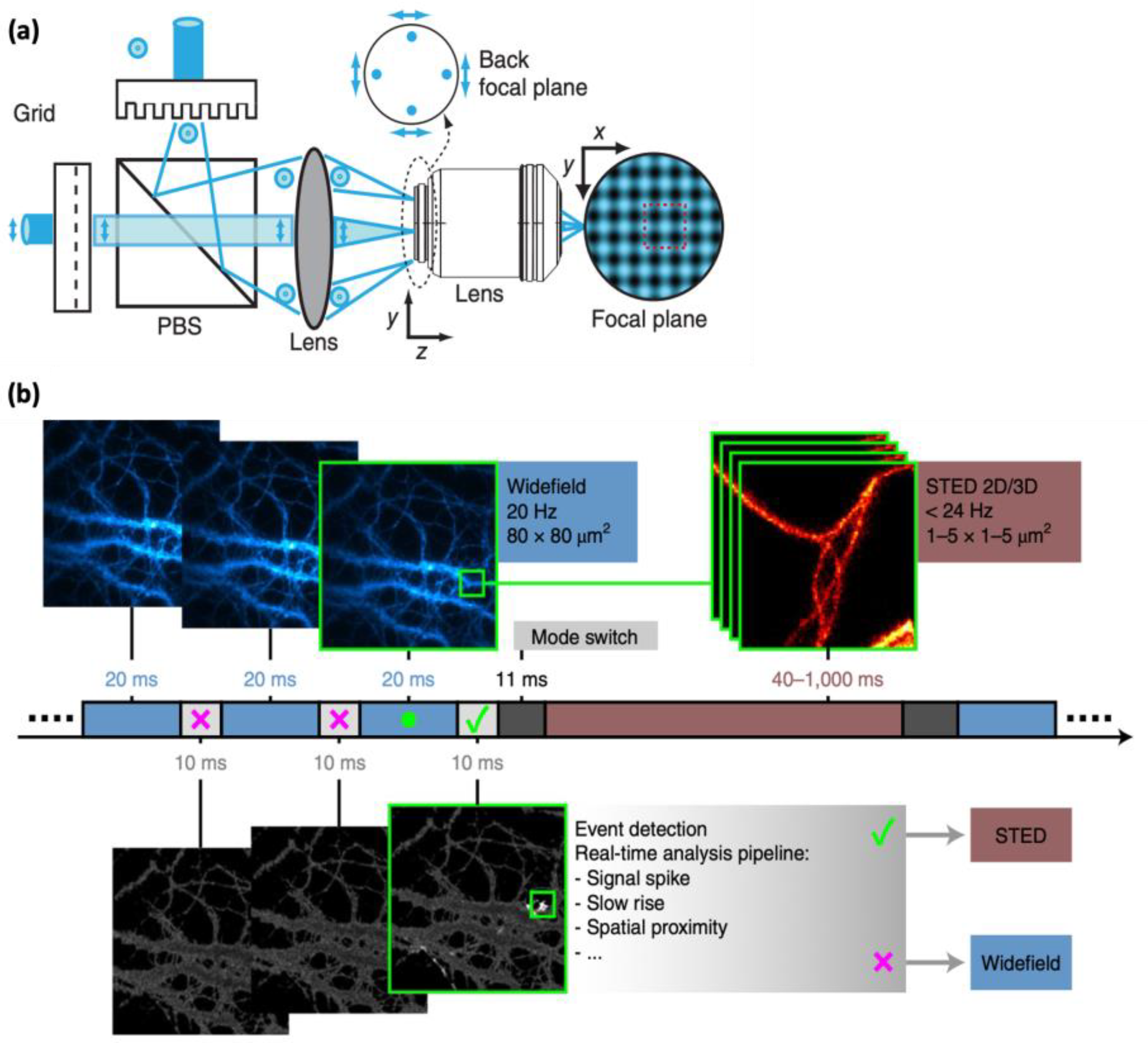
4.3. Event-Triggered STED (etSTED)
5. Labeling Strategy for STED Nanoscopy
5.1. Single-Domain Antibodies and Affibodies
5.2. Genetically Encoded Protein Tags
5.3. Genetic Code Expansion
6. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Abbe, E. Beiträge zur Theorie des Mikroskops und der Mikroskopischen Wahrnehmung. Arch. für Mikrosk. Anat. 1873, 9, 413–468. [Google Scholar] [CrossRef]
- Betzig, E.; Patterson, G.H.; Sougrat, R.; Lindwasser, O.W.; Olenych, S.; Bonifacino, J.S.; Davidson, M.W.; Lippincott-Schwartz, J.; Hess, H.F. Imaging Intracellular Fluorescent Proteins at Nanometer Resolution. Science 2006, 313, 1642–1645. [Google Scholar] [CrossRef] [PubMed]
- Rust, M.J.; Bates, M.; Zhuang, X. Sub-Diffraction-Limit Imaging by Stochastic Optical Reconstruction Microscopy (STORM). Nat. Methods 2006, 3, 793–796. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, M.G.L. Surpassing the Lateral Resolution Limit by a Factor of Two Using Structured Illumination Microscopy. J. Microsc. 2000, 198, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Westphal, V.; Hell, S.W. Nanoscale Resolution in the Focal Plane of an Optical Microscope. Phys. Rev. Lett. 2005, 94, 143903. [Google Scholar] [CrossRef] [PubMed]
- Hell, S.W.; Wichmann, J. Breaking the Diffraction Resolution Limit by Stimulated Emission: Stimulated-Emission-Depletion Fluorescence Microscopy. Opt. Lett. 1994, 19, 780. [Google Scholar] [CrossRef] [PubMed]
- Ahn, Y.; Park, M.; Seo, D. Observation of Reactions in Single Molecules/nanoparticles Using Light Microscopy. Bull. Korean Chem. Soc. 2023, 44, 35–44. [Google Scholar] [CrossRef]
- Sydor, A.M.; Czymmek, K.J.; Puchner, E.M.; Mennella, V. Super-Resolution Microscopy: From Single Molecules to Supramolecular Assemblies. Trends Cell Biol. 2015, 25, 730–748. [Google Scholar] [CrossRef]
- Leutenegger, M.; Eggeling, C.; Hell, S.W. Analytical Description of STED Microscopy Performance. Opt. Express 2010, 18, 26417. [Google Scholar] [CrossRef]
- Harke, B.; Keller, J.; Ullal, C.K.; Westphal, V.; Schönle, A.; Hell, S.W. Resolution Scaling in STED Microscopy. Opt. Express 2008, 16, 4154. [Google Scholar] [CrossRef]
- Rittweger, E.; Han, K.Y.; Irvine, S.E.; Eggeling, C.; Hell, S.W. STED Microscopy Reveals Crystal Colour Centres with Nanometric Resolution. Nat. Photonics 2009, 3, 144–147. [Google Scholar] [CrossRef]
- Ma, Y.; Ha, T. Fight against Background Noise in Stimulated Emission Depletion Nanoscopy. Phys. Biol. 2019, 16, 51002. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-C.; Ma, Y.; Han, K.Y.; Ha, T. Accurate Background Subtraction in STED Nanoscopy by Polarization Switching. ACS Photonics 2019, 6, 1789–1797. [Google Scholar] [CrossRef]
- Hanne, J.; Falk, H.J.; Görlitz, F.; Hoyer, P.; Engelhardt, J.; Sahl, S.J.; Hell, S.W. STED Nanoscopy with Fluorescent Quantum Dots. Nat. Commun. 2015, 6, 7127. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.; Kim, J.; Lee, J.-C. Suppressing Background Noise in STED Optical Nanoscopy. J. Korean Phys. Soc. 2021, 78, 401–407. [Google Scholar] [CrossRef]
- Vicidomini, G.; Moneron, G.; Han, K.Y.; Westphal, V.; Ta, H.; Reuss, M.; Engelhardt, J.; Eggeling, C.; Hell, S.W. Sharper Low-Power STED Nanoscopy by Time Gating. Nat. Methods 2011, 8, 571–573. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Prunsche, B.; Zhou, L.; Nienhaus, K.; Nienhaus, G.U. Background Suppression in Fluorescence Nanoscopy with Stimulated Emission Double Depletion. Nat. Photonics 2017, 11, 163–169. [Google Scholar] [CrossRef]
- Jeong, S.; Kim, J.; Koh, D.; Lee, J. Simultaneously Enhancing the Resolution and Signal-to-Background Ratio in STED Optical Nanoscopy via Differential Depletion. Opt. Express 2023, 31, 37549. [Google Scholar] [CrossRef]
- Alvelid, J.; Damenti, M.; Sgattoni, C.; Testa, I. Event-Triggered STED Imaging. Nat. Methods 2022, 19, 1268–1275. [Google Scholar] [CrossRef]
- Puthukodan, S.; Murtezi, E.; Jacak, J.; Klar, T.A. Localization STED (LocSTED) Microscopy with 15 Nm Resolution. Nanophotonics 2020, 9, 783–792. [Google Scholar] [CrossRef]
- Weber, M.; Leutenegger, M.; Stoldt, S.; Jakobs, S.; Mihaila, T.S.; Butkevich, A.N.; Hell, S.W. MINSTED Fluorescence Localization and Nanoscopy. Nat. Photonics 2021, 15, 361–366. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.; von der Emde, H.; Leutenegger, M.; Gunkel, P.; Sambandan, S.; Khan, T.A.; Keller-Findeisen, J.; Cordes, V.C.; Hell, S.W. MINSTED Nanoscopy Enters the Ångström Localization Range. Nat. Biotechnol. 2022, 41, 569–576. [Google Scholar] [CrossRef] [PubMed]
- Staudt, T.; Engler, A.; Rittweger, E.; Harke, B.; Engelhardt, J.; Hell, S.W. Far-Field Optical Nanoscopy with Reduced Number of State Transition Cycles. Opt. Express 2011, 19, 5644. [Google Scholar] [CrossRef] [PubMed]
- Göttfert, F.; Pleiner, T.; Heine, J.; Westphal, V.; Görlich, D.; Sahl, S.J.; Hell, S.W. Strong Signal Increase in STED Fluorescence Microscopy by Imaging Regions of Subdiffraction Extent. Proc. Natl. Acad. Sci. USA 2017, 114, 2125–2130. [Google Scholar] [CrossRef] [PubMed]
- Heine, J.; Reuss, M.; Harke, B.; D’Este, E.; Sahl, S.J.; Hell, S.W. Adaptive-Illumination STED Nanoscopy. Proc. Natl. Acad. Sci. USA 2017, 114, 9797–9802. [Google Scholar] [CrossRef] [PubMed]
- Danzl, J.G.; Sidenstein, S.C.; Gregor, C.; Urban, N.T.; Ilgen, P.; Jakobs, S.; Hell, S.W. Coordinate-Targeted Fluorescence Nanoscopy with Multiple off States. Nat. Photonics 2016, 10, 122–128. [Google Scholar] [CrossRef]
- Vicidomini, G.; Schönle, A.; Ta, H.; Han, K.Y.; Moneron, G.; Eggeling, C.; Hell, S.W. STED Nanoscopy with Time-Gated Detection: Theoretical and Experimental Aspects. PLoS ONE 2013, 8, e54421. [Google Scholar] [CrossRef]
- Hernández, I.C.; Buttafava, M.; Boso, G.; Diaspro, A.; Tosi, A.; Vicidomini, G. Gated STED Microscopy with Time-Gated Single-Photon Avalanche Diode. Biomed. Opt. Express 2015, 6, 2258. [Google Scholar] [CrossRef]
- Lanzanò, L.; Coto Hernández, I.; Castello, M.; Gratton, E.; Diaspro, A.; Vicidomini, G. Encoding and Decoding Spatio-Temporal Information for Super-Resolution Microscopy. Nat. Commun. 2015, 6, 6701. [Google Scholar] [CrossRef]
- Pelicci, S.; Tortarolo, G.; Vicidomini, G.; Diaspro, A.; Lanzanò, L. Improving SPLIT-STED Super-Resolution Imaging with Tunable Depletion and Excitation Power. J. Phys. D Appl. Phys. 2020, 53, 234003. [Google Scholar] [CrossRef]
- Wang, L.; Chen, B.; Yan, W.; Yang, Z.; Peng, X.; Lin, D.; Weng, X.; Ye, T.; Qu, J. Resolution Improvement in STED Super-Resolution Microscopy at Low Power Using a Phasor Plot Approach. Nanoscale 2018, 10, 16252–16260. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Ye, S.; Peng, X.; Song, J.; Qu, J. Green Emitted CdSe@ZnS Quantum Dots for FLIM and STED Imaging Applications. J. Innov. Opt. Health Sci. 2019, 12, 1–7. [Google Scholar] [CrossRef]
- Han, K.Y.; Ha, T. Dual-Color Three-Dimensional STED Microscopy with a Single High-Repetition-Rate Laser. Opt. Lett. 2015, 40, 2653. [Google Scholar] [CrossRef] [PubMed]
- Curdt, F.; Herr, S.J.; Lutz, T.; Schmidt, R.; Engelhardt, J.; Sahl, S.J.; Hell, S.W. isoSTED Nanoscopy with Intrinsic Beam Alignment. Opt. Express 2015, 23, 30891. [Google Scholar] [CrossRef] [PubMed]
- Siegmund, R.; Werner, F.; Jakobs, S.; Geisler, C.; Egner, A. isoSTED Microscopy with Water-Immersion Lenses and Background Reduction. Biophys. J. 2021, 120, 3303–3314. [Google Scholar] [CrossRef] [PubMed]
- Bingen, P.; Reuss, M.; Engelhardt, J.; Hell, S.W. Parallelized STED Fluorescence Nanoscopy. Opt. Express 2011, 19, 23716. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Przybilla, F.; Mestre, M.; Trebbia, J.-B.; Lounis, B. Large Parallelization of STED Nanoscopy Using Optical Lattices. Opt. Express 2014, 22, 5581. [Google Scholar] [CrossRef] [PubMed]
- Ries, J.; Kaplan, C.; Platonova, E.; Eghlidi, H.; Ewers, H. A Simple, Versatile Method for GFP-Based Super-Resolution Microscopy via Nanobodies. Nat. Methods 2012, 9, 582–584. [Google Scholar] [CrossRef]
- Muyldermans, S. Nanobodies: Natural Single-Domain Antibodies. Annu. Rev. Biochem. 2013, 82, 775–797. [Google Scholar] [CrossRef]
- Pleiner, T.; Bates, M.; Trakhanov, S.; Lee, C.-T.; Schliep, J.E.; Chug, H.; Böhning, M.; Stark, H.; Urlaub, H.; Görlich, D. Nanobodies: Site-Specific Labeling for Super-Resolution Imaging, Rapid Epitope-Mapping and Native Protein Complex Isolation. Elife 2015, 4, e11349. [Google Scholar] [CrossRef]
- Morozova, K.S.; Piatkevich, K.D.; Gould, T.J.; Zhang, J.; Bewersdorf, J.; Verkhusha, V.V. Far-Red Fluorescent Protein Excitable with Red Lasers for Flow Cytometry and Superresolution STED Nanoscopy. Biophys. J. 2010, 99, L13–L15. [Google Scholar] [CrossRef] [PubMed]
- Chu, J.; Haynes, R.D.; Corbel, S.Y.; Li, P.; González-González, E.; Burg, J.S.; Ataie, N.J.; Lam, A.J.; Cranfill, P.J.; Baird, M.A.; et al. Non-Invasive Intravital Imaging of Cellular Differentiation with a Bright Red-Excitable Fluorescent Protein. Nat. Methods 2014, 11, 572–578. [Google Scholar] [CrossRef] [PubMed]
- Wegner, W.; Ilgen, P.; Gregor, C.; Van Dort, J.; Mott, A.C.; Steffens, H.; Willig, K.I. In Vivo Mouse and Live Cell STED Microscopy of Neuronal Actin Plasticity Using Far-Red Emitting Fluorescent Proteins. Sci. Rep. 2017, 7, 11781. [Google Scholar] [CrossRef] [PubMed]
- Hense, A.; Prunsche, B.; Gao, P.; Ishitsuka, Y.; Nienhaus, K.; Ulrich Nienhaus, G. Monomeric Garnet, a Far-Red Fluorescent Protein for Live-Cell STED Imaging. Sci. Rep. 2015, 5, 18006. [Google Scholar] [CrossRef] [PubMed]
- Matela, G.; Gao, P.; Guigas, G.; Eckert, A.F.; Nienhaus, K.; Ulrich Nienhaus, G. A Far-Red Emitting Fluorescent Marker Protein, mGarnet2, for Microscopy and STED Nanoscopy. Chem. Commun. 2017, 53, 979–982. [Google Scholar] [CrossRef] [PubMed]
- Kamper, M.; Ta, H.; Jensen, N.A.; Hell, S.W.; Jakobs, S. Near-Infrared STED Nanoscopy with an Engineered Bacterial Phytochrome. Nat. Commun. 2018, 9, 4762. [Google Scholar] [CrossRef] [PubMed]
- Matlashov, M.E.; Shcherbakova, D.M.; Alvelid, J.; Baloban, M.; Pennacchietti, F.; Shemetov, A.A.; Testa, I.; Verkhusha, V.V. A Set of Monomeric near-Infrared Fluorescent Proteins for Multicolor Imaging across Scales. Nat. Commun. 2020, 11, 239. [Google Scholar] [CrossRef]
- Filonov, G.S.; Piatkevich, K.D.; Ting, L.M.; Zhang, J.; Kim, K.; Verkhusha, V.V. Bright and Stable near-Infrared Fluorescent Protein for in Vivo Imaging. Nat. Biotechnol. 2011, 29, 757–761. [Google Scholar] [CrossRef]
- Shu, X.; Royant, A.; Lin, M.Z.; Aguilera, T.A.; Lev-Ram, V.; Steinbach, P.A.; Tsien, R.Y. Mammalian Expression of Infrared Fluorescent Proteins Engineered from a Bacterial Phytochrome. Science 2009, 324, 804–807. [Google Scholar] [CrossRef]
- Shcherbakova, D.M.; Baloban, M.; Emelyanov, A.V.; Brenowitz, M.; Guo, P.; Verkhusha, V.V. Bright Monomeric near-Infrared Fluorescent Proteins as Tags and Biosensors for Multiscale Imaging. Nat. Commun. 2016, 7, 12405. [Google Scholar] [CrossRef]
- Erdmann, R.S.; Baguley, S.W.; Richens, J.H.; Wissner, R.F.; Xi, Z.; Allgeyer, E.S.; Zhong, S.; Thompson, A.D.; Lowe, N.; Butler, R.; et al. Labeling Strategies Matter for Super-Resolution Microscopy: A Comparison between HaloTags and SNAP-Tags. Cell Chem. Biol. 2019, 26, 584–592.e6. [Google Scholar] [CrossRef] [PubMed]
- Gregor, C.; Grimm, F.; Rehman, J.; Wurm, C.A.; Egner, A. Two-Color Live-Cell STED Nanoscopy by Click Labeling with Cell-Permeable Fluorophores. bioRxiv 2022. [Google Scholar] [CrossRef]
- Kompa, J.; Bruins, J.; Glogger, M.; Wilhelm, J.; Frei, M.S.; Tarnawski, M.; D’Este, E.; Heilemann, M.; Hiblot, J.; Johnsson, K. Exchangeable HaloTag Ligands for Super-Resolution Fluorescence Microscopy. J. Am. Chem. Soc. 2023, 145, 3075–3083. [Google Scholar] [CrossRef] [PubMed]
- Uttamapinant, C.; Howe, J.D.; Lang, K.; Beránek, V.; Davis, L.; Mahesh, M.; Barry, N.P.; Chin, J.W. Genetic Code Expansion Enables Live-Cell and Super-Resolution Imaging of Site-Specifically Labeled Cellular Proteins. J. Am. Chem. Soc. 2015, 137, 4602–4605. [Google Scholar] [CrossRef] [PubMed]
- Kuhlemann, A.; Beliu, G.; Janzen, D.; Petrini, E.M.; Taban, D.; Helmerich, D.A.; Doose, S.; Bruno, M.; Barberis, A.; Villmann, C.; et al. Genetic Code Expansion and Click-Chemistry Labeling to Visualize GABA-A Receptors by Super-Resolution Microscopy. Front. Synaptic Neurosci. 2021, 13, 727406. [Google Scholar] [CrossRef]
- Tortarolo, G.; Castello, M.; Diaspro, A.; Koho, S.; Vicidomini, G. Evaluating Image Resolution in Stimulated Emission Depletion Microscopy. Optica 2018, 5, 32. [Google Scholar] [CrossRef]
- Eggeling, C.; Willig, K.I.; Sahl, S.J.; Hell, S.W. Lens-Based Fluorescence Nanoscopy. Q. Rev. Biophys. 2015, 48, 178–243. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.; Widengren, J.; Lee, J.C. Fluorescent Probes for STED Optical Nanoscopy. Nanomaterials 2021, 12, 21. [Google Scholar] [CrossRef]
- Blom, H.; Widengren, J. Stimulated Emission Depletion Microscopy. Chem. Rev. 2017, 117, 7377–7427. [Google Scholar] [CrossRef]
- Hofmann, M.; Eggeling, C.; Jakobs, S.; Hell, S.W. Breaking the Diffraction Barrier in Fluorescence Microscopy at Low Light Intensities by Using Reversibly Photoswitchable Proteins. Proc. Natl. Acad. Sci. USA 2005, 102, 17565–17569. [Google Scholar] [CrossRef]
- Nahidiazar, L.; Agronskaia, A.V.; Broertjes, J.; Van Den Broek, B.; Jalink, K. Optimizing Imaging Conditions for Demanding Multi-Color Super Resolution Localization Microscopy. PLoS ONE 2016, 11, e0158884. [Google Scholar] [CrossRef] [PubMed]
- Vicidomini, G.; Moneron, G.; Eggeling, C.; Rittweger, E.; Hell, S.W. STED with Wavelengths Closer to the Emission Maximum. Opt. Express 2012, 20, 5225. [Google Scholar] [CrossRef] [PubMed]
- Bordenave, M.D.; Balzarotti, F.; Stefani, F.D.; Hell, S.W. STED Nanoscopy with Wavelengths at the Emission Maximum. J. Phys. D Appl. Phys. 2016, 49, 365102. [Google Scholar] [CrossRef]
- Cordes, T.; Vogelsang, J.; Tinnefeld, P. On the Mechanism of Trolox as Antiblinking and Antibleaching Reagent. J. Am. Chem. Soc. 2009, 131, 5018–5019. [Google Scholar] [CrossRef] [PubMed]
- Vogelsang, J.; Kasper, R.; Steinhauer, C.; Person, B.; Heilemann, M.; Sauer, M.; Tinnefeld, P. A Reducing and Oxidizing System Minimizes Photobleaching and Blinking of Fluorescent Dyes. Angew. Chemie Int. Ed. 2008, 47, 5465–5469. [Google Scholar] [CrossRef] [PubMed]
- Balzarotti, F.; Eilers, Y.; Gwosch, K.C.; Gynnå, A.H.; Westphal, V.; Stefani, F.D.; Elf, J.; Hell, S.W. Nanometer Resolution Imaging and Tracking of Fluorescent Molecules with Minimal Photon Fluxes. Science 2017, 355, 606–612. [Google Scholar] [CrossRef] [PubMed]
- Masullo, L.A.; Steiner, F.; Zähringer, J.; Lopez, L.F.; Bohlen, J.; Richter, L.; Cole, F.; Tinnefeld, P.; Stefani, F.D. Pulsed Interleaved MINFLUX. Nano Lett. 2021, 21, 840–846. [Google Scholar] [CrossRef]
- Gwosch, K.C.; Pape, J.K.; Balzarotti, F.; Hoess, P.; Ellenberg, J.; Ries, J.; Hell, S.W. MINFLUX Nanoscopy Delivers 3D Multicolor Nanometer Resolution in Cells. Nat. Methods 2020, 17, 217–224. [Google Scholar] [CrossRef]
- Deguchi, T.; Iwanski, M.K.; Schentarra, E.-M.; Heidebrecht, C.; Schmidt, L.; Heck, J.; Weihs, T.; Schnorrenberg, S.; Hoess, P.; Liu, S.; et al. Direct Observation of Motor Protein Stepping in Living Cells Using MINFLUX. Science 2023, 379, 1010–1015. [Google Scholar] [CrossRef]
- Digman, M.A.; Caiolfa, V.R.; Zamai, M.; Gratton, E. The Phasor Approach to Fluorescence Lifetime Imaging Analysis. Biophys. J. 2008, 94, L14–L16. [Google Scholar] [CrossRef]
- Benda, A.; Fagul’ová, V.; Deyneka, A.; Enderlein, J.; Hof, M. Fluorescence Lifetime Correlation Spectroscopy Combined with Lifetime Tuning: New Perspectives in Supported Phospholipid Bilayer Research. Langmuir 2006, 22, 9580–9585. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Karedla, N.; Thiele, J.C.; Gregor, I.; Enderlein, J. Fluorescence Lifetime Correlation Spectroscopy: Basics and Applications. Methods 2018, 140–141, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Macmillan, A.; Yang, Y.; Gaus, K. Lifetime Based Axial Contrast Enable Simple 3D-STED Imaging. Methods Appl. Fluoresc. 2022, 10, 35001. [Google Scholar] [CrossRef] [PubMed]
- Sender, V.; Hentrich, K.; Pathak, A.; Tan Qian Ler, A.; Embaie, B.T.; Lundström, S.L.; Gaetani, M.; Bergstrand, J.; Nakamoto, R.; Sham, L.-T.; et al. Capillary Leakage Provides Nutrients and Antioxidants for Rapid Pneumococcal Proliferation in Influenza-Infected Lower Airways. Proc. Natl. Acad. Sci. USA 2020, 117, 31386–31397. [Google Scholar] [CrossRef] [PubMed]
- Tabusi, M.; Thorsdottir, S.; Lysandrou, M.; Narciso, A.R.; Minoia, M.; Srambickal, C.V.; Widengren, J.; Henriques-Normark, B.; Iovino, F. Neuronal Death in Pneumococcal Meningitis Is Triggered by Pneumolysin and RrgA Interactions with β-Actin. PLoS Pathog. 2021, 17, e1009432. [Google Scholar] [CrossRef]
- Pathak, A.; Bergstrand, J.; Sender, V.; Spelmink, L.; Aschtgen, M.S.; Muschiol, S.; Widengren, J.; Henriques-Normark, B. Factor H Binding Proteins Protect Division Septa on Encapsulated Streptococcus pneumoniae against Complement C3b Deposition and Amplification. Nat. Commun. 2018, 9, 3398. [Google Scholar] [CrossRef] [PubMed]
- Iovino, F.; Engelen-Lee, J.Y.; Brouwer, M.; van de Beek, D.; van der Ende, A.; Seron, M.V.; Mellroth, P.; Muschiol, S.; Bergstrand, J.; Widengren, J.; et al. pIgR and PEC AM-1 Bind to Pneumococcal Adhesins RrgA and PspC Mediating Bacterial Brain Invasion. J. Exp. Med. 2017, 214, 1619–1630. [Google Scholar] [CrossRef]
- Rönnlund, D.; Yang, Y.; Blom, H.; Auer, G.; Widengren, J. Fluorescence Nanoscopy of Platelets Resolves Platelet-State Specific Storage, Release and Uptake of Proteins, Opening up Future Diagnostic Applications. Adv. Healthc. Mater. 2012, 1, 707–713. [Google Scholar] [CrossRef]
- Rönnlund, D.; Gad, A.K.B.; Blom, H.; Aspenström, P.; Widengren, J. Spatial Organization of Proteins in Metastasizing Cells. Cytom. Part A 2013, 83, 855–865. [Google Scholar] [CrossRef]
- Rönnlund, D.; Xu, L.; Perols, A.; Gad, A.K.B.; Eriksson Karlström, A.; Auer, G.; Widengren, J. Multicolor Fluorescence Nanoscopy by Photobleaching: Concept, Verification, and Its Application To Resolve Selective Storage of Proteins in Platelets. ACS Nano 2014, 8, 4358–4365. [Google Scholar] [CrossRef]
- Bergstrand, J.; Xu, L.; Miao, X.; Li, N.; Öktem, O.; Franzén, B.; Auer, G.; Lomnytska, M.; Widengren, J. Super-Resolution Microscopy Can Identify Specific Protein Distribution Patterns in Platelets Incubated with Cancer Cells. Nanoscale 2019, 11, 10023–10033. [Google Scholar] [CrossRef] [PubMed]
- Bergstrand, J.; Miao, X.; Srambickal, C.V.; Auer, G.; Widengren, J. Fast, Streamlined Fluorescence Nanoscopy Resolves Rearrangements of SNARE and Cargo Proteins in Platelets Co-Incubated with Cancer Cells. J. Nanobiotechnol. 2022, 20, 292. [Google Scholar] [CrossRef] [PubMed]
- Bethge, P.; Chéreau, R.; Avignone, E.; Marsicano, G.; Nägerl, U.V. Two-Photon Excitation STED Microscopy in Two Colors in Acute Brain Slices. Biophys. J. 2013, 104, 778–785. [Google Scholar] [CrossRef] [PubMed]
- Tønnesen, J.; Nadrigny, F.; Willig, K.I.; Wedlich-Söldner, R.; Nägerl, U.V. Two-Color STED Microscopy of Living Synapses Using A Single Laser-Beam Pair. Biophys. J. 2011, 101, 2545–2552. [Google Scholar] [CrossRef] [PubMed]
- Berning, S.; Willig, K.I.; Steffens, H.; Dibaj, P.; Hell, S.W. Nanoscopy in a Living Mouse Brain. Science 2012, 335, 551. [Google Scholar] [CrossRef] [PubMed]
- Calovi, S.; Soria, F.N.; Tønnesen, J. Super-Resolution STED Microscopy in Live Brain Tissue. Neurobiol. Dis. 2021, 156, 105420. [Google Scholar] [CrossRef] [PubMed]
- Nägerl, U.V.; Willig, K.I.; Hein, B.; Hell, S.W.; Bonhoeffer, T. Live-Cell Imaging of Dendritic Spines by STED Microscopy. Proc. Natl. Acad. Sci. USA 2008, 105, 18982–18987. [Google Scholar] [CrossRef]
- Chmyrov, A.; Keller, J.; Grotjohann, T.; Ratz, M.; D’Este, E.; Jakobs, S.; Eggeling, C.; Hell, S.W. Nanoscopy with More than 100,000 “Doughnuts”. Nat. Methods 2013, 10, 737–740. [Google Scholar] [CrossRef]
- Damenti, M.; Coceano, G.; Pennacchietti, F.; Bodén, A.; Testa, I. STED and Parallelized RESOLFT Optical Nanoscopy of the Tubular Endoplasmic Reticulum and Its Mitochondrial Contacts in Neuronal Cells. Neurobiol. Dis. 2021, 155, 105361. [Google Scholar] [CrossRef]
- Bodén, A.; Pennacchietti, F.; Coceano, G.; Damenti, M.; Ratz, M.; Testa, I. Volumetric Live Cell Imaging with Three-Dimensional Parallelized RESOLFT Microscopy. Nat. Biotechnol. 2021, 39, 609–618. [Google Scholar] [CrossRef]
- Maidorn, M.; Olichon, A.; Rizzoli, S.O.; Opazo, F. Nanobodies Reveal an Extra-Synaptic Population of SNAP-25 and Syntaxin 1A in Hippocampal Neurons. MAbs 2019, 11, 305–321. [Google Scholar] [CrossRef] [PubMed]
- Schneider, A.F.L.; Benz, L.S.; Lehmann, M.; Hackenberger, C.P.R. Cell-Permeable Nanobodies Allow Dual-Color Super-Resolution Microscopy in Untransfected Living Cells. Angew. Chemie-Int. Ed. 2021, 60, 22075–22080. [Google Scholar] [CrossRef] [PubMed]
- Sörensen, J.; Sandberg, D.; Sandström, M.; Wennborg, A.; Feldwisch, J.; Tolmachev, V.; Åström, G.; Lubberink, M.; Garske-Román, U.; Carlsson, J.; et al. First-in-Human Molecular Imaging of HER2 Expression in Breast Cancer Metastases Using the 111In-ABY-025 Affibody Molecule. J. Nucl. Med. 2014, 55, 730–735. [Google Scholar] [CrossRef] [PubMed]
- Garousi, J.; Andersson, K.G.; Mitran, B.; Pichl, M.L.; Stahl, S.; Orlova, A.; Löfblom, J.; Tolmachev, V. PET Imaging of Epidermal Growth Factor Receptor Expression in Tumours Using 89Zr-Labelled ZEGFR:2377 Affibody Molecules. Int. J. Oncol. 2016, 48, 1325–1332. [Google Scholar] [CrossRef] [PubMed]
- Gomes de Castro, M.A.; Wildhagen, H.; Sograte-Idrissi, S.; Hitzing, C.; Binder, M.; Trepel, M.; Engels, N.; Opazo, F. Differential Organization of Tonic and Chronic B Cell Antigen Receptors in the Plasma Membrane. Nat. Commun. 2019, 10, 820. [Google Scholar] [CrossRef] [PubMed]
- Sograte-Idrissi, S.; Schlichthaerle, T.; Duque-Afonso, C.J.; Alevra, M.; Strauss, S.; Moser, T.; Jungmann, R.; Rizzoli, S.O.; Opazo, F. Circumvention of Common Labelling Artefacts Using Secondary Nanobodies. Nanoscale 2020, 12, 10226–10239. [Google Scholar] [CrossRef] [PubMed]
- Griffin, B.A.; Adams, S.R.; Tsien, R.Y. Specific Covalent Labeling of Recombinant Protein Molecules Inside Live Cells. Science 1998, 281, 269–272. [Google Scholar] [CrossRef]
- Keppler, A.; Gendreizig, S.; Gronemeyer, T.; Pick, H.; Vogel, H.; Johnsson, K. A General Method for the Covalent Labeling of Fusion Proteins with Small Molecules in Vivo. Nat. Biotechnol. 2003, 21, 86–89. [Google Scholar] [CrossRef]
- Los, G.V.; Darzins, A.; Zimprich, C.; Karassina, N.; Learish, R.; Mcdougall, M.G.; Lance, P.; Friedman-ohana, R.; Wood, M.; Vidugiris, G.; et al. HaloTagTM Interchangeable Labeling Technology for Cell Imaging, Protein Capture and Immobilization. Promega Cell Notes 2005, 11, 2–6. [Google Scholar]
- Gautier, A.; Juillerat, A.; Heinis, C.; Corrêa, I.R.; Kindermann, M.; Beaufils, F.; Johnsson, K. An Engineered Protein Tag for Multiprotein Labeling in Living Cells. Chem. Biol. 2008, 15, 128–136. [Google Scholar] [CrossRef]
- Gould, T.J.; Burke, D.; Bewersdorf, J.; Booth, M.J. Adaptive Optics Enables 3D STED Microscopy in Aberrating Specimens. Opt. Express 2012, 20, 20998–21009. [Google Scholar] [CrossRef]
- Cimini, V.; Mellini, M.; Rampioni, G.; Sbroscia, M.; Leoni, L.; Barbieri, M.; Gianani, I. Adaptive Tracking of Enzymatic Reactions with Quantum Light. Opt. Express 2019, 27, 35245. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeong, S.; Koh, D.; Gwak, E.; Srambickal, C.V.; Seo, D.; Widengren, J.; Lee, J.-C. Pushing the Resolution Limit of Stimulated Emission Depletion Optical Nanoscopy. Int. J. Mol. Sci. 2024, 25, 26. https://doi.org/10.3390/ijms25010026
Jeong S, Koh D, Gwak E, Srambickal CV, Seo D, Widengren J, Lee J-C. Pushing the Resolution Limit of Stimulated Emission Depletion Optical Nanoscopy. International Journal of Molecular Sciences. 2024; 25(1):26. https://doi.org/10.3390/ijms25010026
Chicago/Turabian StyleJeong, Sejoo, Dongbin Koh, Eunha Gwak, Chinmaya V. Srambickal, Daeha Seo, Jerker Widengren, and Jong-Chan Lee. 2024. "Pushing the Resolution Limit of Stimulated Emission Depletion Optical Nanoscopy" International Journal of Molecular Sciences 25, no. 1: 26. https://doi.org/10.3390/ijms25010026