
The Proteomic Analysis of Cancer-Related Alterations in the Human Unfoldome


 , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
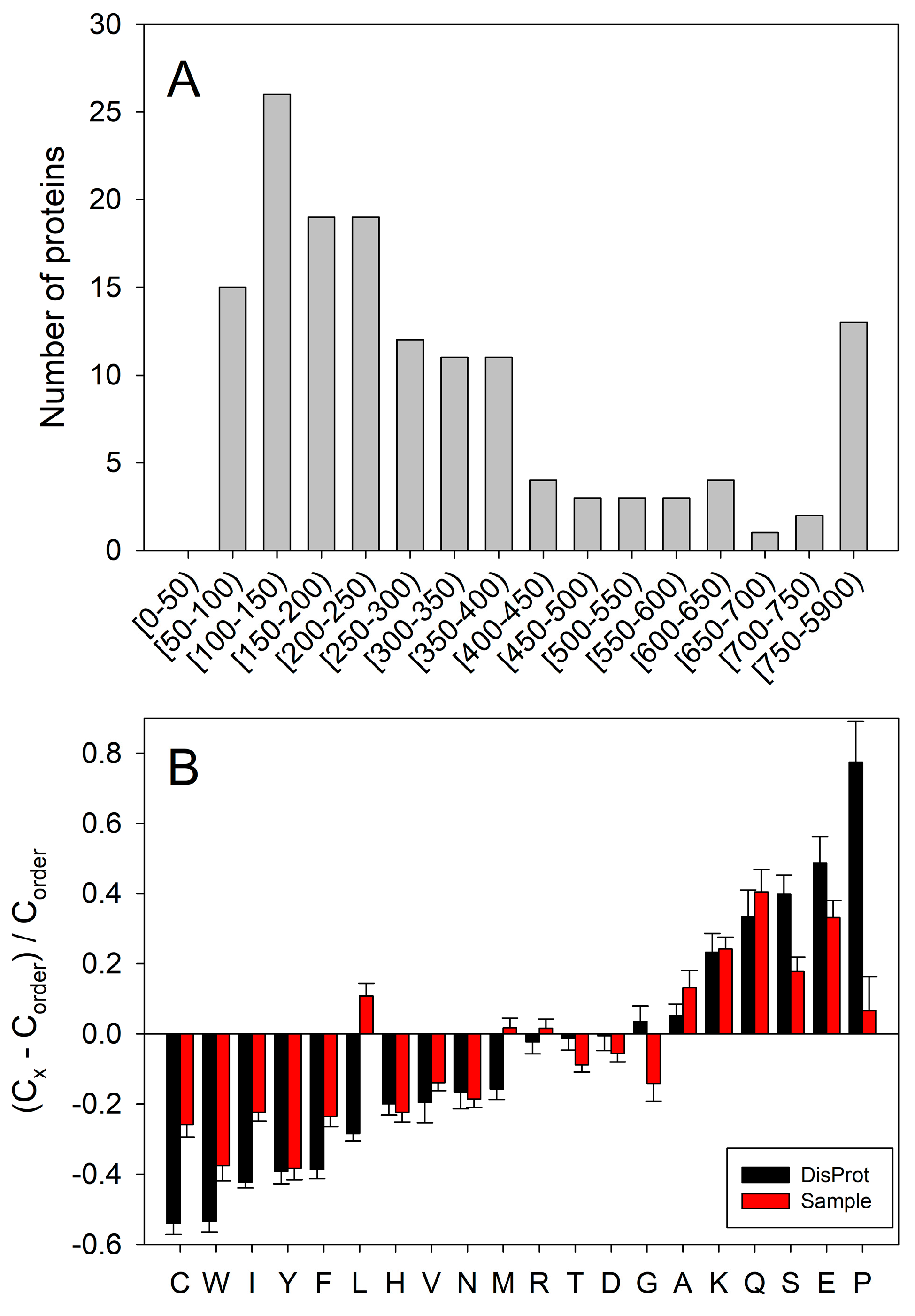
2.1. Proteomics Analysis
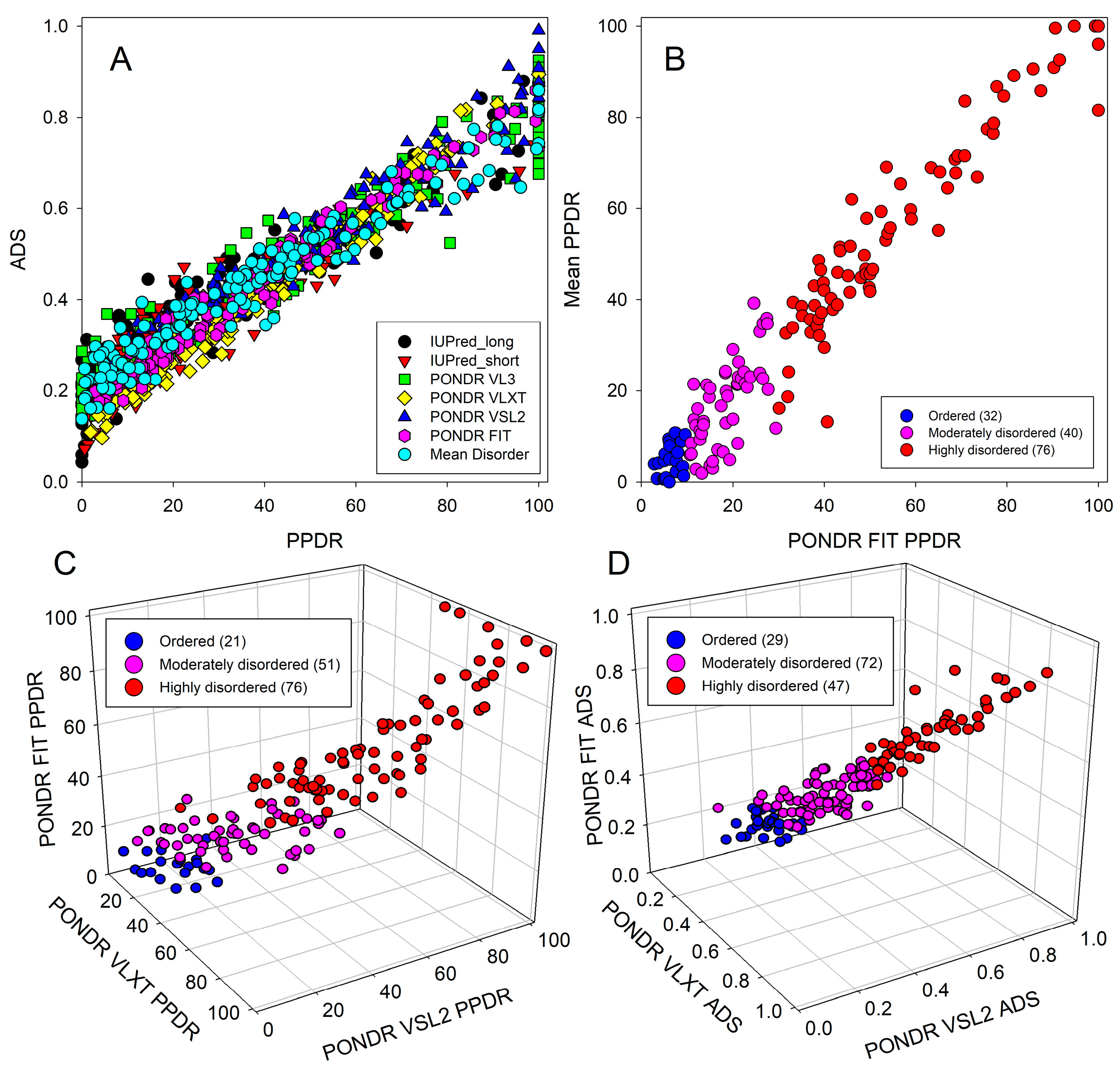
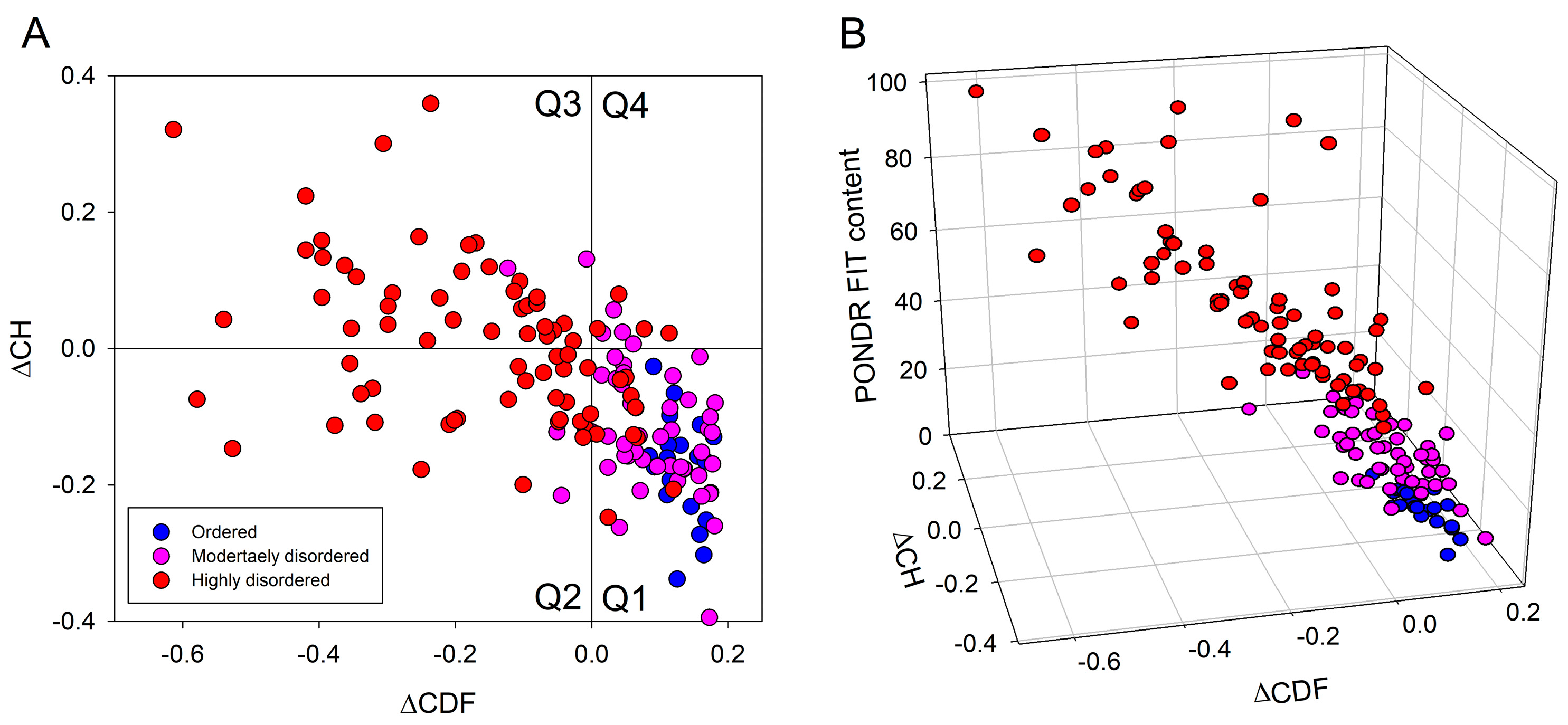
2.2. In Silico Protein Disorder Analysis
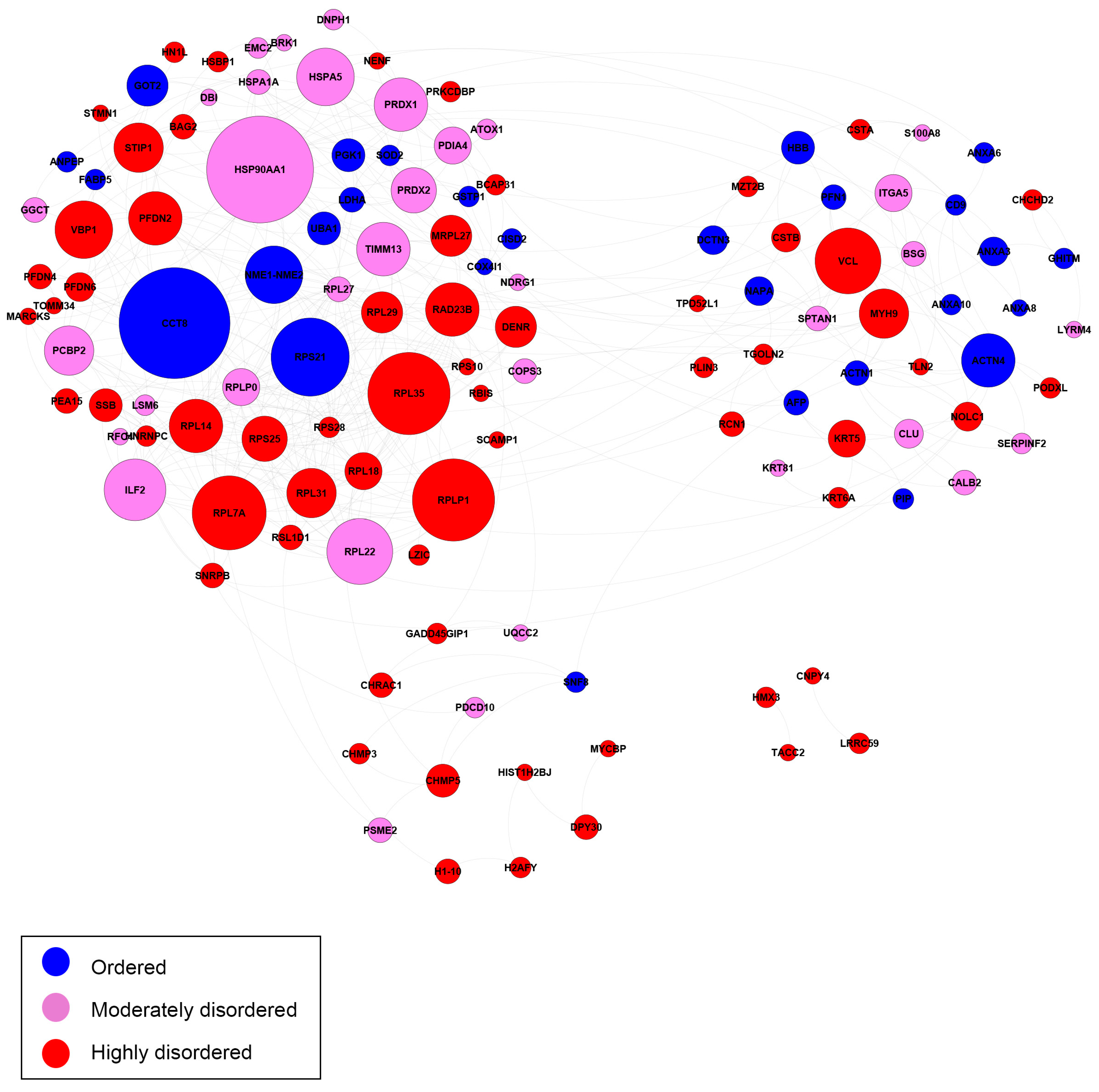
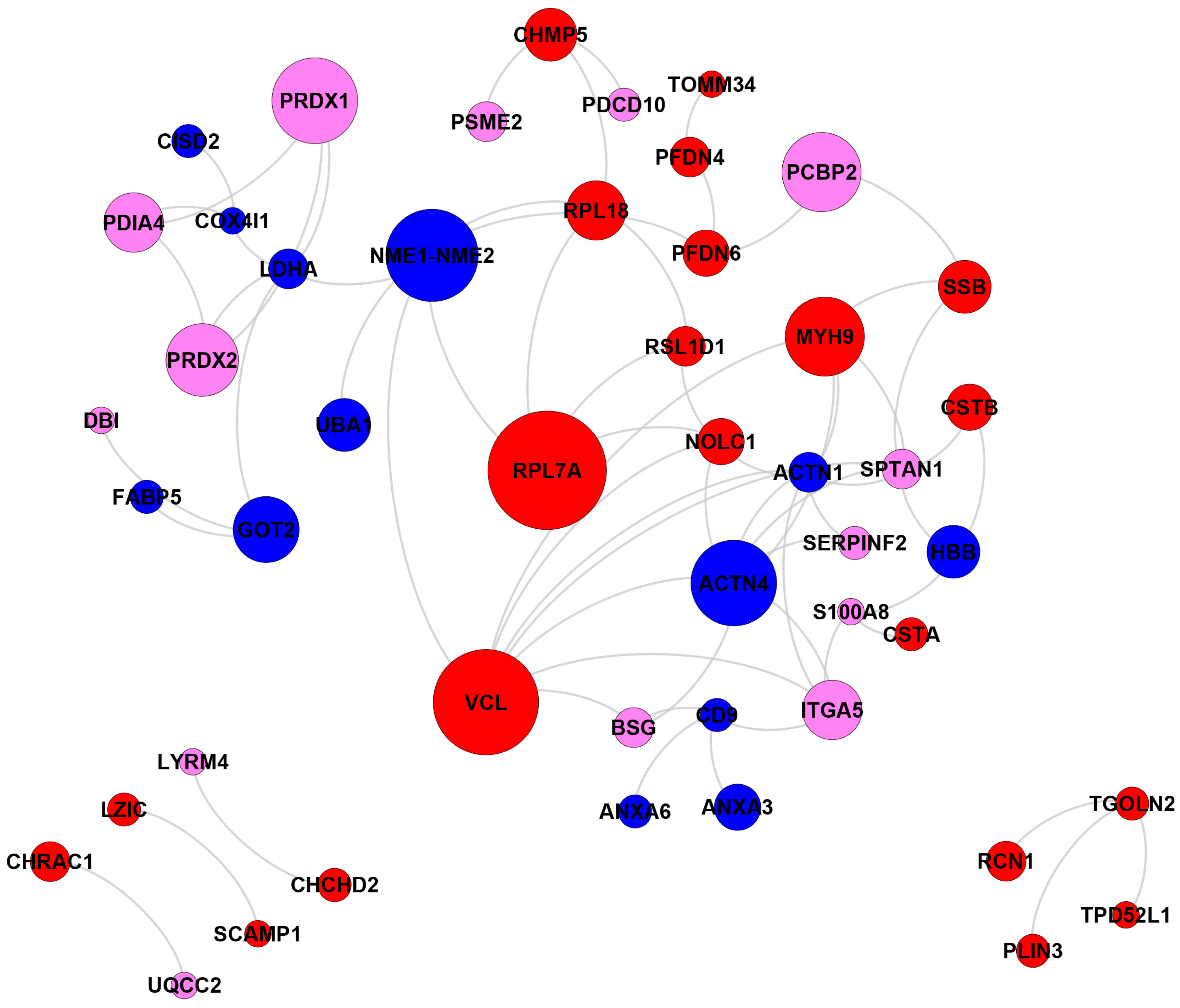
2.3. Biological Network and Pathway Analyses
3. Materials and Methods
3.1. Cell Culture
3.2. Sample Preparation
3.3. Nano-LC-MS/MS Analysis
3.4. Proteins Identification and Quantification
3.5. Protein Annotation, Biological Network and Pathway Analyses
3.6. Compositional Profiling of the Members of Human Cancer-Related Unfoldome
3.7. Per-Residue Intrinsic Disorder Analysis
3.8. Binary and CH-CDF Intrinsic Disorder Analysis
3.9. Evaluation of the Interactability within the Human Cancer-Related Unfoldome
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wright, P.E.; Dyson, H.J. Intrinsically unstructured proteins: Re-assessing the protein structure-function paradigm. J. Mol. Biol. 1999, 293, 321–331. [Google Scholar] [CrossRef]
- Uversky, V.N.; Gillespie, J.R.; Fink, A.L. Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins 2000, 41, 415–427. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K.; Lawson, J.D.; Brown, C.J.; Williams, R.M.; Romero, P.; Oh, J.S.; Oldfield, C.J.; Campen, A.M.; Ratliff, C.M.; Hipps, K.W.; et al. Intrinsically disordered protein. J. Mol. Graph. Model. 2001, 19, 26–59. [Google Scholar] [CrossRef]
- Dunker, A.K.; Obradovic, Z. The protein trinity--linking function and disorder. Nat. Biotechnol. 2001, 19, 805–806. [Google Scholar] [CrossRef] [PubMed]
- Tompa, P. Intrinsically unstructured proteins. Trends Biochem. Sci. 2002, 27, 527–533. [Google Scholar] [CrossRef]
- Uversky, V.N. Natively unfolded proteins: A point where biology waits for physics. Protein Sci. 2002, 11, 739–756. [Google Scholar] [CrossRef]
- Iakoucheva, L.M.; Brown, C.J.; Lawson, J.D.; Obradovic, Z.; Dunker, A.K. Intrinsic disorder in cell-signaling and cancer-associated proteins. J. Mol. Biol. 2002, 323, 573–584. [Google Scholar] [CrossRef]
- Dunker, A.K.; Brown, C.J.; Lawson, J.D.; Iakoucheva, L.M.; Obradovic, Z. Intrinsic disorder and protein function. Biochemistry 2002, 41, 6573–6582. [Google Scholar] [CrossRef]
- Dunker, A.K.; Brown, C.J.; Obradovic, Z. Identification and functions of usefully disordered proteins. Adv. Protein Chem. 2002, 62, 25–49. [Google Scholar]
- Dyson, H.J.; Wright, P.E. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 2005, 6, 197–208. [Google Scholar] [CrossRef]
- Tompa, P. The interplay between structure and function in intrinsically unstructured proteins. FEBS Lett. 2005, 579, 3346–3354. [Google Scholar] [CrossRef] [PubMed]
- Radivojac, P.; Iakoucheva, L.M.; Oldfield, C.J.; Obradovic, Z.; Uversky, V.N.; Dunker, A.K. Intrinsic disorder and functional proteomics. Biophys. J. 2007, 92, 1439–1456. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K.; Oldfield, C.J.; Meng, J.; Romero, P.; Yang, J.Y.; Chen, J.W.; Vacic, V.; Obradovic, Z.; Uversky, V.N. The unfoldomics decade: An update on intrinsically disordered proteins. BMC Genom. 2008, 9 (Suppl. 2), S1–S26. [Google Scholar] [CrossRef] [PubMed]
- Galea, C.A.; Wang, Y.; Sivakolundu, S.G.; Kriwacki, R.W. Regulation of cell division by intrinsically unstructured proteins: Intrinsic flexibility, modularity, and signaling conduits. Biochemistry 2008, 47, 7598–7609. [Google Scholar] [CrossRef] [PubMed]
- Tompa, P. Unstructural biology coming of age. Curr. Opin. Struct. Biol. 2011, 21, 419–425. [Google Scholar] [CrossRef]
- Hong, S.; Choi, S.; Kim, R.; Koh, J. Mechanisms of Macromolecular Interactions Mediated by Protein Intrinsic Disorder. Mol. Cells 2020, 43, 899–908. [Google Scholar] [CrossRef]
- Pajkos, M.; Dosztanyi, Z. Functions of intrinsically disordered proteins through evolutionary lenses. Prog. Mol. Biol. Transl. Sci. 2021, 183, 45–74. [Google Scholar] [CrossRef]
- Malagrino, F.; Pennacchietti, V.; Santorelli, D.; Pagano, L.; Nardella, C.; Diop, A.; Toto, A.; Gianni, S. On the Effects of Disordered Tails, Supertertiary Structure and Quinary Interactions on the Folding and Function of Protein Domains. Biomolecules 2022, 12, 209. [Google Scholar] [CrossRef]
- Cermakova, K.; Hodges, H.C. Interaction modules that impart specificity to disordered protein. Trends Biochem. Sci. 2023, 48, 477–490. [Google Scholar] [CrossRef]
- Davey, N.E.; Simonetti, L.; Ivarsson, Y. The next wave of interactomics: Mapping the SLiM-based interactions of the intrinsically disordered proteome. Curr. Opin. Struct. Biol. 2023, 80, 102593. [Google Scholar] [CrossRef]
- Evans, R.; Ramisetty, S.; Kulkarni, P.; Weninger, K. Illuminating Intrinsically Disordered Proteins with Integrative Structural Biology. Biomolecules 2023, 13, 124. [Google Scholar] [CrossRef]
- Fenton, M.; Gregory, E.; Daughdrill, G. Protein disorder and autoinhibition: The role of multivalency and effective concentration. Curr. Opin. Struct. Biol. 2023, 83, 102705. [Google Scholar] [CrossRef] [PubMed]
- Handa, T.; Kundu, D.; Dubey, V.K. Perspectives on evolutionary and functional importance of intrinsically disordered proteins. Int. J. Biol. Macromol. 2023, 224, 243–255. [Google Scholar] [CrossRef] [PubMed]
- Holehouse, A.S.; Kragelund, B.B. The molecular basis for cellular function of intrinsically disordered protein regions. Nat. Rev. Mol. Cell. Biol. 2023. [Google Scholar] [CrossRef]
- Moses, D.; Ginell, G.M.; Holehouse, A.S.; Sukenik, S. Intrinsically disordered regions are poised to act as sensors of cellular chemistry. Trends Biochem. Sci. 2023, 48, 1019–1034. [Google Scholar] [CrossRef] [PubMed]
- Oldfield, C.J.; Dunker, A.K. Intrinsically disordered proteins and intrinsically disordered protein regions. Annu. Rev. Biochem. 2014, 83, 553–584. [Google Scholar] [CrossRef] [PubMed]
- Deiana, A.; Forcelloni, S.; Porrello, A.; Giansanti, A. Intrinsically disordered proteins and structured proteins with intrinsically disordered regions have different functional roles in the cell. PLoS ONE 2019, 14, e0217889. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Unusual biophysics of intrinsically disordered proteins. Biochim. Biophys. Acta 2013, 1834, 932–951. [Google Scholar] [CrossRef]
- Dunker, A.K.; Obradovic, Z.; Romero, P.; Garner, E.C.; Brown, C.J. Intrinsic protein disorder in complete genomes. Genome Inform. Ser. Workshop Genome Inform. 2000, 11, 161–171. [Google Scholar]
- Ward, J.J.; Sodhi, J.S.; McGuffin, L.J.; Buxton, B.F.; Jones, D.T. Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J. Mol. Biol. 2004, 337, 635–645. [Google Scholar] [CrossRef]
- Oldfield, C.J.; Cheng, Y.; Cortese, M.S.; Brown, C.J.; Uversky, V.N.; Dunker, A.K. Comparing and combining predictors of mostly disordered proteins. Biochemistry 2005, 44, 1989–2000. [Google Scholar] [CrossRef]
- Tompa, P.; Dosztanyi, Z.; Simon, I. Prevalent structural disorder in E. coli and S. cerevisiae proteomes. J. Proteome Res. 2006, 5, 1996–2000. [Google Scholar] [CrossRef]
- Lu, Y.; Yang, Y.; Zhu, G.; Zeng, H.; Fan, Y.; Guo, F.; Xu, D.; Wang, B.; Chen, D.; Ge, G. Emerging Pharmacotherapeutic Strategies to Overcome Undruggable Proteins in Cancer. Int. J. Biol. Sci. 2023, 19, 3360–3382. [Google Scholar] [CrossRef]
- Santofimia-Castano, P.; Rizzuti, B.; Xia, Y.; Abian, O.; Peng, L.; Velazquez-Campoy, A.; Neira, J.L.; Iovanna, J. Targeting intrinsically disordered proteins involved in cancer. Cell. Mol. Life Sci. 2020, 77, 1695–1707. [Google Scholar] [CrossRef]
- Kulkarni, V.; Kulkarni, P. Intrinsically disordered proteins and phenotypic switching: Implications in cancer. Prog. Mol. Biol. Transl. Sci. 2019, 166, 63–84. [Google Scholar] [CrossRef]
- Garg, N.; Kumar, P.; Gadhave, K.; Giri, R. The dark proteome of cancer: Intrinsic disorderedness and functionality of HIF-1alpha along with its interacting proteins. Prog. Mol. Biol. Transl. Sci. 2019, 166, 371–403. [Google Scholar] [CrossRef]
- Nussinov, R.; Jang, H.; Tsai, C.J.; Liao, T.J.; Li, S.; Fushman, D.; Zhang, J. Intrinsic protein disorder in oncogenic KRAS signaling. Cell. Mol. Life Sci. 2017, 74, 3245–3261. [Google Scholar] [CrossRef]
- Kumar, D.; Sharma, N.; Giri, R. Therapeutic Interventions of Cancers Using Intrinsically Disordered Proteins as Drug Targets: c-Myc as Model System. Cancer Inform. 2017, 16, 1176935117699408. [Google Scholar] [CrossRef] [PubMed]
- Mooney, S.M.; Jolly, M.K.; Levine, H.; Kulkarni, P. Phenotypic plasticity in prostate cancer: Role of intrinsically disordered proteins. Asian J. Androl. 2016, 18, 704–710. [Google Scholar] [CrossRef] [PubMed]
- Russo, A.; Manna, S.L.; Novellino, E.; Malfitano, A.M.; Marasco, D. Molecular signaling involving intrinsically disordered proteins in prostate cancer. Asian J. Androl. 2016, 18, 673–681. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Intrinsic disorder in proteins associated with neurodegenerative diseases. Front. Biosci. (Landmark Ed.) 2009, 14, 5188–5238. [Google Scholar] [CrossRef] [PubMed]
- Birol, M.; Melo, A.M. Untangling the Conformational Polymorphism of Disordered Proteins Associated With Neurodegeneration at the Single-Molecule Level. Front. Mol. Neurosci. 2019, 12, 309. [Google Scholar] [CrossRef] [PubMed]
- Ayyadevara, S.; Ganne, A.; Balasubramaniam, M.; Shmookler Reis, R.J. Intrinsically disordered proteins identified in the aggregate proteome serve as biomarkers of neurodegeneration. Metab. Brain Dis. 2022, 37, 147–152. [Google Scholar] [CrossRef]
- Di Liegro, C.M.; Schiera, G.; Schiro, G.; Di Liegro, I. RNA-Binding Proteins as Epigenetic Regulators of Brain Functions and Their Involvement in Neurodegeneration. Int. J. Mol. Sci. 2022, 23, 14622. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, P.; Zweckstetter, M. Role of aberrant phase separation in pathological protein aggregation. Curr. Opin. Struct. Biol. 2023, 82, 102678. [Google Scholar] [CrossRef]
- Tsoi, P.S.; Quan, M.D.; Ferreon, J.C.; Ferreon, A.C.M. Aggregation of Disordered Proteins Associated with Neurodegeneration. Int. J. Mol. Sci. 2023, 24, 3380. [Google Scholar] [CrossRef]
- Cieplak, M.; Mioduszewski, L.; Chwastyk, M. Contact-Based Analysis of Aggregation of Intrinsically Disordered Proteins. Methods Mol. Biol. 2022, 2340, 105–120. [Google Scholar] [CrossRef]
- Mei, Y.; Glover, K.; Su, M.; Sinha, S.C. Conformational flexibility of BECN1: Essential to its key role in autophagy and beyond. Protein Sci. 2016, 25, 1767–1785. [Google Scholar] [CrossRef]
- Du, Z.; Uversky, V.N. A Comprehensive Survey of the Roles of Highly Disordered Proteins in Type 2 Diabetes. Int. J. Mol. Sci. 2017, 18, 2010. [Google Scholar] [CrossRef]
- Nguyen, P.H.; Ramamoorthy, A.; Sahoo, B.R.; Zheng, J.; Faller, P.; Straub, J.E.; Dominguez, L.; Shea, J.E.; Dokholyan, N.V.; De Simone, A.; et al. Amyloid Oligomers: A Joint Experimental/Computational Perspective on Alzheimer’s Disease, Parkinson’s Disease, Type II Diabetes, and Amyotrophic Lateral Sclerosis. Chem. Rev. 2021, 121, 2545–2647. [Google Scholar] [CrossRef]
- Usher, E.T.; Showalter, S.A. Biophysical insights into glucose-dependent transcriptional regulation by PDX1. J. Biol. Chem. 2022, 298, 102623. [Google Scholar] [CrossRef]
- Tovo-Rodrigues, L.; Recamonde-Mendoza, M.; Paixao-Cortes, V.R.; Bruxel, E.M.; Schuch, J.B.; Friedrich, D.C.; Rohde, L.A.; Hutz, M.H. The role of protein intrinsic disorder in major psychiatric disorders. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2016, 171, 848–860. [Google Scholar] [CrossRef]
- Cheng, Y.; LeGall, T.; Oldfield, C.J.; Dunker, A.K.; Uversky, V.N. Abundance of intrinsic disorder in protein associated with cardiovascular disease. Biochemistry 2006, 45, 10448–10460. [Google Scholar] [CrossRef]
- Vacic, V.; Iakoucheva, L.M. Disease mutations in disordered regions--exception to the rule? Mol. Biosyst. 2012, 8, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Tarnovskaya, S.; Kiselev, A.; Kostareva, A.; Frishman, D. Structural consequences of mutations associated with idiopathic restrictive cardiomyopathy. Amino Acids 2017, 49, 1815–1829. [Google Scholar] [CrossRef] [PubMed]
- Johnston, J.R.; Landim-Vieira, M.; Marques, M.A.; de Oliveira, G.A.P.; Gonzalez-Martinez, D.; Moraes, A.H.; He, H.; Iqbal, A.; Wilnai, Y.; Birk, E.; et al. The intrinsically disordered C terminus of troponin T binds to troponin C to modulate myocardial force generation. J. Biol. Chem. 2019, 294, 20054–20069. [Google Scholar] [CrossRef]
- Theophall, G.G.; Ramirez, L.M.S.; Premo, A.; Reverdatto, S.; Manigrasso, M.B.; Yepuri, G.; Burz, D.S.; Ramasamy, R.; Schmidt, A.M.; Shekhtman, A. Disruption of the productive encounter complex results in dysregulation of DIAPH1 activity. J. Biol. Chem. 2023, 299, 105342. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Oldfield, C.J.; Dunker, A.K. Intrinsically disordered proteins in human diseases: Introducing the D2 concept. Annu. Rev. Biophys. 2008, 37, 215–246. [Google Scholar] [CrossRef]
- Goh, K.I.; Cusick, M.E.; Valle, D.; Childs, B.; Vidal, M.; Barabasi, A.L. The human disease network. Proc. Natl. Acad. Sci. USA 2007, 104, 8685–8690. [Google Scholar] [CrossRef]
- Midic, U.; Oldfield, C.J.; Dunker, A.K.; Obradovic, Z.; Uversky, V.N. Protein disorder in the human diseasome: Unfoldomics of human genetic diseases. BMC Genom. 2009, 10 (Suppl. 1), S12. [Google Scholar] [CrossRef]
- Williams, R.M.; Obradovi, Z.; Mathura, V.; Braun, W.; Garner, E.C.; Young, J.; Takayama, S.; Brown, C.J.; Dunker, A.K. The Protein Non-Folding Problem: Amino Acid Determinants of Intrinsic Order and Disorder. In Biocomputing 2001—Proceedings of the Pacific Symposium; World Scientific Publishing: Singapore, 2001; pp. 89–100. [Google Scholar]
- Timm, D.E.; Vissavajjhala, P.; Ross, A.H.; Neet, K.E. Spectroscopic and chemical studies of the interaction between nerve growth factor (NGF) and the extracellular domain of the low affinity NGF receptor. Protein Sci. 1992, 1, 1023–1031. [Google Scholar] [CrossRef]
- Kim, T.D.; Ryu, H.J.; Cho, H.I.; Yang, C.H.; Kim, J. Thermal behavior of proteins: Heat-resistant proteins and their heat-induced secondary structural changes. Biochemistry 2000, 39, 14839–14846. [Google Scholar] [CrossRef]
- Uversky, V.N. Intrinsically disordered proteins and their environment: Effects of strong denaturants, temperature, pH, counter ions, membranes, binding partners, osmolytes, and macromolecular crowding. Protein J. 2009, 28, 305–325. [Google Scholar] [CrossRef]
- Konno, T.; Tanaka, N.; Kataoka, M.; Takano, E.; Maki, M. A circular dichroism study of preferential hydration and alcohol effects on a denatured protein, pig calpastatin domain I. Biochim. Biophys. Acta 1997, 1342, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Lynn, A.; Chandra, S.; Malhotra, P.; Chauhan, V.S. Heme binding and polymerization by Plasmodium falciparum histidine rich protein II: Influence of pH on activity and conformation. FEBS Lett. 1999, 459, 267–271. [Google Scholar] [CrossRef]
- Johansson, J.; Gudmundsson, G.H.; Rottenberg, M.E.; Berndt, K.D.; Agerberth, B. Conformation-dependent antibacterial activity of the naturally occurring human peptide LL-37. J. Biol. Chem. 1998, 273, 3718–3724. [Google Scholar] [CrossRef] [PubMed]
- Cortese, M.S.; Baird, J.P.; Uversky, V.N.; Dunker, A.K. Uncovering the unfoldome: Enriching cell extracts for unstructured proteins by Acid treatment. J. Proteome Res. 2005, 4, 1610–1618. [Google Scholar] [CrossRef]
- Csizmok, V.; Szollosi, E.; Friedrich, P.; Tompa, P. A novel two-dimensional electrophoresis technique for the identification of intrinsically unstructured proteins. Mol. Cell Proteom. 2006, 5, 265–273. [Google Scholar] [CrossRef]
- Galea, C.A.; Pagala, V.R.; Obenauer, J.C.; Park, C.G.; Slaughter, C.A.; Kriwacki, R.W. Proteomic studies of the intrinsically unstructured mammalian proteome. J. Proteome Res. 2006, 5, 2839–2848. [Google Scholar] [CrossRef]
- Kaddour, H.; Lyu, Y.; Welch, J.L.; Paromov, V.; Mandape, S.N.; Sakhare, S.S.; Pandhare, J.; Stapleton, J.T.; Pratap, S.; Dash, C.; et al. Proteomics Profiling of Autologous Blood and Semen Exosomes from HIV-infected and Uninfected Individuals Reveals Compositional and Functional Variabilities. Mol. Cell. Proteom. 2020, 19, 78–100. [Google Scholar] [CrossRef]
- Kaddour, H.; Kopcho, S.; Lyu, Y.; Shouman, N.; Paromov, V.; Pratap, S.; Dash, C.; Kim, E.Y.; Martinson, J.; McKay, H.; et al. HIV-infection and cocaine use regulate semen extracellular vesicles proteome and miRNAome in a manner that mediates strategic monocyte haptotaxis governed by miR-128 network. Cell. Mol. Life Sci. 2021, 79, 5. [Google Scholar] [CrossRef] [PubMed]
- Schirmer, E.C.; Yates, J.R., 3rd; Gerace, L. MudPIT: A powerful proteomics tool for discovery. Discov. Med. 2003, 3, 38–39. [Google Scholar] [PubMed]
- Cagney, G.; Park, S.; Chung, C.; Tong, B.; O’Dushlaine, C.; Shields, D.C.; Emili, A. Human tissue profiling with multidimensional protein identification technology. J. Proteome Res. 2005, 4, 1757–1767. [Google Scholar] [CrossRef] [PubMed]
- Kislinger, T.; Gramolini, A.O.; MacLennan, D.H.; Emili, A. Multidimensional protein identification technology (MudPIT): Technical overview of a profiling method optimized for the comprehensive proteomic investigation of normal and diseased heart tissue. J. Am. Soc. Mass. Spectrom. 2005, 16, 1207–1220. [Google Scholar] [CrossRef] [PubMed]
- Delahunty, C.M.; Yates, J.R., 3rd. MudPIT: Multidimensional protein identification technology. Biotechniques 2007, 43, 563, 565, 567 passim. [Google Scholar] [PubMed]
- Link, A.J.; Washburn, M.P. Analysis of protein composition using multidimensional chromatography and mass spectrometry. Curr. Protoc. Protein Sci. 2014, 78, 23.1.1–23.1.25. [Google Scholar] [CrossRef]
- Hoek, K.L.; Samir, P.; Howard, L.M.; Niu, X.; Prasad, N.; Galassie, A.; Liu, Q.; Allos, T.M.; Floyd, K.A.; Guo, Y.; et al. A cell-based systems biology assessment of human blood to monitor immune responses after influenza vaccination. PLoS ONE 2015, 10, e0118528. [Google Scholar] [CrossRef] [PubMed]
- Samir, P.; Rahul; Slaughter, J.C.; Link, A.J. Environmental Interactions and Epistasis Are Revealed in the Proteomic Responses to Complex Stimuli. PLoS ONE 2015, 10, e0134099. [Google Scholar] [CrossRef]
- Gerbasi, V.R.; Browne, C.M.; Samir, P.; Shen, B.; Sun, M.; Hazelbaker, D.Z.; Galassie, A.C.; Frank, J.; Link, A.J. Critical Role for Saccharomyces cerevisiae Asc1p in Translational Initiation at Elevated Temperatures. Proteomics 2018, 18, e1800208. [Google Scholar] [CrossRef]
- Samir, P.; Browne, C.M.; Rahul; Sun, M.; Shen, B.; Li, W.; Frank, J.; Link, A.J. Identification of Changing Ribosome Protein Compositions using Mass Spectrometry. Proteomics 2018, 18, e1800217. [Google Scholar] [CrossRef]
- Sherman, B.T.; Hao, M.; Qiu, J.; Jiao, X.; Baseler, M.W.; Lane, H.C.; Imamichi, T.; Chang, W. DAVID: A web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 2022, 50, W216–W221. [Google Scholar] [CrossRef]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Tan, Q.; Kir, J.; Liu, D.; Bryant, D.; Guo, Y.; Stephens, R.; Baseler, M.W.; Lane, H.C.; et al. DAVID Bioinformatics Resources: Expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res. 2007, 35, W169–W175. [Google Scholar] [CrossRef]
- Xin, L.; Qiao, R.; Chen, X.; Tran, H.; Pan, S.; Rabinoviz, S.; Bian, H.; He, X.; Morse, B.; Shan, B.; et al. A streamlined platform for analyzing tera-scale DDA and DIA mass spectrometry data enables highly sensitive immunopeptidomics. Nat. Commun. 2022, 13, 3108. [Google Scholar] [CrossRef]
- Vacic, V.; Uversky, V.; Dunker, A.K.; Lonardi, S. Composition Profiler: A tool for discovery and visualization of amino acid composition differences. BMC Bioinform. 2007, 8, 211. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Sickmeier, M.; Hamilton, J.A.; LeGall, T.; Vacic, V.; Cortese, M.S.; Tantos, A.; Szabo, B.; Tompa, P.; Chen, J.; Uversky, V.N.; et al. DisProt: The Database of Disordered Proteins. Nucleic Acids Res. 2007, 35, D786–D793. [Google Scholar] [CrossRef] [PubMed]
- Dayhoff, G.W., 2nd; Uversky, V.N. Rapid prediction and analysis of protein intrinsic disorder. Protein Sci. 2022, 31, e4496. [Google Scholar] [CrossRef] [PubMed]
- Rajagopalan, K.; Mooney, S.M.; Parekh, N.; Getzenberg, R.H.; Kulkarni, P. A majority of the cancer/testis antigens are intrinsically disordered proteins. J. Cell Biochem. 2011, 112, 3256–3267. [Google Scholar] [CrossRef]
- Warde-Farley, D.; Donaldson, S.L.; Comes, O.; Zuberi, K.; Badrawi, R.; Chao, P.; Franz, M.; Grouios, C.; Kazi, F.; Lopes, C.T.; et al. The GeneMANIA prediction server: Biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010, 38, W214–W220. [Google Scholar] [CrossRef] [PubMed]
- Olokpa, E.; Mandape, S.N.; Pratap, S.; Stewart, M.V. Metformin regulates multiple signaling pathways within castration-resistant human prostate cancer cells. BMC Cancer 2022, 22, 1025. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Franceschini, A.; Kuhn, M.; Simonovic, M.; Roth, A.; Minguez, P.; Doerks, T.; Stark, M.; Muller, J.; Bork, P.; et al. The STRING database in 2011: Functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2011, 39, D561–D568. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING database in 2023: Protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2023, 51, D638–D646. [Google Scholar] [CrossRef] [PubMed]
- Snel, B.; Lehmann, G.; Bork, P.; Huynen, M.A. STRING: A web-server to retrieve and display the repeatedly occurring neighbourhood of a gene. Nucleic Acids Res. 2000, 28, 3442–3444. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, Z.; Huang, Y.; Wei, W.; Ning, S.; Li, J.; Liang, X.; Liu, K.; Zhang, L. Plasma HSP90AA1 Predicts the Risk of Breast Cancer Onset and Distant Metastasis. Front. Cell Dev. Biol. 2021, 9, 639596. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Wang, C.; Chen, S.; Liu, J.; Fu, Y.; Luo, Y. Extracellular Hsp90alpha and clusterin synergistically promote breast cancer epithelial-to-mesenchymal transition and metastasis via LRP1. J. Cell Sci. 2019, 132, jcs.228213. [Google Scholar] [CrossRef]
- Zhang, S.; Shao, J.; Su, F. Prognostic significance of STIP1 expression in human cancer: A meta-analysis. Clin. Chim. Acta 2018, 486, 168–176. [Google Scholar] [CrossRef]
- Zhang, Z.; Ren, H.; Yang, L.; Zhang, X.; Liang, W.; Wu, H.; Huang, L.; Kang, J.; Xu, J.; Zhai, E.; et al. Aberrant expression of stress-induced phosphoprotein 1 in colorectal cancer and its clinicopathologic significance. Hum. Pathol. 2018, 79, 135–143. [Google Scholar] [CrossRef]
- Luo, X.; Liu, Y.; Ma, S.; Liu, L.; Xie, R.; Li, M.; Shen, P.; Wang, S. STIP1 is over-expressed in hepatocellular carcinoma and promotes the growth and migration of cancer cells. Gene 2018, 662, 110–117. [Google Scholar] [CrossRef]
- Guan, Y.; Zhu, X.; Liang, J.; Wei, M.; Huang, S.; Pan, X. Upregulation of HSPA1A/HSPA1B/HSPA7 and Downregulation of HSPA9 Were Related to Poor Survival in Colon Cancer. Front. Oncol. 2021, 11, 749673. [Google Scholar] [CrossRef]
- Cheang, M.C.; Voduc, D.; Bajdik, C.; Leung, S.; McKinney, S.; Chia, S.K.; Perou, C.M.; Nielsen, T.O. Basal-like breast cancer defined by five biomarkers has superior prognostic value than triple-negative phenotype. Clin. Cancer Res. 2008, 14, 1368–1376. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Ke, S.; Liu, Z.; Shao, H.; He, M.; Guo, J. HSPA5 Promotes the Proliferation, Metastasis and Regulates Ferroptosis of Bladder Cancer. Int. J. Mol. Sci. 2023, 24, 5144. [Google Scholar] [CrossRef] [PubMed]
- Cerezo, M.; Rocchi, S. New anti-cancer molecules targeting HSPA5/BIP to induce endoplasmic reticulum stress, autophagy and apoptosis. Autophagy 2017, 13, 216–217. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, M.S.; Holmgren, L.; Shing, Y.; Chen, C.; Rosenthal, R.A.; Moses, M.; Lane, W.S.; Cao, Y.; Sage, E.H.; Folkman, J. Angiostatin: A novel angiogenesis inhibitor that mediates the suppression of metastases by a Lewis lung carcinoma. Cell 1994, 79, 315–328. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Luo, Y.; Zhang, D.; Wang, X.; Zhang, P.; Li, H.; Ejaz, S.; Liang, S. PGK1-mediated cancer progression and drug resistance. Am. J. Cancer Res. 2019, 9, 2280–2302. [Google Scholar] [PubMed]
- Kim, Y.S.; Gupta Vallur, P.; Phaeton, R.; Mythreye, K.; Hempel, N. Insights into the Dichotomous Regulation of SOD2 in Cancer. Antioxidants 2017, 6, 86. [Google Scholar] [CrossRef] [PubMed]
- Fukai, T.; Ushio-Fukai, M. Superoxide dismutases: Role in redox signaling, vascular function, and diseases. Antioxid. Redox Signal 2011, 15, 1583–1606. [Google Scholar] [CrossRef]
- Zheng, X.; Xu, H.; Gong, L.; Cao, D.; Jin, T.; Wang, Y.; Pi, J.; Yang, Y.; Yi, X.; Liao, D.; et al. Vinculin orchestrates prostate cancer progression by regulating tumor cell invasion, migration, and proliferation. Prostate 2021, 81, 347–356. [Google Scholar] [CrossRef]
- Li, H.; Wang, C.; Lan, L.; Behrens, A.; Tomaschko, M.; Ruiz, J.; Su, Q.; Zhao, G.; Yuan, C.; Xiao, X.; et al. High expression of vinculin predicts poor prognosis and distant metastasis and associates with influencing tumor-associated NK cell infiltration and epithelial-mesenchymal transition in gastric cancer. Aging 2021, 13, 5197–5225. [Google Scholar] [CrossRef]
- Zhu, Y.; Lin, H.; Li, Z.; Wang, M.; Luo, J. Modulation of expression of ribosomal protein L7a (rpL7a) by ethanol in human breast cancer cells. Breast Cancer Res. Treat. 2001, 69, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Pamidimukkala, N.; Puts, G.S.; Kathryn Leonard, M.; Snyder, D.; Dabernat, S.; De Fabo, E.C.; Noonan, F.P.; Slominski, A.; Merlino, G.; Kaetzel, D.M. Nme1 and Nme2 genes exert metastasis-suppressor activities in a genetically engineered mouse model of UV-induced melanoma. Br. J. Cancer 2021, 124, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Khan, I.; Gril, B.; Steeg, P.S. Metastasis Suppressors NME1 and NME2 Promote Dynamin 2 Oligomerization and Regulate Tumor Cell Endocytosis, Motility, and Metastasis. Cancer Res. 2019, 79, 4689–4702. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Li, J.J.; Deng, T.; Li, B.H.; Geng, P.L.; Zeng, X.T. Actinin-4 as a Diagnostic Biomarker in Serum of Breast Cancer Patients. Med. Sci. Monit. 2019, 25, 3298–3302. [Google Scholar] [CrossRef] [PubMed]
- Hsu, K.S.; Kao, H.Y. Alpha-actinin 4 and tumorigenesis of breast cancer. Vitam. Horm. 2013, 93, 323–351. [Google Scholar] [CrossRef]
- Nikolopoulos, S.N.; Spengler, B.A.; Kisselbach, K.; Evans, A.E.; Biedler, J.L.; Ross, R.A. The human non-muscle alpha-actinin protein encoded by the ACTN4 gene suppresses tumorigenicity of human neuroblastoma cells. Oncogene 2000, 19, 380–386. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Kawashima, M.; Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 2023, 51, D587–D592. [Google Scholar] [CrossRef]
- Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 2019, 28, 1947–1951. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Sato, Y.; Furumichi, M.; Tanabe, M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012, 40, D109–D114. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Kanehisa, M. A database for post-genome analysis. Trends Genet. 1997, 13, 375–376. [Google Scholar] [CrossRef]
- Madden, E.; Logue, S.E.; Healy, S.J.; Manie, S.; Samali, A. The role of the unfolded protein response in cancer progression: From oncogenesis to chemoresistance. Biol. Cell 2019, 111, 1–17. [Google Scholar] [CrossRef]
- Kyuno, D.; Takasawa, A.; Kikuchi, S.; Takemasa, I.; Osanai, M.; Kojima, T. Role of tight junctions in the epithelial-to-mesenchymal transition of cancer cells. Biochim. Biophys. Acta Biomembr. 2021, 1863, 183503. [Google Scholar] [CrossRef]
- Le Bras, G.F.; Taubenslag, K.J.; Andl, C.D. The regulation of cell-cell adhesion during epithelial-mesenchymal transition, motility and tumor progression. Cell Adhes. Migr. 2012, 6, 365–373. [Google Scholar] [CrossRef]
- Anczukow, O.; Krainer, A.R. Splicing-factor alterations in cancers. RNA 2016, 22, 1285–1301. [Google Scholar] [CrossRef] [PubMed]
- Miyagi, T.; Tatsumi, T.; Takehara, T.; Kanto, T.; Kuzushita, N.; Sugimoto, Y.; Jinushi, M.; Kasahara, A.; Sasaki, Y.; Hori, M.; et al. Impaired expression of proteasome subunits and human leukocyte antigens class I in human colon cancer cells. J. Gastroenterol. Hepatol. 2003, 18, 32–40. [Google Scholar] [CrossRef]
- Min, L.; Xu, H.; Wang, J.; Qu, L.; Jiang, B.; Zeng, Y.; Meng, L.; Jin, H.; Shou, C. N-alpha-acetyltransferase 10 protein is a negative regulator of 28S proteasome through interaction with PA28beta. FEBS Lett. 2013, 587, 1630–1637. [Google Scholar] [CrossRef]
- Link, A.J.; Eng, J.; Schieltz, D.M.; Carmack, E.; Mize, G.J.; Morris, D.R.; Garvik, B.M.; Yates, J.R., 3rd. Direct analysis of protein complexes using mass spectrometry. Nat. Biotechnol. 1999, 17, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhao, S.; He, Y.; Zheng, N.; Yan, X.; Wang, J. Pipeline for Targeted Meta-Proteomic Analyses to Assess the Diversity of Cattle Rumen Microbial Urease. Front. Microbiol. 2020, 11, 573414. [Google Scholar] [CrossRef] [PubMed]
- Sakaya, G.R.; Parada, C.A.; Eichler, R.A.; Yamaki, V.N.; Navon, A.; Heimann, A.S.; Figueiredo, E.G.; Ferro, E.S. Peptidomic profiling of cerebrospinal fluid from patients with intracranial saccular aneurysms. J. Proteom. 2021, 240, 104188. [Google Scholar] [CrossRef] [PubMed]
- Shahbazy, M.; Ramarathinam, S.H.; Illing, P.T.; Jappe, E.C.; Faridi, P.; Croft, N.P.; Purcell, A.W. Benchmarking Bioinformatics Pipelines in Data-Independent Acquisition Mass Spectrometry for Immunopeptidomics. Mol. Cell. Proteom. 2023, 22, 100515. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Bastian, M.; Heymann, S.; Jacomy, M. Gephi: An Open Source Software for Exploring and Manipulating Networks. In Proceedings of the International Aaai Conference on Web And Social Media, San Jose, CA, USA, 17–20 May 2009; pp. 361–362. [Google Scholar]
- Liao, Y.; Wang, J.; Jaehnig, E.J.; Shi, Z.; Zhang, B. WebGestalt 2019: Gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 2019, 47, W199–W205. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Kirov, S.; Snoddy, J. WebGestalt: An integrated system for exploring gene sets in various biological contexts. Nucleic Acids Res. 2005, 33, W741–W748. [Google Scholar] [CrossRef]
- Romero, P.; Obradovic, Z.; Li, X.; Garner, E.C.; Brown, C.J.; Dunker, A.K. Sequence complexity of disordered protein. Proteins 2001, 42, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Peng, K.; Radivojac, P.; Vucetic, S.; Dunker, A.K.; Obradovic, Z. Length-dependent prediction of protein intrinsic disorder. BMC Bioinform. 2006, 7, 208. [Google Scholar] [CrossRef] [PubMed]
- Peng, K.; Vucetic, S.; Radivojac, P.; Brown, C.J.; Dunker, A.K.; Obradovic, Z. Optimizing long intrinsic disorder predictors with protein evolutionary information. J. Bioinform. Comput. Biol. 2005, 3, 35–60. [Google Scholar] [CrossRef]
- Xue, B.; Dunbrack, R.L.; Williams, R.W.; Dunker, A.K.; Uversky, V.N. PONDR-FIT: A meta-predictor of intrinsically disordered amino acids. Biochim. Biophys. Acta 2010, 1804, 996–1010. [Google Scholar] [CrossRef]
- Dosztanyi, Z.; Csizmok, V.; Tompa, P.; Simon, I. IUPred: Web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics 2005, 21, 3433–3434. [Google Scholar] [CrossRef]
- Dosztanyi, Z.; Csizmok, V.; Tompa, P.; Simon, I. The pairwise energy content estimated from amino acid composition discriminates between folded and intrinsically unstructured proteins. J. Mol. Biol. 2005, 347, 827–839. [Google Scholar] [CrossRef]
- Mohan, A.; Sullivan, W.J., Jr.; Radivojac, P.; Dunker, A.K.; Uversky, V.N. Intrinsic disorder in pathogenic and non-pathogenic microbes: Discovering and analyzing the unfoldomes of early-branching eukaryotes. Mol. Biosyst. 2008, 4, 328–340. [Google Scholar] [CrossRef]
- Kyte, J.; Doolittle, R.F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982, 157, 105–132. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paromov, V.; Uversky, V.N.; Cooley, A.; Liburd, L.E., II; Mukherjee, S.; Na, I.; Dayhoff, G.W., II; Pratap, S. The Proteomic Analysis of Cancer-Related Alterations in the Human Unfoldome. Int. J. Mol. Sci. 2024, 25, 1552. https://doi.org/10.3390/ijms25031552
Paromov V, Uversky VN, Cooley A, Liburd LE II, Mukherjee S, Na I, Dayhoff GW II, Pratap S. The Proteomic Analysis of Cancer-Related Alterations in the Human Unfoldome. International Journal of Molecular Sciences. 2024; 25(3):1552. https://doi.org/10.3390/ijms25031552
Chicago/Turabian StyleParomov, Victor, Vladimir N. Uversky, Ayorinde Cooley, Lincoln E. Liburd, II, Shyamali Mukherjee, Insung Na, Guy W. Dayhoff, II, and Siddharth Pratap. 2024. "The Proteomic Analysis of Cancer-Related Alterations in the Human Unfoldome" International Journal of Molecular Sciences 25, no. 3: 1552. https://doi.org/10.3390/ijms25031552