
Bone Health Impairment in Patients with Hemoglobinopathies: From Biological Bases to New Possible Therapeutic Strategies

 , , ,
, , ,
Abstract
:1. Introduction
2. Bone Metabolism: Cells and Pathways Involved in Its Modulation
2.1. Bone Remodeling: A Finely Regulated Process
- -
- Bone matrix resorption by OCs;
- -
- Transition phase between bone matrix resorption and deposition, in which the main protagonists are macrophage-like cells;
- -
- Bone matrix deposition by OBs [42].
2.2. Bone Metabolism Impairment and Osteoporosis in Some Pathophysiological Situations, the Alteration of the Bone Itself and the Subsequent Impairment of Its Own Function Is Caused by the Dysregulation of Different Molecular Pathways, Involved in the Metabolism of Osteoblasts, Osteoclasts, and Osteocyte, Thus Leading to a Variety of Bone Metabolic Diseases
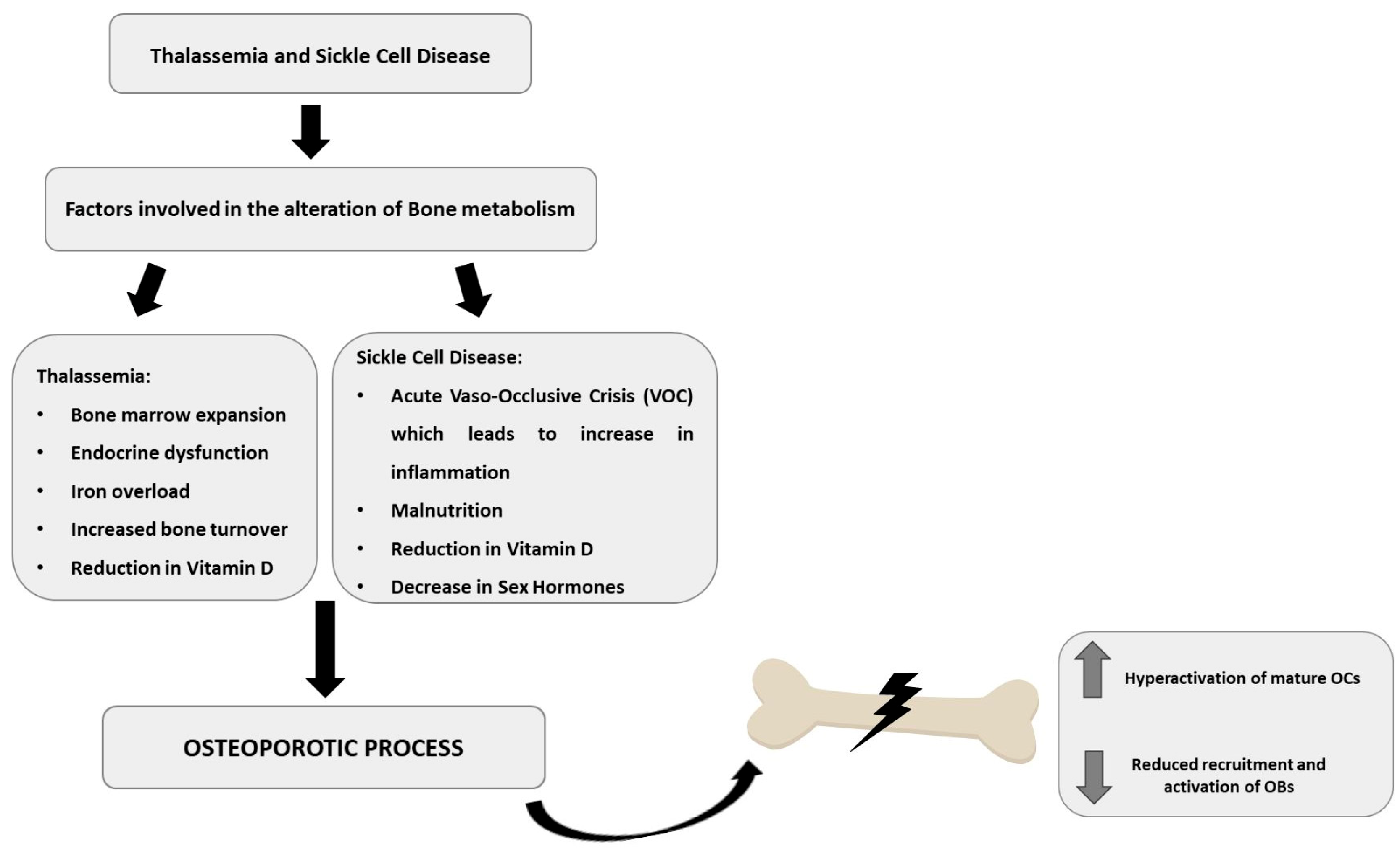
3. Bone Metabolism Alteration in Hemoglobinopathies
3.1. Bone Metabolism Alteration in Thalassemic Patients
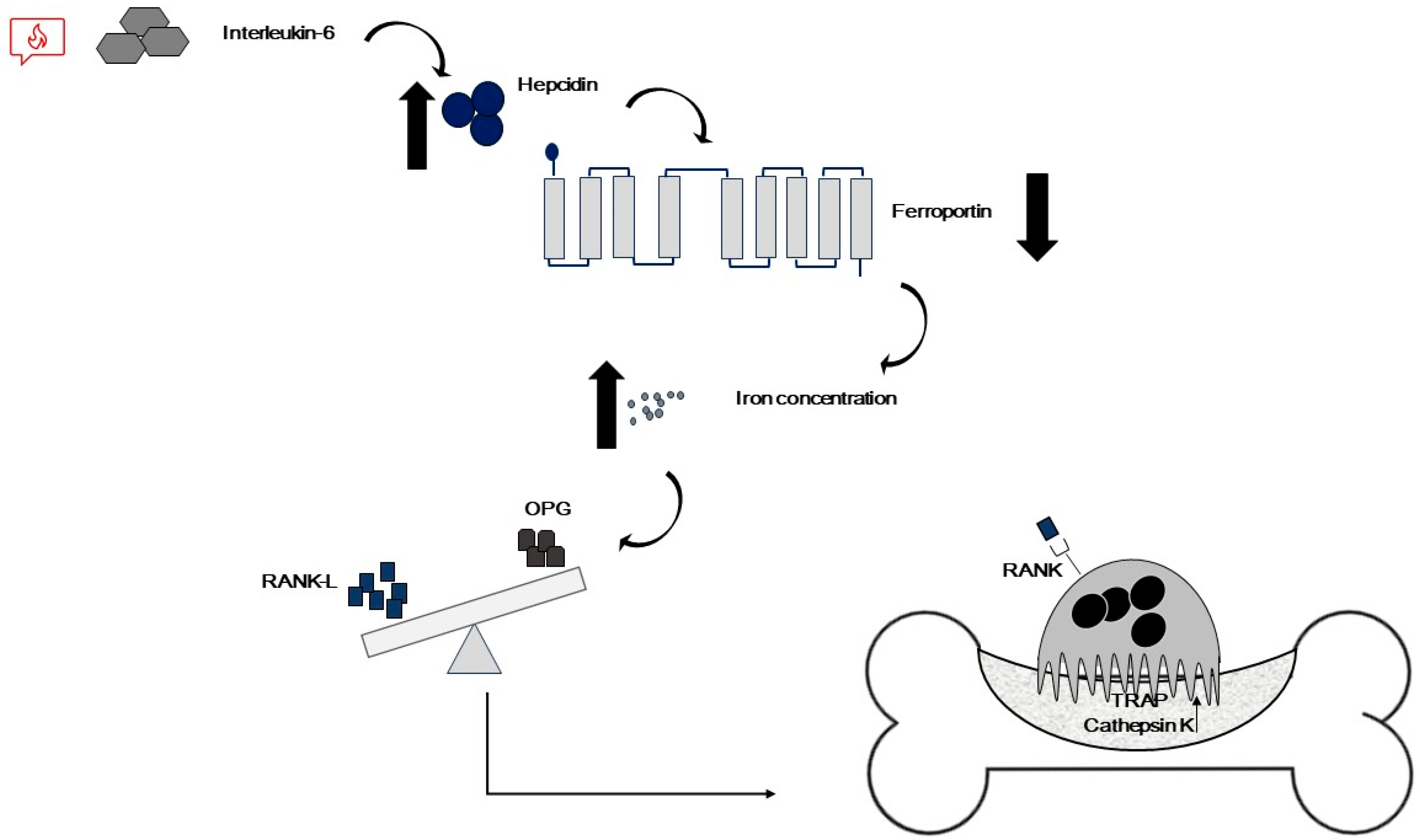
3.1.1. TM-Associated OP: Involved Cells and Pathways
3.1.2. TM-Associated OP: Genetic Factors
3.2. Bone Metabolism Alteration in Sickle Cell Patients
4. Therapeutic Intervention and Novel Evidence for the Treatment of Osteoporosis in Patients with Hemoglobinopathies
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Harteveld, C.L.; Achour, A.; Arkesteijn, S.J.G.; Ter Huurne, J.; Verschuren, M.; Bhagwandien-Bisoen, S.; Schaap, R.; Vijfhuizen, L.; El Idrissi, H.; Koopmann, T.T. The hemoglobinopathies, molecular disease mechanisms and diagnostics. Int. J. Lab. Hematol. 2022, 44 (Suppl. S1), 28–36. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, M.H.; Forget, B.G.; Higgs, D.R.; Weatherall, D.J. Disorders of Hemoglobin: Genetics, Pathophysiology, and Clinical Management, 2nd ed.; Cambridge University Press: Cambridge, UK, 2009; p. 18. [Google Scholar]
- Williams, T.N.; Weatherall, D.J. World distribution, population genetics, and health burden of the hemoglobinopathies. Cold Spring Harb. Perspect. Med. 2012, 2, a011692. [Google Scholar] [CrossRef] [PubMed]
- Kattamis, A.; Kwiatkowski, J.L.; Aydinok, Y. Thalassaemia. Lancet 2022, 399, 2310–2324. [Google Scholar] [CrossRef] [PubMed]
- Taher, A.T.; Weatherall, D.J.; Cappellini, M.D. Thalassaemia. Lancet 2018, 391, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Taher, A.; Vichinsky, E.; Musallam, K.; Cappellini, M.D.; Viprakasit, V. Guidelines for the Management of Non Transfusion Dependent Thalassaemia (NTDT); Weatherall, D., Ed.; Thalassaemia International Federation: Nicosia, Cyprus, 2013. [Google Scholar]
- Musallam, K.M.; Rivella, S.; Vichinsky, E.; Rachmilewitz, E.A. Non-transfusion-dependent thalassemias. Haematologica 2013, 98, 833–844. [Google Scholar] [CrossRef] [PubMed]
- Musallam, K.M.; Taher, A.T.; Rachmilewitz, E.A. β-Thalassemia Intermedia: A Clinical Perspective. CSH Perspect. Med. 2012, 2, a013482. [Google Scholar] [CrossRef] [PubMed]
- Farashi, S.; Harteveld, C.L. Molecular basis of alpha-thalassemia. Blood Cell Mol. Dis. 2018, 70, 43–53. [Google Scholar] [CrossRef]
- De Sanctis, V.; Soliman, A.T.; Elsedfy, H.; Yassin, M.; Canatan, D.; Kilinc, Y.; Sobti, P.; Skordis, N.; Karimi, M.; Raiola, G.; et al. Osteoporosis in thalassemia major: An update and the I-CET 2013 recommendations for surveillance and treatment. Pediatr. Endocrinol. Rev. 2013, 11, 167–180. [Google Scholar]
- Dede, A.D.; Trovas, G.; Chronopoulos, E.; Triantafyllopoulos, I.K.; Dontas, I.; Papaioannou, N.; Tournis, S. Thalassemia-associated osteoporosis: A systematic review on treatment and brief overview of the disease. Osteoporos. Int. 2016, 27, 3409–3425. [Google Scholar] [CrossRef]
- Wong, P.; Fuller, P.J.; Gillespie, M.T.; Milat, F. Bone Disease in Thalassemia: A Molecular and Clinical Overview. Endocr. Rev. 2016, 37, 320–346. [Google Scholar] [CrossRef]
- Fung, E.B.; Harmatz, P.; Milet, M.; Ballas, S.K.; De Castro, L.; Hagar, W.; Owen, W.; Olivieri, N.; Smith-Whitley, K.; Darbari, D.; et al. Morbidity and mortality in chronically transfused subjects with thalassemia and sickle cell disease: A report from the multi-center study of iron overload. Am. J. Hematol. 2007, 82, 255–265. [Google Scholar] [CrossRef]
- Goodnough, L.T.; Panigrahi, A.K. Blood Transfusion Therapy. Med. Clin. N. Am. 2017, 101, 431–447. [Google Scholar] [CrossRef]
- Punzo, F.; Tortora, C.; Argenziano, M.; Casale, M.; Perrotta, S.; Rossi, F. Iron chelating properties of Eltrombopag: Investigating its role in thalassemia-induced osteoporosis. PLoS ONE 2018, 13, e0208102. [Google Scholar] [CrossRef]
- Fibach, E.; Rachmilewitz, E.A. Iron overload in hematological disorders. Presse Med. 2017, 46, E296–E305. [Google Scholar] [CrossRef]
- Taher, A.T.; Saliba, A.N. Iron overload in thalassemia: Different organs at different rates. Hematol.-Am. Soc. Hematol. 2017, 2017, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Rossi, F.; Perrotta, S.; Bellini, G.; Luongo, L.; Tortora, C.; Siniscalco, D.; Francese, M.; Torella, M.; Nobili, B.; Di Marzo, V.; et al. Iron overload causes osteoporosis in thalassemia major patients through interaction with transient receptor potential vanilloid type 1 (TRPV1) channels. Haematologica 2014, 99, 1876–1884. [Google Scholar] [CrossRef]
- Mahachoklertwattana, P.; Sirikulchayanonta, V.; Chuansumrit, A.; Karnsombat, P.; Choubtum, L.; Sriphrapradang, A.; Domrongkitchaiporn, S.; Sirisriro, R.; Rajatanavin, R. Bone histomorphometry in children and adolescents with beta-thalassemia disease: Iron-associated focal osteomalacia. J. Clin. Endocr. Metab. 2003, 88, 3966–3972. [Google Scholar] [CrossRef] [PubMed]
- Daher, R.; Karim, Z. Iron metabolism: State of the art. Transfus. Clin. Biol. 2017, 24, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Dev, S.; Babitt, J.L. Overview of iron metabolism in health and disease. Hemodial. Int. 2017, 21 (Suppl. S1), S6–S20. [Google Scholar] [CrossRef] [PubMed]
- Maggio, A.; Filosa, A.; Vitrano, A.; Aloj, G.; Kattamis, A.; Ceci, A.; Fucharoen, S.; Cianciulli, P.; Grady, R.W.; Prossomariti, L.; et al. Iron chelation therapy in thalassemia major: A systematic review with meta-analyses of 1520 patients included on randomized clinical trials. Blood Cell Mol. Dis. 2011, 47, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Voskaridou, E.; Kyrtsonis, M.C.; Terpos, E.; Skordili, M.; Theodoropoulos, I.; Bergele, A.; Diamanti, E.; Kalovidouris, A.; Loutradi, A.; Loukopoulos, D. Bone resorption is increased in young adults with thalassaemia major. Br. J. Haematol. 2001, 112, 36–41. [Google Scholar] [CrossRef]
- Morabito, N.; Gaudio, A.; Lasco, A.; Atteritano, M.; Pizzoleo, M.A.; Cincotta, M.; La Rosa, M.; Guarino, R.; Meo, A.; Frisina, N. Osteoprotegerin and RANKL in the pathogenesis of thalassemia-induced osteoporosis: New pieces of the puzzle. J. Bone Miner. Res. 2004, 19, 722–727. [Google Scholar] [CrossRef]
- Voskaridou, E.; Terpos, E. Osteoprotegerin to soluble receptor activator of nuclear factor kappa-B ligand ratio is reduced in patients with thalassaemia-related osteoporosis who receive vitamin D-3. Eur. J. Haematol. 2005, 74, 359–361. [Google Scholar] [CrossRef]
- Rossi, F.; Bellini, G.; Luongo, L.; Torella, M.; Mancusi, S.; De Petrocellis, L.; Petrosino, S.; Siniscalco, D.; Orlando, P.; Scafuro, M.; et al. The endovanilloid/endocannabinoid system: A new potential target for osteoporosis therapy. Bone 2011, 48, 997–1007. [Google Scholar] [CrossRef]
- Aydin, H.M.; Piskin, E. Cathepsin K/TRAP: Can they be used to induce osteogenesis? Med. Hypotheses 2009, 72, 464–465. [Google Scholar] [CrossRef]
- Gagliardi, I.; Celico, M.; Gamberini, M.R.; Pontrelli, M.; Fortini, M.; Carnevale, A.; Napoli, N.; Zatelli, M.C.; Ambrosio, M.R. Efficacy and Safety of Teriparatide in Beta-Thalassemia Major Associated Osteoporosis: A Real-Life Experience. Calcif. Tissue Int. 2022, 111, 56–65. [Google Scholar] [CrossRef]
- Giusti, A. Bisphosphonates in the management of thalassemia-associated osteoporosis: A systematic review of randomised controlled trials. J. Bone Miner. Metab. 2014, 32, 606–615. [Google Scholar] [CrossRef] [PubMed]
- Rossini, M.; Adami, S.; Bertoldo, F.; Diacinti, D.; Gatti, D.; Giannini, S.; Giusti, A.; Malavolta, N.; Minisola, S.; Osella, G.; et al. Guidelines for the diagnosis, prevention and management of osteoporosis. Reumatismo 2016, 68, 1–39. [Google Scholar] [CrossRef] [PubMed]
- Terpos, E.; Voskaridou, E. Treatment options for thalassemia patients with osteoporosis. Ann. N. Y. Acad. Sci. 2010, 1202, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Bab, I.; Zimmer, A.; Melamed, E. Cannabinoids and the skeleton: From marijuana to reversal of bone loss. Ann. Med. 2009, 41, 560–567. [Google Scholar] [CrossRef] [PubMed]
- Bab, I.; Ofek, O.; Tam, J.; Rehnelt, J.; Zimmer, A. Endocannabinoids and the regulation of bone metabolism. J. Neuroendocrinol. 2008, 20, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Idris, A.I.; Hof, R.J.V.; Greig, I.R.; Ridge, S.A.; Baker, D.; Ross, R.A.; Ralston, S.H. Regulation of bone mass, bone loss and osteoclast activity by cannabinoid receptors. Nat. Med. 2005, 11, 774–779. [Google Scholar] [CrossRef] [PubMed]
- Rossi, F.; Siniscalco, D.; Luongo, L.; De Petrocellis, L.; Bellini, G.; Petrosino, S.; Torella, M.; Santoro, C.; Nobili, B.; Perrotta, S.; et al. The endovanilloid/endocannabinoid system in human osteoclasts: Possible involvement in bone formation and resorption. Bone 2009, 44, 476–484. [Google Scholar] [CrossRef] [PubMed]
- Rossi, F.; Tortora, C.; Punzo, F.; Bellini, G.; Argenziano, M.; Di Paola, A.; Torella, M.; Perrotta, S. The Endocannabinoid/Endovanilloid System in Bone: From Osteoporosis to Osteosarcoma. Int. J. Mol. Sci. 2019, 20, 1919. [Google Scholar] [CrossRef] [PubMed]
- Brandow, A.M.; Liem, R.I. Advances in the diagnosis and treatment of MAJOR cell disease. J. Hematol. Oncol. 2022, 15, 20. [Google Scholar] [CrossRef] [PubMed]
- De Franceschi, L.; Gabbiani, D.; Giusti, A.; Forni, G.; Stefanoni, F.; Pinto, V.M.; Sartori, G.; Balocco, M.; Dal Zotto, C.; Valenti, M.T.; et al. Development of Algorithm for Clinical Management of Sickle Cell Bone Disease: Evidence for a Role of Vertebral Fractures in Patient Follow-up. J. Clin. Med. 2020, 9, 1601. [Google Scholar] [CrossRef] [PubMed]
- Giordano, P.; Urbano, F.; Lassandro, G.; Faienza, M.F. Mechanisms of Bone Impairment in Sickle Bone Disease. Int. J. Environ. Res. Public. Health 2021, 18, 1832. [Google Scholar] [CrossRef] [PubMed]
- Nolan, V.G.; Nottage, K.A.; Cole, E.W.; Hankins, J.S.; Gurney, J.G. Prevalence of Vitamin D Deficiency in Sickle Cell Disease: A Systematic Review. PLoS ONE 2015, 10, e0119908. [Google Scholar] [CrossRef]
- Dermience, M.; Lognay, G.; Mathieu, F.; Goyens, P. Effects of thirty elements on bone metabolism. J. Trace Elem. Med. Biol. 2015, 32, 86–106. [Google Scholar] [CrossRef]
- Montaseri, A.; Giampietri, C.; Rossi, M.; Riccioli, A.; Del Fattore, A.; Filippini, A. The Role of Autophagy in Osteoclast Differentiation and Bone Resorption Function. Biomolecules 2020, 10, 1398. [Google Scholar] [CrossRef]
- Ciosek, Z.; Kot, K.; Kosik-Bogacka, D.; Lanocha-Arendarczyk, N.; Rotter, I. The Effects of Calcium, Magnesium, Phosphorus, Fluoride, and Lead on Bone Tissue. Biomolecules 2021, 11, 506. [Google Scholar] [CrossRef] [PubMed]
- Florencio-Silva, R.; Sasso, G.R.D.; Sasso-Cerri, E.; Simoes, M.J.; Cerri, P.S. Biology of Bone Tissue: Structure, Function, and Factors That Influence Bone Cells. Biomed. Res. Int. 2015, 2015, 421746. [Google Scholar] [CrossRef] [PubMed]
- Capulli, M.; Paone, R.; Rucci, N. Osteoblast and osteocyte: Games without frontiers. Arch. Biochem. Biophys. 2014, 561, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Ponzetti, M.; Rucci, N. Osteoblast Differentiation and Signaling: Established Concepts and Emerging Topics. Int. J. Mol. Sci. 2021, 22, 6651. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, K.; Zhou, X.; Kunkel, G.; Zhang, Z.P.; Deng, J.M.; Behringer, R.R.; de Crombrugghe, B. The novel zinc finger-containing transcription factor Osterix is required for osteoblast differentiation and bone formation. Cell 2002, 108, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Franz-Odendaal, T.A.; Hall, B.K.; Witten, P.E. Buried alive: How osteoblasts become osteocytes. Dev. Dyn. 2006, 235, 176–190. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.R.; Shibata, Y.; Zhu, T.M.; Zhou, J.; Zhang, J.M. Osteocytes in bone aging: Advances, challenges, and future perspectives. Ageing Res. Rev. 2022, 77, 101608. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Calle, J.; Bellido, T. The Osteocyte as a Signaling Cell. Physiol. Rev. 2022, 102, 379–410. [Google Scholar] [CrossRef]
- Karthik, V.; Guntur, A.R. Energy Metabolism of Osteocytes. Curr. Osteoporos. Rep. 2021, 19, 444–451. [Google Scholar] [CrossRef]
- Goldring, S.R. The osteocyte: Key player in regulating bone turnover. Rmd Open 2015, 1, e000049. [Google Scholar] [CrossRef]
- Yao, Z.; Getting, S.J.; Locke, I.C. Regulation of TNF-Induced Osteoclast Differentiation. Cells 2021, 11, 132. [Google Scholar] [CrossRef] [PubMed]
- Lozano, C.; Duroux-Richard, I.; Firat, H.; Schordan, E.; Apparailly, F. MicroRNAs: Key Regulators to Understand Osteoclast Differentiation? Front. Immunol. 2019, 10, 375. [Google Scholar] [CrossRef] [PubMed]
- Crockett, J.C.; Mellis, D.J.; Scott, D.I.; Helfrich, M.H. New knowledge on critical osteoclast formation and activation pathways from study of rare genetic diseases of osteoclasts: Focus on the RANK/RANKL axis. Osteoporos. Int. 2011, 22, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.M.; Lin, C.; Stavre, Z.; Greenblatt, M.B.; Shim, J.H. Osteoblast-Osteoclast Communication and Bone Homeostasis. Cells 2020, 9, 2073. [Google Scholar] [CrossRef]
- Muruganandan, S.; Ionescu, A.M.; Sinal, C.J. At the Crossroads of the Adipocyte and Osteoclast Differentiation Programs: Future Therapeutic Perspectives. Int. J. Mol. Sci. 2020, 21, 2277. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, J.A.; Partridge, N.C. Physiological Bone Remodeling: Systemic Regulation and Growth Factor Involvement. Physiology 2016, 31, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Wang, H.; Li, Y.C.; Song, L. Lipid metabolism within the bone micro-environment is closely associated with bone metabolism in physiological and pathophysiological stages. Lipids Health Dis. 2022, 21, 5. [Google Scholar] [CrossRef] [PubMed]
- Tu, K.N.; Lie, J.D.; Wan, C.K.V.; Cameron, M.; Austel, A.G.; Nguyen, J.K.; Van, K.; Hyun, D. Osteoporosis: A Review of Treatment Options. Pharm. Ther. 2018, 43, 92–104. [Google Scholar]
- Cooper, C.; Campion, G.; Melton, L.J. 3rd. Hip fractures in the elderly: A world-wide projection. Osteoporos. Int. 1992, 2, 285–289. [Google Scholar] [CrossRef]
- Reginster, J.Y.; Burlet, N. Osteoporosis: A still increasing prevalence. Bone 2006, 38 (Suppl. S1), 4–9. [Google Scholar] [CrossRef]
- Hudec, S.M.; Camacho, P.M. Secondary causes of osteoporosis. Endocr. Pract. 2013, 19, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Lespessailles, E.; Chapurlat, R. High fracture risk patients with glucocorticoid-induced osteoporosis should get an anabolic treatment first. Osteoporos. Int. 2020, 31, 1829–1834. [Google Scholar] [CrossRef] [PubMed]
- Marcucci, G.; Brandi, M.L. Rare causes of osteoporosis. Clin. Cases Miner. Bone Metab. 2015, 12, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Aspray, T.J.; Hill, T.R. Osteoporosis and the Ageing Skeleton. Subcell. Biochem. 2019, 91, 453–476. [Google Scholar]
- Bijelic, R.; Milicevic, S.; Balaban, J. Risk Factors for Osteoporosis in Postmenopausal Women. Med. Arch. 2017, 71, 25–28. [Google Scholar] [CrossRef]
- Matzkin, E.G.; DeMaio, M.; Charles, J.F.; Franklin, C.C. Diagnosis and Treatment of Osteoporosis: What Orthopaedic Surgeons Need to Know. J. Am. Acad. Orthop. Surg. 2019, 27, E902–E912. [Google Scholar] [CrossRef] [PubMed]
- Haas, A.V.; LeBoff, M.S. Osteoanabolic Agents for Osteoporosis. J. Endocr. Soc. 2018, 2, 922–932. [Google Scholar] [CrossRef]
- Chen, J.S.; Sambrook, P.N. Antiresorptive therapies for osteoporosis: A clinical overview. Nat. Rev. Endocrinol. 2012, 8, 81–91. [Google Scholar] [CrossRef]
- Rossi, F.; Tortora, C.; Paoletta, M.; Marrapodi, M.M.; Argenziano, M.; Di Paola, A.; Pota, E.; Di Pinto, D.; Di Martino, M.; Iolascon, G. Osteoporosis in Childhood Cancer Survivors: Physiopathology, Prevention, Therapy and Future Perspectives. Cancers 2022, 14, 4349. [Google Scholar] [CrossRef]
- Liu, P.; Wang, W.Z.; Li, Z.; Li, Y.; Yu, X.P.; Tu, J.; Zhang, Z.D. Ferroptosis: A New Regulatory Mechanism in Osteoporosis. Oxidative Med. Cell. Longev. 2022, 2022, 2634431. [Google Scholar] [CrossRef]
- Chauhan, W.; Shoaib, S.; Fatma, R.; Zaka-Ur-Rab, Z.; Afzal, M. Beta-thalassemia and the advent of new interventions beyond transfusion and iron chelation. Br. J. Clin. Pharmacol. 2022, 88, 3610–3626. [Google Scholar] [CrossRef] [PubMed]
- Di Paola, A.; Tortora, C.; Argenziano, M.; Marrapodi, M.M.; Rossi, F. Emerging Roles of the Iron Chelators in Inflammation. Int. J. Mol. Sci. 2022, 23, 7977. [Google Scholar] [CrossRef]
- Ginzburg, Y.Z. Hepcidin-ferroportin axis in health and disease. Vitam. Horm. 2019, 110, 17–45. [Google Scholar] [PubMed]
- Kim, S.Y.; Yoo, D.M.; Min, C.Y.; Choi, H.G. Association between Osteoporosis and Low Hemoglobin Levels: A Nested Case-Control Study Using a National Health Screening Cohort. Int. J. Environ. Res. Public Health 2021, 18, 8598. [Google Scholar] [CrossRef] [PubMed]
- Evangelidis, P.; Venou, T.M.; Fani, B.; Vlachaki, E.; Gavriilaki, E.; Res, I.H. Endocrinopathies in Hemoglobinopathies: What Is the Role of Iron? Int. J. Mol. Sci. 2023, 24, 16263. [Google Scholar] [CrossRef] [PubMed]
- Morabito, N.; Russo, G.T.; Gaudio, A.; Lasco, A.; Catalano, A.; Morini, E.; Franchina, F.; Maisano, D.; La Rosa, M.; Plota, M.; et al. The “lively” cytokines network in β-Thalassernia Major-related osteoporosis. Bone 2007, 40, 1588–1594. [Google Scholar] [CrossRef] [PubMed]
- Bolarin, D.M.; Azinge, E.C. Osteocalcin and specific markers of bone resorption in sickle cell disease. Acta Physiol. Hung. 2010, 97, 290–296. [Google Scholar] [CrossRef]
- Abd El-Moneim, E.S.; Zolaly, M.A.; Al-Hawsawi, Z.M.; Abdelmoneim, A.A.; Abosdera, M.M. Age-related changes in biochemical bone profile in thalassemic children. Pediatr. Neonatol. 2018, 59, 189–197. [Google Scholar] [CrossRef]
- Voskaridou, E.; Papassotiriou, L.; Premetis, E.; Meletis, J.; Loukopoulos, D.; Terpos, E. Osteoporosis in sickle cell/β-thalassemia is primarily caused by an imbalance at the RANKL/OPG pathway. Blood 2005, 106, 3173. [Google Scholar] [CrossRef]
- Adewoye, A.H.; Chen, T.C.; Ma, Q.; McMahon, L.; Mathieu, J.; Malabanan, A.; Steinberg, M.H.; Holick, M.F. Sickle cell bone disease: Response to vitamin D and calcium. Am. J. Hematol. 2008, 83, 271–274. [Google Scholar] [CrossRef]
- Nouraie, M.; Cheng, K.; Niu, X.M.; Moore-King, E.; Fadojutimi-Akinsi, M.F.; Minniti, C.P.; Sable, C.; Rana, S.; Dham, N.; Campbell, A.; et al. Predictors of osteoclast activity in patients with sickle cell disease. Haematologica 2011, 96, 1092–1098. [Google Scholar] [CrossRef]
- Salama, O.S.; Al-Tonbary, Y.A.; Shahin, R.A.; Eldeen, O.A.S. Unbalanced bone turnover in children with β-thalassemia. Hematology 2006, 11, 197–202. [Google Scholar] [CrossRef]
- Fung, E.B.; Kawchak, D.A.; Zemel, B.S.; Rovner, A.J.; Ohene-Frempong, K.; Stallings, V.A. Markers of bone turnover are associated with growth and development in young subjects with sickle cell anemia. Pediatr. Blood Cancer 2008, 50, 620–623. [Google Scholar] [CrossRef]
- Angelopoulos, N.G.; Goula, A.; Katounda, E.; Rombopoulos, G.; Kaltzidou, V.; Kaltsas, D.; Malaktari, S.; Athanasiou, V.; Tolis, G. Circulating osteoprotegerin and receptor activator of NF-kappaB ligand system in patients with beta-thalassemia major. J. Bone Miner. Metab. 2007, 25, 60–67. [Google Scholar] [CrossRef]
- Çelik, T.; Sangün, Ö.; Ünal, S.; Balci, A.; Motor, S. Assessment of biochemical bone markers of osteoporosis in children with thalassemia major. Ital. J. Pediatr. 2022, 48, 105. [Google Scholar] [CrossRef]
- Toumba, M.; Skordis, N. Osteoporosis syndrome in thalassaemia major: An overview. J. Osteoporos. 2010, 2010, 537673. [Google Scholar] [CrossRef] [PubMed]
- Baldini, M.; Forti, S.; Orsatti, A.; Marcon, A.; Ulivieri, F.M.; Airaghi, L.; Zanaboni, L.; Cappellini, M.D. Thalassemic osteopathy: A new marker of bone deposition. Blood Cell Mol. Dis. 2014, 52, 91–94. [Google Scholar] [CrossRef] [PubMed]
- Pirinççioğlu, A.G.; Söker, D.G. Parathyroid Functions in Thalassemia Major Patients. Ann. Clin. Endocrinol. Metab. 2017, 1, 15–19. [Google Scholar] [CrossRef]
- Goyal, M.; Abrol, P.; Lal, H. Parathyroid and calcium status in patients with thalassemia. Indian J. Clin. Biochem. IJCB 2010, 25, 385–387. [Google Scholar] [CrossRef] [PubMed]
- Elshal, M.F.; Bernawi, A.E.; Al-Ghamdy, M.A.; Jalal, J.A. The association of bone mineral density and parathyroid hormone with serum magnesium in adult patients with sickle-cell anaemia. Arch. Med. Sci. 2012, 8, 270–276. [Google Scholar] [CrossRef]
- Manolopoulos, P.P.; Lavranos, G.; Mamais, I.; Angouridis, A.; Giannakou, K.; Johnson, E.O. Vitamin D and bone health status in beta thalassemia patients-systematic review. Osteoporos. Int. 2021, 32, 1031–1040. [Google Scholar] [CrossRef]
- Soe, H.H.; Abas, A.B.; Than, N.N.; Ni, H.; Singh, J.; Said, A.R.; Osunkwo, I. Vitamin D supplementation for sickle cell disease. Cochrane Database Syst. Rev. 2017, 1, CD010858. [Google Scholar] [CrossRef]
- Lasco, A.; Morabito, N.; Gaudio, A.; Crisafulli, A.; Meo, A.; Denuzzo, G.; Frisina, N. Osteoporosis and β-thalassemia major: Role of the IGF-I/IGFBP-III axis. J. Endocrinol. Investig. 2002, 25, 338–344. [Google Scholar] [CrossRef]
- Luporini, S.M.; Bendit, I.; Manhani, R.; Bracco, O.L.; Manzella, L.; Giannella-Neto, D. Growth hormone and insulin-like growth factor I axis and growth of children with different sickle cell anemia haplotypes. J. Pediatr. Hematol. Oncol. 2001, 23, 357–363. [Google Scholar] [CrossRef]
- Anapliotou, M.L.; Kastanias, I.T.; Psara, P.; Evangelou, E.A.; Liparaki, M.; Dimitriou, P. The contribution of hypogonadism to the development of osteoporosis in thalassaemia major: New therapeutic approaches. Clin. Endocrinol. 1995, 42, 279–287. [Google Scholar] [CrossRef]
- Gaudio, A.; Morabito, N.; Catalano, A.; Rapisarda, R.; Xourafa, A.; Lasco, A. Pathogenesis of Thalassemia Major-associated Osteoporosis: A Review with Insights from Clinical Experience. J. Clin. Res. Pediatr. Endocrinol. 2019, 11, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Thavonlun, S.; Houngngam, N.; Kingpetch, K.; Numkarunarunrote, N.; Santisitthanon, P.; Buranasupkajorn, P.; Pongchaiyakul, C.; Sutcharitchan, P.; Wattanachanya, L. Association of osteoporosis and sarcopenia with fracture risk in transfusion-dependent thalassemia. Sci. Rep. 2023, 13, 16413. [Google Scholar] [CrossRef] [PubMed]
- Teawtrakul, N.; Chansai, S.; Yamsri, S.; Chansung, K.; Wanitpongpun, C.; Lanamtieng, T.; Phiphitaporn, P.; Fucharoen, S.; Pongchaiyakul, C. The association of growth differentiation factor-15 levels and osteoporosis in patients with thalassemia. Am. J. Med. Sci. 2023, 366, 96–101. [Google Scholar] [CrossRef] [PubMed]
- Baldan, A.; Giusti, A.; Bosi, C.; Malaventura, C.; Musso, M.; Forni, G.L.; Volpato, S.; Zuliani, G.; Borgna-Pignatti, C. Klotho, a new marker for osteoporosis and muscle strength in β-thalassemia major. Blood Cell Mol. Dis. 2015, 55, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Voskaridou, E.; Christoulas, D.; Plata, E.; Bratengeier, C.; Anastasilakis, A.D.; Komninaka, V.; Kaliontzi, D.; Gkotzamanidou, M.; Polyzos, S.A.; Dimopoulou, M.; et al. High Circulating Sclerostin is Present in Patients with Thalassemia-associated Osteoporosis and Correlates with Bone Mineral Density. Horm. Metab. Res. 2012, 44, 909–913. [Google Scholar] [CrossRef] [PubMed]
- Piriyakhuntorn, P.; Tantiworawit, A.; Niprapan, P.; Thonusin, C.; Kaewchur, T.; Chattipakorn, N.; Chattipakorn, S. Alterations of Plasma Metabolomics Profile in Thalassemia Patients with Low Bone Mineral Density. Blood 2023, 142 (Suppl. S1), 3852. [Google Scholar] [CrossRef]
- Borgna-Pignatti, C.; Cappellini, M.D.; De Stefano, P.; Del Vecchio, G.C.; Forni, G.L.; Gamberini, M.R.; Ghilardi, R.; Origa, R.; Piga, A.; Romeo, M.A.; et al. Survival and complications in thalassemia. Ann. N. Y. Acad. Sci. 2005, 1054, 40–47. [Google Scholar] [CrossRef] [PubMed]
- De Sanctis, V.; Soliman, A.T.; Elsefdy, H.; Soliman, N.; Bedair, E.; Fiscina, B.; Kattamis, C. Bone disease in beta thalassemia patients: Past, present and future perspectives. Metabolism 2018, 80, 66–79. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, A.; Swe, K.M.M.; Sinha, N.K.; Osunkwo, I. Treatment for osteoporosis in people with beta-thalassaemia. Cochrane Database Syst. Rev. 2016, CD010429. [Google Scholar] [CrossRef]
- Pellegrino, F.; Zatelli, M.C.; Bondanelli, M.; Carnevale, A.; Cittanti, C.; Fortini, M.; Gamberini, M.R.; Giganti, M.; Ambrosio, M.R. Dual-energy X-ray absorptiometry pitfalls in Thalassemia Major. Endocrine 2019, 65, 469–482. [Google Scholar] [CrossRef] [PubMed]
- Gaudio, A.; Xourafa, A.; Rapisarda, R.; Zanoli, L.; Signorelli, S.S.; Castellino, P. Hematological Diseases and Osteoporosis. Int. J. Mol. Sci. 2020, 21, 3538. [Google Scholar] [CrossRef]
- Napoli, N.; Carmina, E.; Bucchieri, S.; Sferrazza, C.; Rini, G.B.; Di Fede, G. Low serum levels of 25-hydroxy vitamin D in adults affected by thalassemia major or intermedia. Bone 2006, 38, 888–892. [Google Scholar] [CrossRef]
- Carmina, E.; Di Fede, G.; Napoli, N.; Renda, G.; Vitale, G.; Lo Pinto, C.; Bruno, D.; Malizia, R.; Rini, G.B. Hypogonadism and hormone replacement therapy on bone mass of adult women with thalassemia major. Calcif. Tissue Int. 2004, 74, 68–71. [Google Scholar] [CrossRef]
- Casale, M.; Forni, G.L.; Cassinerio, E.; Pasquali, D.; Origa, R.; Serra, M.; Campisi, S.; Peluso, A.; Renni, R.; Cattoni, A.; et al. Risk factors for endocrine complications in transfusion-dependent thalassemia patients on chelation therapy with deferasirox: A risk assessment study from a multi-center nation-wide cohort. Haematologica 2022, 107, 467–477. [Google Scholar] [CrossRef]
- Chatterjee, R.; Shah, F.T.; Davis, B.A.; Byers, M.; Sooranna, D.; Bajoria, R.; Pringle, J.; Porter, J.B. Prospective study of histomorphometry, biochemical bone markers and bone densitometric response to pamidronate in beta-thalassaemia presenting with osteopenia-osteoporosis syndrome. Br. J. Haematol. 2012, 159, 462–471. [Google Scholar] [CrossRef]
- Jeney, V. Clinical Impact and Cellular Mechanisms of Iron Overload-Associated Bone Loss. Front. Pharmacol. 2017, 8, 77. [Google Scholar] [CrossRef] [PubMed]
- Casale, M.; Citarella, S.; Filosa, A.; De Michele, E.; Palmieri, F.; Ragozzino, A.; Amendola, G.; Pugliese, U.; Tartaglione, I.; Della Rocca, F.; et al. Endocrine function and bone disease during long-term chelation therapy with deferasirox in patients with beta-thalassemia major. Am. J. Hematol. 2014, 89, 1102–1106. [Google Scholar] [CrossRef] [PubMed]
- Khandros, E.; Kwiatkowski, J.L. Beta Thalassemia Monitoring and New Treatment Approaches. Hematol. Oncol. Clin. 2019, 33, 339–353. [Google Scholar] [CrossRef] [PubMed]
- Naderi, M.; Sadeghi-Bojd, S.; Valeshabad, A.K.; Jahantigh, A.; Alizadeh, S.; Dorgalaleh, A.; Tabibian, S.; Bamedi, T. A prospective study of tubular dysfunction in pediatric patients with Beta thalassemia major receiving deferasirox. Pediatr. Hematol. Oncol. 2013, 30, 748–754. [Google Scholar] [CrossRef]
- Quinn, C.T.; Johnson, V.L.; Kim, H.Y.; Trachtenberg, F.; Vogiatzi, M.G.; Kwiatkowski, J.L.; Neufeld, E.J.; Fung, E.; Oliveri, N.; Kirby, M.; et al. Renal dysfunction in patients with thalassaemia. Br. J. Haematol. 2011, 153, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Wong, P.; Polkinghorne, K.; Kerr, P.G.; Doery, J.C.G.; Gillespie, M.T.; Larmour, I.; Fuller, P.J.; Bowden, D.K.; Milat, F. Deferasirox at therapeutic doses is associated with dose-dependent hypercalciuria. Bone 2016, 85, 55–58. [Google Scholar] [CrossRef]
- Chan, Y.L.; Pang, L.M.; Chik, K.W.; Cheng, J.C.Y.; Li, C.K. Patterns of bone diseases in transfusion-dependent homozygous thalassaemia major: Predominance of osteoporosis and desferrioxamine-induced bone dysplasia. Pediatr. Radiol. 2002, 32, 492–497. [Google Scholar] [CrossRef]
- Olivieri, N.F.; Koren, G.; Harris, J.; Khattak, S.; Freedman, M.H.; Templeton, D.M.; Bailey, J.D.; Reilly, B.J. Growth Failure and Bony Changes Induced by Deferoxamine. Am. J. Pediat Hematol. 1992, 14, 48–56. [Google Scholar] [CrossRef]
- Wonke, B.; Jensen, C.; Hanslip, J.J.; Prescott, E.; Lalloz, M.; Layton, M.; Erten, S.; Tuck, S.; Agnew, J.E.; Raja, K.; et al. Genetic and acquired predisposing factors and treatment of osteoporosis in thalassaemia major. J. Pediatr. Endocrinol. Metab. 1998, 11, 795–801. [Google Scholar]
- Ferrara, M.; Matarese, S.M.R.; Francese, M.; Borrelli, B.; Coppola, A.; Coppola, L.; Esposito, L. Effect of VDR polymorphisms on growth and bone mineral density in homozygous beta thalassaemia. Br. J. Haematol. 2002, 117, 436–440. [Google Scholar] [CrossRef]
- Langdahl, B.L.; Carstens, M.; Stenkjaer, L.; Eriksen, E.F. Polymorphisms in the transforming growth factor beta 1 gene and osteoporosis. Bone 2003, 32, 297–310. [Google Scholar] [CrossRef]
- Cong, Y.; Ru, J.Y.; Bao, N.R.; Guo, T.; Zhao, J.N. A single nucleotide polymorphism in the TGF-beta 1 gene (rs1982073 C > T) may contribute to increased risks of bone fracture, osteoporosis, and osteoarthritis: A meta-analysis. Clin. Rheumatol. 2016, 35, 973–985. [Google Scholar] [CrossRef] [PubMed]
- Ragab, S.M.; Badr, E.A.; Ibrahim, A.S. Evaluation of Glutathione-S-Transferase P1 Polymorphism and its Relation to Bone Mineral Density in Egyptian Children and Adolescents with Beta-Thalassemia Major. Mediterr. J. Hematol. Infect. Dis. 2016, 8, e2016004. [Google Scholar] [CrossRef] [PubMed]
- Howard, J.; Davies, S.C. Sickle cell disease in North Europe. Scand. J. Clin. Lab. Investig. 2007, 67, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Eskiocak, O.; Yilmaz, M.O.; Ilhan, G. Metabolic bone diseases in sickle cell anemia patients and evaluation of associated factors. Am. J. Med. Sci. 2022, 363, 490–494. [Google Scholar] [CrossRef] [PubMed]
- Kosaraju, V.; Harwani, A.; Partovi, S.; Bhojwani, N.; Garg, V.; Ayyappan, S.; Kosmas, C.; Robbin, M. Imaging of musculoskeletal manifestations in sickle cell disease patients. Br. J. Radiol. 2017, 90, 20160130. [Google Scholar] [CrossRef] [PubMed]
- Ejindu, V.C.; Hine, A.L.; Mashayekhi, M.; Shorvon, P.J.; Misra, R.R. Musculoskeletal manifestations of sickle cell disease. Radiographics 2007, 27, 1005–1021. [Google Scholar] [CrossRef]
- Lee, C.N.Y.; Lam, S.C.; Tsang, A.Y.K.; Ng, B.T.Y.; Leung, J.C.Y.; Chong, A.C.Y. Preliminary investigation on prevalence of osteoporosis and osteopenia: Should we tune our focus on healthy adults? Jpn. J. Nurs. Sci. 2015, 12, 232–248. [Google Scholar] [CrossRef]
- Conti, V.; Russomanno, G.; Corbi, G.; Toro, G.; Simeon, V.; Filippelli, W.; Ferrara, N.; Grimaldi, M.; D’Argenio, V.; Maffulli, N.; et al. A Polymorphism at the Translation Start Site of the Vitamin D Receptor Gene Is Associated with the Response to Anti-Osteoporotic Therapy in Postmenopausal Women from Southern Italy. Int. J. Mol. Sci. 2015, 16, 5452–5466. [Google Scholar] [CrossRef]
- Aicale, R.; Tarantino, D.; Maccauro, G.; Peretti, G.M.; Maffulli, N. Genetics in Orthopaedic Practice. J. Biol. Regul. Homeost. Agents 2019, 33, 103–117. [Google Scholar]
- Barden, E.M.; Kawchak, D.A.; Ohene-Frempong, K.; Stallings, V.A.; Zemel, B.S. Body composition in children with sickle cell disease. Am. J. Clin. Nutr. 2002, 76, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Buison, A.M.; Kawchak, D.A.; Schall, J.I.; Ohene-Frempong, K.; Stallings, V.A.; Leonard, M.B.; Zemel, B.S. Bone area and bone mineral content deficits in children with sickle cell disease. Pediatrics 2005, 116, 943–949. [Google Scholar] [CrossRef] [PubMed]
- Chawla, A.; Sprinz, P.G.; Welch, J.; Heeney, M.; Usmani, N.; Pashankar, F.; Kavanagh, P. Weight Status of Children with Sickle Cell Disease. Pediatrics 2013, 131, E1168–E1173. [Google Scholar] [CrossRef] [PubMed]
- Sarrai, M.; Duroseau, H.; D’Augustine, J.; Moktan, S.; Bellevue, R. Bone mass density in adults with sickle cell disease. Br. J. Haematol. 2007, 136, 666–672. [Google Scholar] [CrossRef]
- Miller, R.G.; Segal, J.B.; Ashar, B.H.; Leung, S.; Ahmed, S.; Siddique, S.; Rice, T.; Lanzkron, S. High prevalence and correlates of low bone mineral density in young adults with sickle cell disease. Am. J. Hematol. 2006, 81, 236–241. [Google Scholar] [CrossRef]
- Sadat-Ali, M.; Al Elq, A.H. Sickle cell anaemia: Is it a cause for secondary osteoporosis? West. Afr. J. Med. 2007, 26, 134–137. [Google Scholar]
- Arlet, J.B.; Courbebaisse, M.; Chatellier, G.; Eladari, D.; Souberbielle, J.C.; Friedlander, G.; de Montalembert, M.; Prié, D.; Pouchot, J.; Ribeil, J.A. Relationship between vitamin D deficiency and bone fragility in sickle cell disease: A cohort study of 56 adults. Bone 2013, 52, 206–211. [Google Scholar] [CrossRef]
- Moreau, R.; Malu, D.T.; Dumais, M.; Dalko, E.; Gaudreault, V.; Roméro, H.; Martineau, C.; Kevorkova, O.; Dardon, J.S.; Dodd, E.L.; et al. Alterations in Bone and Erythropoiesis in Hemolytic Anemia: Comparative Study in Bled, Phenylhydrazine-Treated and -Infected Mice. PLoS ONE 2012, 7, e46101. [Google Scholar] [CrossRef]
- Giusti, A.; Pinto, V.; Forni, G.L.; Pilotto, A. Management of beta-thalassemia-associated osteoporosis. Ann. N. Y. Acad. Sci. 2016, 1368, 73–81. [Google Scholar] [CrossRef]
- Yavropoulou, M.P.; Anastasilakis, A.D.; Tzoulis, P.; Tournis, S.; Rigatou, E.; Kassi, E.; Kattamis, A.; Makras, P. Approach to the management of beta thalassemia major associated osteoporosis—A long-standing relationship revisited. Acta Biomed. 2022, 93, e2022305. [Google Scholar]
- Ruggiero, S.L.; Dodson, T.B.; Fantasia, J.; Goodday, R.; Aghaloo, T.; Mehrotra, B.; O’Ryan, F. American Association of Oral and Maxillofacial Surgeons position paper on medication-related osteonecrosis of the jaw—2014 update. J. Oral. Maxillofac. Surg. 2014, 72, 1938–1956. [Google Scholar] [CrossRef]
- He, L.H.; Liu, M.; He, Y.; Xiao, E.; Zhao, L.; Zhang, T.; Yang, H.Q.; Zhang, Y. TRPV1 deletion impaired fracture healing and inhibited osteoclast and osteoblast differentiation. Sci. Rep. 2017, 7, srep42385. [Google Scholar] [CrossRef] [PubMed]
- Ofek, O.; Karsak, M.; Leclerc, N.; Fogel, M.; Frenkel, B.; Wright, K.; Tam, J.; Attar-Namdar, M.; Kram, V.; Shohami, E.; et al. Peripheral cannabinoid receptor, CB2, regulates bone mass. Proc. Natl. Acad. Sci. USA 2006, 103, 696–701. [Google Scholar] [CrossRef] [PubMed]
- Kanaya, K.; Iba, K.; Dohke, T.; Okazaki, S.; Yamashita, T. TRPV1, ASICs and P2X2/3 expressed in bone cells simultaneously regulate bone metabolic markers in ovariectomized mice. J. Musculoskelet. Neuronal Interact. 2016, 16, 145–151. [Google Scholar] [PubMed]
- Idris, A.I.; Ralston, S.H. Role of cannabinoids in the regulation of bone remodeling. Front. Endocrinol. 2012, 3, 136. [Google Scholar] [CrossRef]
- Idris, A.I. The promise and dilemma of cannabinoid therapy: Lessons from animal studies of bone disease. Bonekey Rep. 2012, 1, 224. [Google Scholar] [CrossRef]
- Idris, A.I.; Ralston, S.H. Cannabinoids and Bone: Friend or Foe? Calcif. Tissue Int. 2010, 87, 285–297. [Google Scholar] [CrossRef]
- Idris, A.I.; Sophocleous, A.; Landao-Bassonga, E.; van’t Hof, R.J.; Ralston, S.H. Regulation of Bone Mass, Osteoclast Function, and Ovariectomy-Induced Bone Loss by the Type 2 Cannabinoid Receptor. Endocrinology 2008, 149, 5619–5626. [Google Scholar] [CrossRef]
- Grimbly, C.; Escagedo, P.D.; Jaremko, J.L.; Bruce, A.; Alos, N.; Robinson, M.E.; Konji, V.N.; Page, M.; Scharke, M.; Simpson, E.; et al. Sickle cell bone disease and response to intravenous bisphosphonates in children. Osteoporos. Int. 2022, 33, 2397–2408. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Marker | Change | Disease | Values | References |
|---|---|---|---|---|
| Urinary Deoxypyridinoline (D-PYR) | Increase | TM | (pmol/μmol ur.creat.) aH: 14.8 ± 5.6 aP: 27.8 ± 8.7 | Morabito, 2007 [78] |
| SCD | (nM/mM) aH: 5.4 ± 2.7 aP: 11.1 ± 4.4 | Bolarin, 2010 [79] | ||
| Osteocalcin | Decrease | TM | (pmol/mL) aH: 5.6 ± 1.9 aP: 2.4 ± 1.1 (ng/mL) aH: 5.6 ± 1.9 aP: 2.4 ± 1.1 cH: 13.69 ± 0.92 cP: 5.68 ± 3.22 | Morabito, 2007 [78] Morabito, 2004 [24] Abd El-Moneim, 2018 [80] |
| SCD | (ng/mL) aH: 12.57 (2.48–30.19) aP: 10.93 (1.04–43.24) (ng/mul) aP: 12.3 ± 3.7 | Voskaridou, 2005 [81] Adewoye, 2008 [82] | ||
| Tartrate-resistant acid phosphatase 5b | Increase | TM | (U/L) aH: 0.91 ± 0.08 aP: 1.01 ± 0.32 | Abd El-Moneim, 2018 [80] |
| SCD | aH: 2.4 (2.0–2.9) aP: 4.4 (3.3–5.7) | Nouraie, 2011 [83] | ||
| Alkaline phosphatase | Increase | TM | (µg/L) a,cH: 171.3 ± 41.7 a,cP: 194.1 ± 37.1 | Salama, 2006 [84] |
| SCD | (U/L) aH: 20.99 (9.45–27.51) aP: 32.83 (17.25–61.43) aH: 41.33 ± 12.4 aP: 85.60 ± 33.3 | Voskaridou, 2005 [81] Fung, 2008 [85] | ||
| sRANKL/OPG | Increase | TM | aH: 1.25 ± 0.2 aP: 2.50 ± 0.2 a,cH: 0.9 a,cP: 1.38 cH: 33.24 cP: 46.09 | Morabito, 2004 [24] Angelopulos, 2007 [86] Celik, 2022 [87] |
| sRANK | Increase | TM | (pmol/L) aH: 4.5 ± 1.2 aP: 8.1 ± 2.8 | Morabito, 2004 [24] Morabito, 2007 [78] |
| OPG | Decrease | TM | (pmol/L) cH: 2.43 ± 0.57 cP: 2.01 ± 0.66 | Celik, 2022 [87] |
| Osteoclastic Markers | Increase | TM | a,cNA | Salama, 2006 [84] Toumba, 2010 [88] |
| Amino-terminal pro-peptide of type I procollagen (P1NP) | Decrease | TM | aUN | Baldini, 2014 [89] |
| C-telopeptide of type-I collagen | Decrease | B/S | (ng/mL) aH: 45.98 (5.04–124) aP: 135.73 (39.93–246.1) | Voskaridou, 2005 [81] |
| PTH | Decrease | TM | (µg/mL) cH: 75.2 ± 31.3 cP: 35.3 ± 15.2 cH: 44.3 ± 5.63 cP: 32.2 ± 0.96 | Pirinççioğlu, 2017 [90] Goyal, 2010 [91] |
| SCD | (pmol/L) aH: 4.11 ± 1.46 aP: 2.22 ± 1.26 | Elshal, 2012 [92] | ||
| Vitamin D | Decrease | TM/SCD | a,cNA | Manolopoulos, 2021 * [93] Soe, 2017 * [94] |
| Serum IGF1 | Decrease | TM | (nmol/mL) aH: 35.25 ± 8.33 aP: 21.07 ± 5.12 (ng/mL) aH: 185.8 ± 27.7 aP: 55.8 ± 16.0 | Giordano, 2021 * [39] Lasco, 2002 [95] Morabito, 2004 [24] |
| SCD | (nmol/mL) cH: 42.88 ± 4.33 cP: 18.09 ± 3.88 | Luporini, 2001 [96] | ||
| IGFBP-3 | Decrease | TM | (mg/mL) aH: 2.5 ± 0.1 aP: 1.9 ± 0.4 | Lasco, 2002 [95] |
| Sexual Hormones (i.e., Estradiol) | Decrease | TM | (pg/mL) aH: 90.8 (62.2–136.1) aP: 13.8 (5.0–45.7) | Anapliotou, 1995 [97] Gaudio, 2019 * [98] Thavonlun, 2023 [99] |
| IL-6 | Increase | TM | (pg/mL) aH: 6.2 ± 3.7 aP: 8.1 ± 3.3 | Morabito, 2007 [78] Gaudio, 2019 * [98] |
| IL-1α | Increase | TM | (pg/mL) aH: 5.6 ± 2.5 aP:13.2 ± 4.1 | Morabito, 2007 [78] Gaudio, 2019 * [98] |
| TNF-α | Increase | TM | (pg/mL) aH: 6.4 ± 2.1 aP:11.4 ± 5.3 | Morabito, 2007 [78] |
| GDF15 | Increase | TM | aUN | Teawtrakul, 2023 [100] |
| Klotho | Decrease | TM | (pg/mL) aH: 618.2 ± 141.1 aP: 558.7 ± 160.5 | Baldan, 2015 [101] |
| Sclerostin | Increase | TM | (pg/mL) aH: 250 (0–720) aP: 605 (22–1227) | Voskaridou, 2012 [102] |
| Arachidonic Acid | Increase | TM | (nM) aH: 8 aP: 52 | Piriyakhuntorn, 2023 [103] |
| Glutamate | Increase | TM | (µM) aH: 8.3 aP: 24.8 | Piriyakhuntorn, 2023 [103] |
| Glutamine | Decrease | TM | (AU) aH: 11.4 aP: 10.0 | Piriyakhuntorn, 2023 [103] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Paola, A.; Marrapodi, M.M.; Di Martino, M.; Giliberti, G.; Di Feo, G.; Rana, D.; Ahmed, S.; Argenziano, M.; Rossi, F.; Roberti, D. Bone Health Impairment in Patients with Hemoglobinopathies: From Biological Bases to New Possible Therapeutic Strategies. Int. J. Mol. Sci. 2024, 25, 2902. https://doi.org/10.3390/ijms25052902
Di Paola A, Marrapodi MM, Di Martino M, Giliberti G, Di Feo G, Rana D, Ahmed S, Argenziano M, Rossi F, Roberti D. Bone Health Impairment in Patients with Hemoglobinopathies: From Biological Bases to New Possible Therapeutic Strategies. International Journal of Molecular Sciences. 2024; 25(5):2902. https://doi.org/10.3390/ijms25052902
Chicago/Turabian StyleDi Paola, Alessandra, Maria Maddalena Marrapodi, Martina Di Martino, Giulia Giliberti, Giuseppe Di Feo, Deeksha Rana, Shakeel Ahmed, Maura Argenziano, Francesca Rossi, and Domenico Roberti. 2024. "Bone Health Impairment in Patients with Hemoglobinopathies: From Biological Bases to New Possible Therapeutic Strategies" International Journal of Molecular Sciences 25, no. 5: 2902. https://doi.org/10.3390/ijms25052902