
Identification of Serum Biomarkers to Monitor Therapeutic Response in Intestinal-Type Gastric Cancer

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
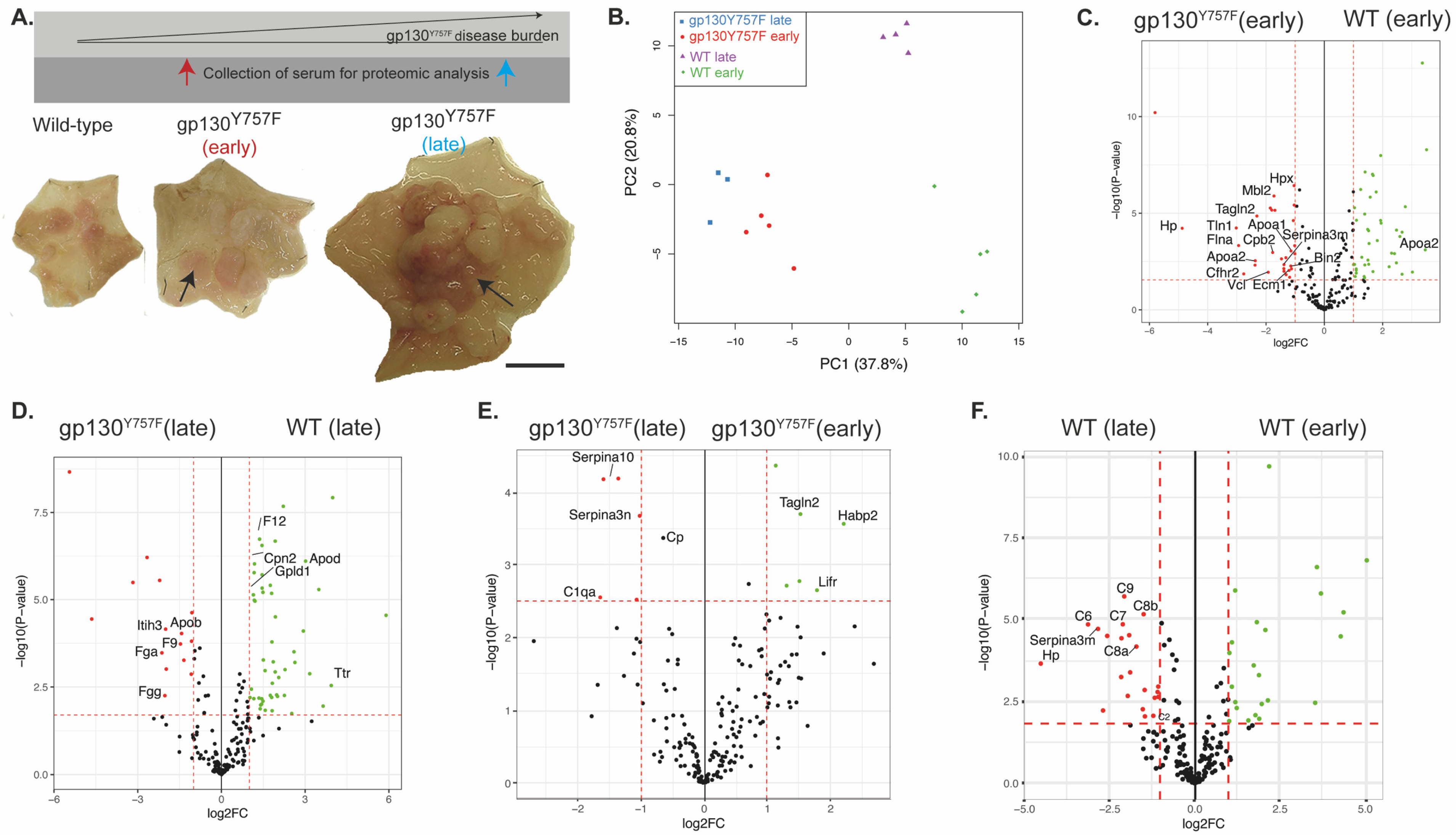
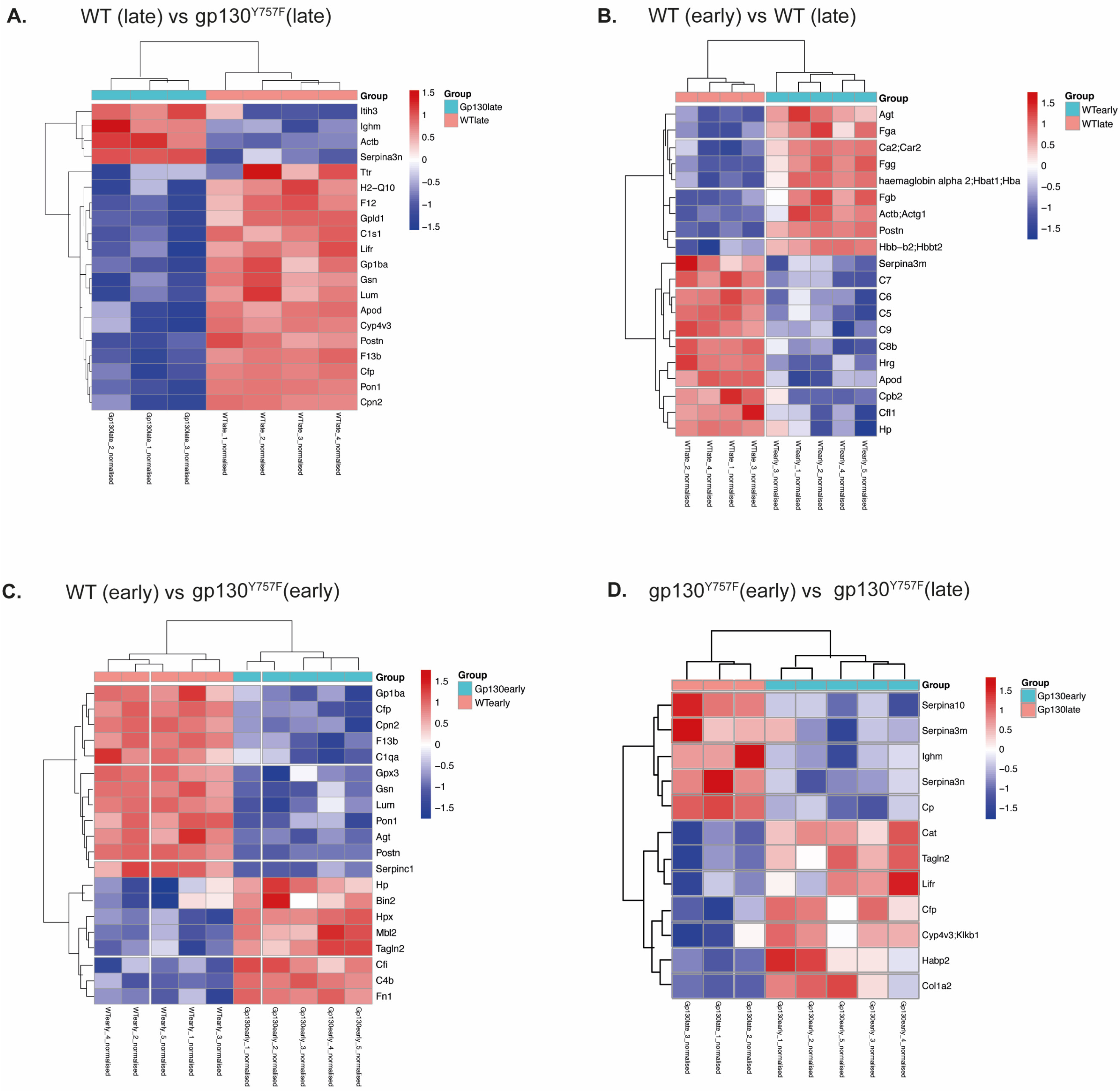
2.1. gp130Y757F Mice Have a Distinct Serum Proteome
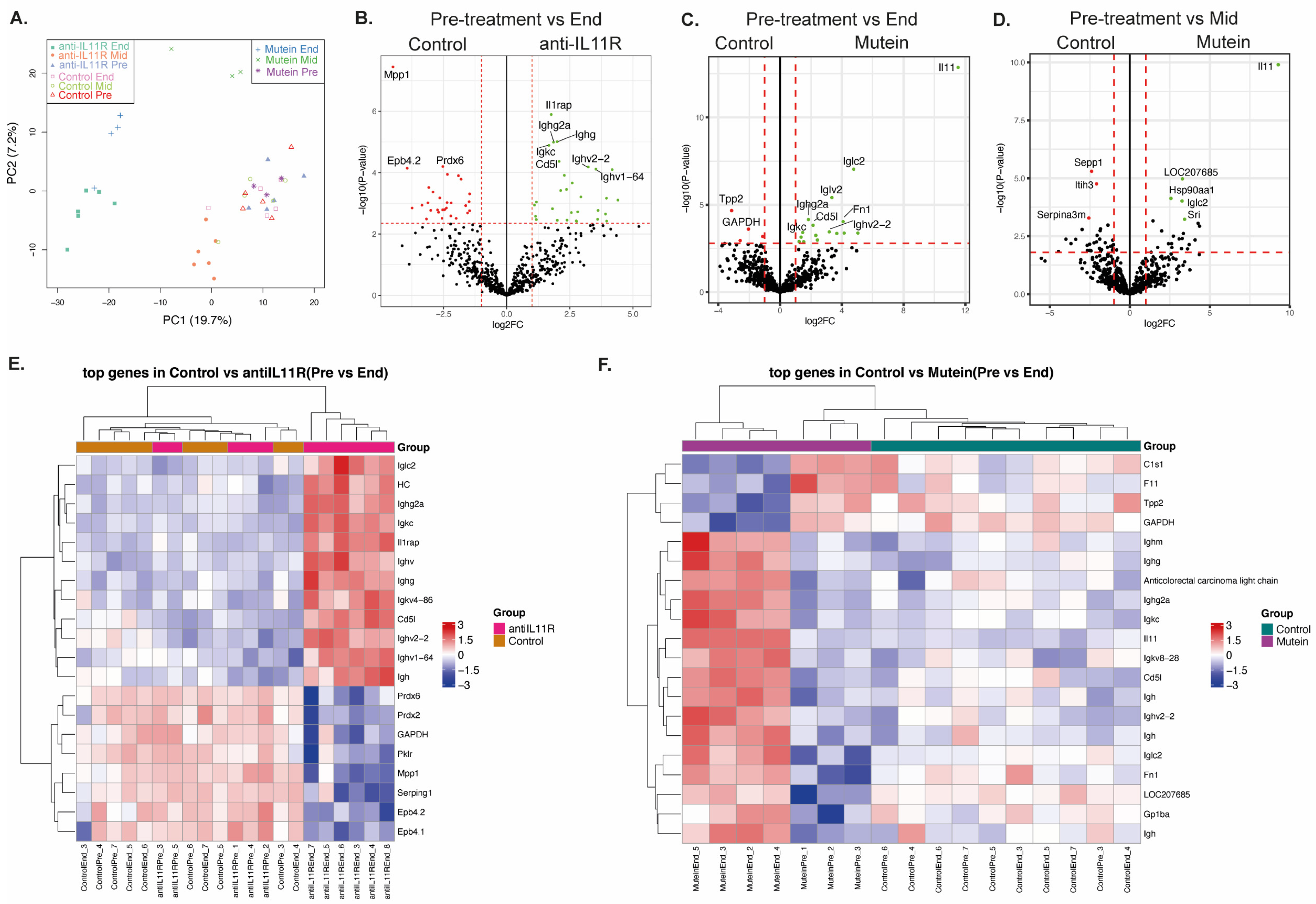
2.2. Therapeutic Modulation of IL-11 Signaling Alters the Serum Proteome
3. Discussion
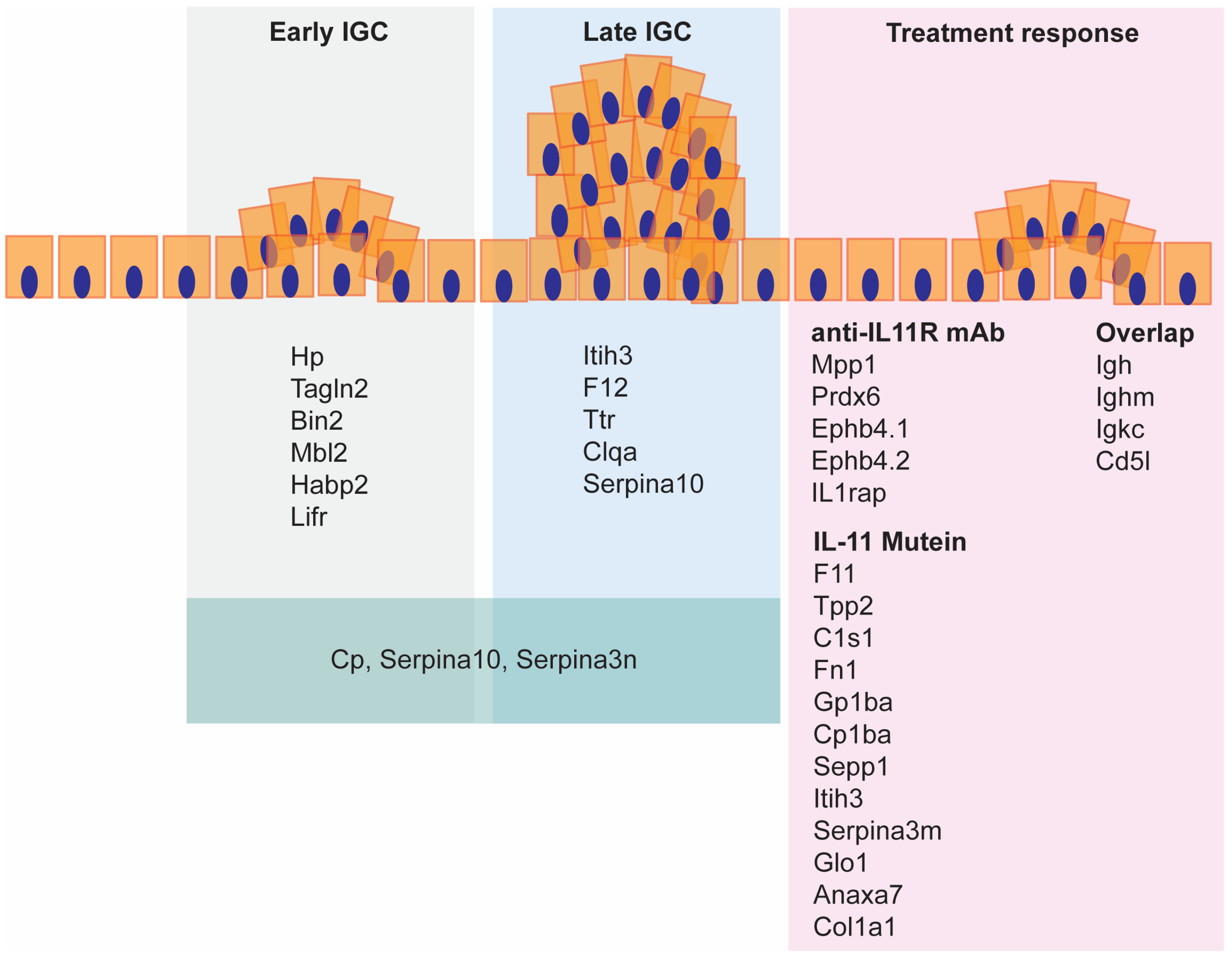
3.1. Serum Biomarkers of Advanced Disease
3.2. Comparison of Two Treatment Modalities That Block IL-11 Signaling
3.3. Serum Biomarkers of Tumor Regression
3.4. Strengths and Limitations
4. Materials and Methods
4.1. Mice
4.2. Gene Expression
4.3. Proteomics
4.3.1. Immunoaffinity Chromatography
4.3.2. Proteomics Sample Preparation
4.3.3. High pH Peptide Fractionation
4.3.4. Mass Spectrometry Experimental Design
4.3.5. Data Processing and Statistical Analysis
4.3.6. Differential Expression and Enrichment Analysis
4.4. Conclusions and Future Prospects
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef]
- Morgan, E.; Arnold, M.; Camargo, M.C.; Gini, A.; Kunzmann, A.T.; Matsuda, T.; Meheus, F.; Verhoeven, R.H.A.; Vignat, J.; Laversanne, M.; et al. The current and future incidence and mortality of gastric cancer in 185 countries, 2020–2040: A population-based modelling study. EClinicalMedicine 2022, 47, 101404. [Google Scholar] [CrossRef]
- Joshi, S.S.; Badgwell, B.D. Current treatment and recent progress in gastric cancer. CA Cancer J. Clin. 2021, 71, 264–279. [Google Scholar] [CrossRef]
- Orditura, M.; Galizia, G.; Sforza, V.; Gambardella, V.; Fabozzi, A.; Laterza, M.M.; Andreozzi, F.; Ventriglia, J.; Savastano, B.; Mabilia, A.; et al. Treatment of gastric cancer. World J. Gastroenterol. 2014, 20, 1635–1649. [Google Scholar] [CrossRef]
- Ye, D.M.; Xu, G.; Ma, W.; Li, Y.; Luo, W.; Xiao, Y.; Liu, Y.; Zhang, Z. Significant function and research progress of biomarkers in gastric cancer. Oncol. Lett. 2020, 19, 17–29. [Google Scholar] [CrossRef]
- Wu, D.; Zhang, P.; Ma, J.; Xu, J.; Yang, L.; Xu, W.; Que, H.; Chen, M.; Xu, H. Serum biomarker panels for the diagnosis of gastric cancer. Cancer Med. 2019, 8, 1576–1583. [Google Scholar] [CrossRef]
- Tatsuta, M.; Itoh, T.; Okuda, S.; Yamamura, H.; Baba, M.; Tamura, H. Carcinoembryonic antigen in gastric juice as an aid in diagnosis of early gastric cancer. Cancer 1980, 46, 2686–2692. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Zhong, X.; Song, Y.; Shi, J.; Wu, Z.; Guo, Z.; Sun, J.; Wang, Z. Prognostic Biomarkers for Gastric Cancer: An Umbrella Review of the Evidence. Front. Oncol. 2019, 9, 1321. [Google Scholar] [CrossRef]
- Ishigami, S.; Natsugoe, S.; Hokita, S.; Che, X.; Tokuda, K.; Nakajo, A.; Iwashige, H.; Tokushige, M.; Watanabe, T.; Takao, S.; et al. Clinical importance of preoperative carcinoembryonic antigen and carbohydrate antigen 19-9 levels in gastric cancer. J. Clin. Gastroenterol. 2001, 32, 41–44. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.K.; Sun, X.Q.; Mou, D.Z. Value of Combined Detection of Serum CEA, CA72-4, CA19-9 and TSGF in the Diagnosis of Gastric Cancer. Asian Pac. J. Cancer Prev. 2015, 16, 3867–3870. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, B.J.; Grail, D.; Nheu, T.; Najdovska, M.; Wang, B.; Waring, P.; Inglese, M.; McLoughlin, R.M.; Jones, S.A.; Topley, N.; et al. Hyperactivation of Stat3 in gp130 mutant mice promotes gastric hyperproliferation and desensitizes TGF-beta signaling. Nat. Med. 2005, 11, 845–852. [Google Scholar] [CrossRef] [PubMed]
- Eissmann, M.F.; Dijkstra, C.; Jarnicki, A.; Phesse, T.; Brunnberg, J.; Poh, A.R.; Etemadi, N.; Tsantikos, E.; Thiem, S.; Huntington, N.D.; et al. IL-33-mediated mast cell activation promotes gastric cancer through macrophage mobilization. Nat. Commun. 2019, 10, 2735. [Google Scholar] [CrossRef]
- Kim, D.Y.; Cha, S.T.; Ahn, D.H.; Kang, H.Y.; Kwon, C.I.; Ko, K.H.; Hwang, S.G.; Park, P.W.; Rim, K.S.; Hong, S.P. STAT3 expression in gastric cancer indicates a poor prognosis. J. Gastroenterol. Hepatol. 2009, 24, 646–651. [Google Scholar] [CrossRef]
- Kim, D.K.; Oh, S.Y.; Kwon, H.C.; Lee, S.; Kwon, K.A.; Kim, B.G.; Kim, S.G.; Kim, S.H.; Jang, J.S.; Kim, M.C.; et al. Clinical significances of preoperative serum interleukin-6 and C-reactive protein level in operable gastric cancer. BMC Cancer 2009, 9, 155. [Google Scholar] [CrossRef] [PubMed]
- Putoczki, T.L.; Thiem, S.; Loving, A.; Busuttil, R.A.; Wilson, N.J.; Ziegler, P.K.; Nguyen, P.M.; Preaudet, A.; Farid, R.; Edwards, K.M.; et al. Interleukin-11 is the dominant IL-6 family cytokine during gastrointestinal tumorigenesis and can be targeted therapeutically. Cancer Cell 2013, 24, 257–271. [Google Scholar] [CrossRef] [PubMed]
- Humphries, J.M.; Penno, M.A.; Weiland, F.; Klingler-Hoffmann, M.; Zuber, A.; Boussioutas, A.; Ernst, M.; Hoffmann, P. Identification and validation of novel candidate protein biomarkers for the detection of human gastric cancer. Biochim. Biophys. Acta 2014, 1844, 1051–1058. [Google Scholar] [CrossRef]
- Penno, M.A.; Klingler-Hoffmann, M.; Brazzatti, J.A.; Boussioutas, A.; Putoczki, T.; Ernst, M.; Hoffmann, P. 2D-DIGE analysis of sera from transgenic mouse models reveals novel candidate protein biomarkers for human gastric cancer. J. Proteom. 2012, 77, 40–58. [Google Scholar] [CrossRef]
- Gaya da Costa, M.; Poppelaars, F.; van Kooten, C.; Mollnes, T.E.; Tedesco, F.; Wurzner, R.; Trouw, L.A.; Truedsson, L.; Daha, M.R.; Roos, A.; et al. Age and Sex-Associated Changes of Complement Activity and Complement Levels in a Healthy Caucasian Population. Front. Immunol. 2018, 9, 2664. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Kopchick, J.J. Plasma biomarkers of mouse aging. Age 2011, 33, 291–307. [Google Scholar] [CrossRef]
- Winship, A.L.; Van Sinderen, M.; Donoghue, J.; Rainczuk, K.; Dimitriadis, E. Targeting Interleukin-11 Receptor-alpha Impairs Human Endometrial Cancer Cell Proliferation and Invasion In Vitro and Reduces Tumor Growth and Metastasis In Vivo. Mol. Cancer Ther. 2016, 15, 720–730. [Google Scholar] [CrossRef]
- Teramura, M.; Kobayashi, S.; Yoshinaga, K.; Iwabe, K.; Mizoguchi, H. Effect of interleukin 11 on normal and pathological thrombopoiesis. Cancer Chemother. Pharmacol. 1996, 38 (Suppl. S1), S99–S102. [Google Scholar] [CrossRef] [PubMed]
- Takemura, K.; Hirokawa, K.; Esaki, Y.; Mishima, Y. Distribution of immunoglobulins and secretory component in gastric cancer of the aged. Cancer 1990, 66, 2168–2173. [Google Scholar] [CrossRef] [PubMed]
- Poh, A.R.; O’Donoghue, R.J.; Ernst, M.; Putoczki, T.L. Mouse models for gastric cancer: Matching models to biological questions. J. Gastroenterol. Hepatol. 2016, 31, 1257–1272. [Google Scholar] [CrossRef] [PubMed]
- Cook, S.A. Understanding interleukin 11 as a disease gene and therapeutic target. Biochem. J. 2023, 480, 1987–2008. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Zheng, W.; Hou, J.; Peng, H.; Zhuo, H. An Essential NRP1-Mediated Role for Tagln2 in Gastric Cancer Angiogenesis. Front. Oncol. 2021, 11, 653246. [Google Scholar] [CrossRef]
- Zhao, Z.; Lu, L.; Li, W. TAGLN2 promotes the proliferation, invasion, migration and epithelial-mesenchymal transition of colorectal cancer cells by activating STAT3 signaling through ANXA2. Oncol. Lett. 2021, 22, 737. [Google Scholar] [CrossRef] [PubMed]
- Baccarelli, A.; Hou, L.; Chen, J.; Lissowska, J.; El-Omar, E.M.; Grillo, P.; Giacomini, S.M.; Yaeger, M.; Bernig, T.; Zatonski, W.; et al. Mannose-binding lectin-2 genetic variation and stomach cancer risk. Int. J. Cancer 2006, 119, 1970–1975. [Google Scholar] [CrossRef]
- Rong, Y.; Jin, D.; Hou, C.; Hu, J.; Wu, W.; Ni, X.; Wang, D.; Lou, W. Proteomics analysis of serum protein profiling in pancreatic cancer patients by DIGE: Up-regulation of mannose-binding lectin 2 and myosin light chain kinase 2. BMC Gastroenterol. 2010, 10, 68. [Google Scholar] [CrossRef] [PubMed]
- Chong, P.K.; Lee, H.; Zhou, J.; Liu, S.C.; Loh, M.C.; Wang, T.T.; Chan, S.P.; Smoot, D.T.; Ashktorab, H.; So, J.B.; et al. ITIH3 is a potential biomarker for early detection of gastric cancer. J. Proteome Res. 2010, 9, 3671–3679. [Google Scholar] [CrossRef]
- Luo, J.H.; Zhang, X.X.; Sun, W.H. F12 as a reliable diagnostic and prognostic biomarker associated with immune infiltration in papillary thyroid cancer. Aging 2022, 14, 3687–3704. [Google Scholar] [CrossRef]
- Dimberg, J.; Strom, K.; Lofgren, S.; Zar, N.; Hugander, A.; Matussek, A. Expression of the serine protease inhibitor serpinA3 in human colorectal adenocarcinomas. Oncol. Lett. 2011, 2, 413–418. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Tian, J.; Qu, C.; Peng, Y.; Lei, J.; Li, K.; Zong, B.; Sun, L.; Liu, S. Overexpression of SERPINA3 promotes tumor invasion and migration, epithelial-mesenchymal-transition in triple-negative breast cancer cells. Breast Cancer 2021, 28, 859–873. [Google Scholar] [CrossRef] [PubMed]
- Metcalfe, R.D.; Putoczki, T.L.; Griffin, M.D.W. Structural Understanding of Interleukin 6 Family Cytokine Signaling and Targeted Therapies: Focus on Interleukin 11. Front. Immunol. 2020, 11, 1424. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, L.A.; Putoczki, T.L.; Mielke, L.A.; Low, J.T.; Lin, A.; Preaudet, A.; Herold, M.J.; Yaprianto, K.; Tai, L.; Kueh, A.; et al. Loss of NF-kappaB1 Causes Gastric Cancer with Aberrant Inflammation and Expression of Immune Checkpoint Regulators in a STAT-1-Dependent Manner. Immunity 2018, 48, 570–583.e8. [Google Scholar] [CrossRef] [PubMed]
- Low, J.T.; Christie, M.; Ernst, M.; Dumoutier, L.; Preaudet, A.; Ni, Y.; Griffin, M.D.W.; Mielke, L.A.; Strasser, A.; Putoczki, T.L.; et al. Loss of NFKB1 Results in Expression of Tumor Necrosis Factor and Activation of Signal Transducer and Activator of Transcription 1 to Promote Gastric Tumorigenesis in Mice. Gastroenterology 2020, 159, 1444–1458.e15. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.; Hu, S.; Zheng, Z.; Tong, H. Contribution of interaction between genetic variants of interleukin-11 and Helicobacter pylori infection to the susceptibility of gastric cancer. OncoTargets Ther. 2019, 12, 7459–7466. [Google Scholar] [CrossRef] [PubMed]
- Calcagno, D.Q.; Takeno, S.S.; Gigek, C.O.; Leal, M.F.; Wisnieski, F.; Chen, E.S.; Araujo, T.M.; Lima, E.M.; Melaragno, M.I.; Demachki, S.; et al. Identification of IL11RA and MELK amplification in gastric cancer by comprehensive genomic profiling of gastric cancer cell lines. World J. Gastroenterol. 2016, 22, 9506–9514. [Google Scholar] [CrossRef] [PubMed]
- Biernatowska, A.; Augoff, K.; Podkalicka, J.; Tabaczar, S.; Gajdzik-Nowak, W.; Czogalla, A.; Sikorski, A.F. MPP1 directly interacts with flotillins in erythrocyte membrane—Possible mechanism of raft domain formation. Biochim. Biophys. Acta Biomembr. 2017, 1859, 2203–2212. [Google Scholar] [CrossRef]
- Huang, W.S.; Huang, C.Y.; Hsieh, M.C.; Kuo, Y.H.; Tung, S.Y.; Shen, C.H.; Hsieh, Y.Y.; Teng, C.C.; Lee, K.C.; Lee, K.F.; et al. Expression of PRDX6 Correlates with Migration and Invasiveness of Colorectal Cancer Cells. Cell. Physiol. Biochem. 2018, 51, 2616–2630. [Google Scholar] [CrossRef]
- Meng, M.; Liu, S.; Wang, C.; Gu, X.; Linghu, E.; Xue, X. Mass spectrum analysis of membrane proteins reveals that CASK, CD36 and EPB42 are differentially expressed in pancreatic adenocarcinoma. Oncol. Lett. 2020, 20, 376. [Google Scholar] [CrossRef]
- Lv, Q.; Xia, Q.; Li, A.; Wang, Z. The Potential Role of IL1RAP on Tumor Microenvironment-Related Inflammatory Factors in Stomach Adenocarcinoma. Technol. Cancer Res. Treat. 2021, 20, 1533033821995282. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Sun, Y.; Wang, X.; Wang, J.; Zhu, Y. The expressions of EphB4 and ephrinB2 in lung adenocarcinomas: A high level of the EphB4 protein is associated with lymph node metastasis. Int. J. Clin. Exp. Pathol. 2019, 12, 3447–3452. [Google Scholar]
- Usukura, K.; Kasamatsu, A.; Okamoto, A.; Kouzu, Y.; Higo, M.; Koike, H.; Sakamoto, Y.; Ogawara, K.; Shiiba, M.; Tanzawa, H.; et al. Tripeptidyl peptidase II in human oral squamous cell carcinoma. J. Cancer Res. Clin. Oncol. 2013, 139, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Sebastian, A.; Hum, N.R.; Martin, K.A.; Gilmore, S.F.; Peran, I.; Byers, S.W.; Wheeler, E.K.; Coleman, M.A.; Loots, G.G. Single-Cell Transcriptomic Analysis of Tumor-Derived Fibroblasts and Normal Tissue-Resident Fibroblasts Reveals Fibroblast Heterogeneity in Breast Cancer. Cancers 2020, 12, 1307. [Google Scholar] [CrossRef]
- Li, Y.; Xu, C.; Wang, B.; Xu, F.; Ma, F.; Qu, Y.; Jiang, D.; Li, K.; Feng, J.; Tian, S.; et al. Proteomic characterization of gastric cancer response to chemotherapy and targeted therapy reveals new therapeutic strategies. Nat. Commun. 2022, 13, 5723. [Google Scholar] [CrossRef]
- Cheng, W.L.; Tsai, M.M.; Tsai, C.Y.; Huang, Y.H.; Chen, C.Y.; Chi, H.C.; Tseng, Y.H.; Chao, I.W.; Lin, W.C.; Wu, S.M.; et al. Glyoxalase-I is a novel prognosis factor associated with gastric cancer progression. PLoS ONE 2012, 7, e34352. [Google Scholar] [CrossRef]
- Li, J.; Ding, Y.; Li, A. Identification of COL1A1 and COL1A2 as candidate prognostic factors in gastric cancer. World J. Surg. Oncol. 2016, 14, 297. [Google Scholar] [CrossRef]
- Nguyen, P.M.; Dagley, L.F.; Preaudet, A.; Lam, N.; Giam, M.; Fung, K.Y.; Aizel, K.; van Duijneveldt, G.; Tan, C.W.; Hirokawa, Y.; et al. Loss of Bcl-G, a Bcl-2 family member, augments the development of inflammation-associated colorectal cancer. Cell Death Differ. 2020, 27, 742–757. [Google Scholar] [CrossRef]
- Cox, J.; Neuhauser, N.; Michalski, A.; Scheltema, R.A.; Olsen, J.V.; Mann, M. Andromeda: A peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 2011, 10, 1794–1805. [Google Scholar] [CrossRef]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef]
- Lazar, C.; Gatto, L.; Ferro, M.; Bruley, C.; Burger, T. Accounting for the Multiple Natures of Missing Values in Label-Free Quantitative Proteomics Data Sets to Compare Imputation Strategies. J. Proteome Res. 2016, 15, 1116–1125. [Google Scholar] [CrossRef] [PubMed]
- Poulos, R.C.; Hains, P.G.; Shah, R.; Lucas, N.; Xavier, D.; Manda, S.S.; Anees, A.; Koh, J.M.S.; Mahboob, S.; Wittman, M.; et al. Strategies to enable large-scale proteomics for reproducible research. Nat. Commun. 2020, 11, 3793. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dagley, L.F.; Yousef, J.; Preaudet, A.; Loving, A.; Webb, A.I.; Ernst, M.; Putoczki, T.L. Identification of Serum Biomarkers to Monitor Therapeutic Response in Intestinal-Type Gastric Cancer. Int. J. Mol. Sci. 2024, 25, 3129. https://doi.org/10.3390/ijms25063129
Dagley LF, Yousef J, Preaudet A, Loving A, Webb AI, Ernst M, Putoczki TL. Identification of Serum Biomarkers to Monitor Therapeutic Response in Intestinal-Type Gastric Cancer. International Journal of Molecular Sciences. 2024; 25(6):3129. https://doi.org/10.3390/ijms25063129
Chicago/Turabian StyleDagley, Laura F., Jumana Yousef, Adele Preaudet, Andrea Loving, Andrew I. Webb, Matthias Ernst, and Tracy L. Putoczki. 2024. "Identification of Serum Biomarkers to Monitor Therapeutic Response in Intestinal-Type Gastric Cancer" International Journal of Molecular Sciences 25, no. 6: 3129. https://doi.org/10.3390/ijms25063129