
In Silico Description of the Direct Inhibition Mechanism of Endothelial Lipase by ANGPTL3
 , , , , , , and
, , , , , , and
Abstract
:1. Introduction
2. Results
2.1. Ab Initio Modeling of the N-Terminal Domain of ANGPTL3
2.2. Homology Modeling of EL
2.3. ANGPTL3∷EL Docking
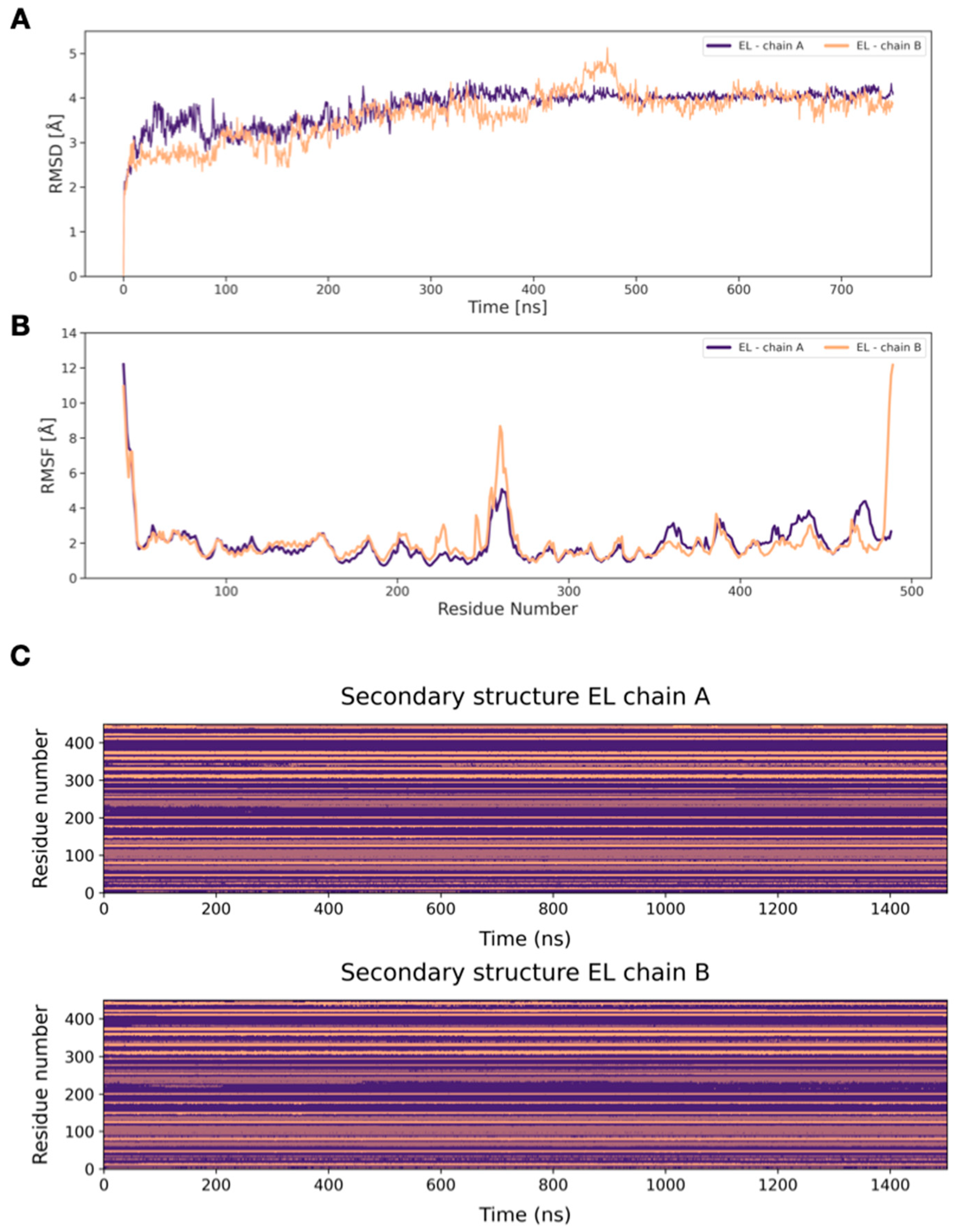
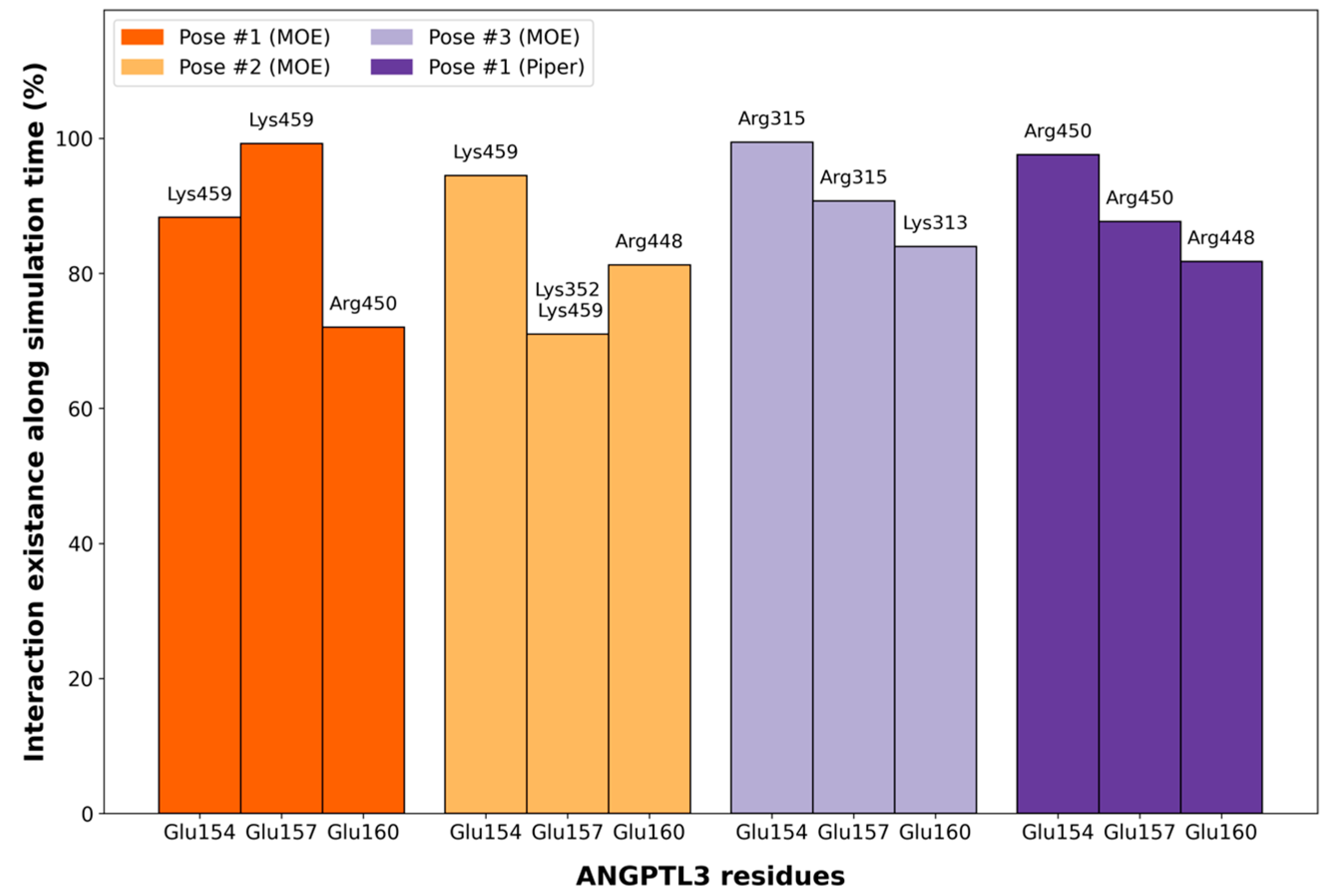
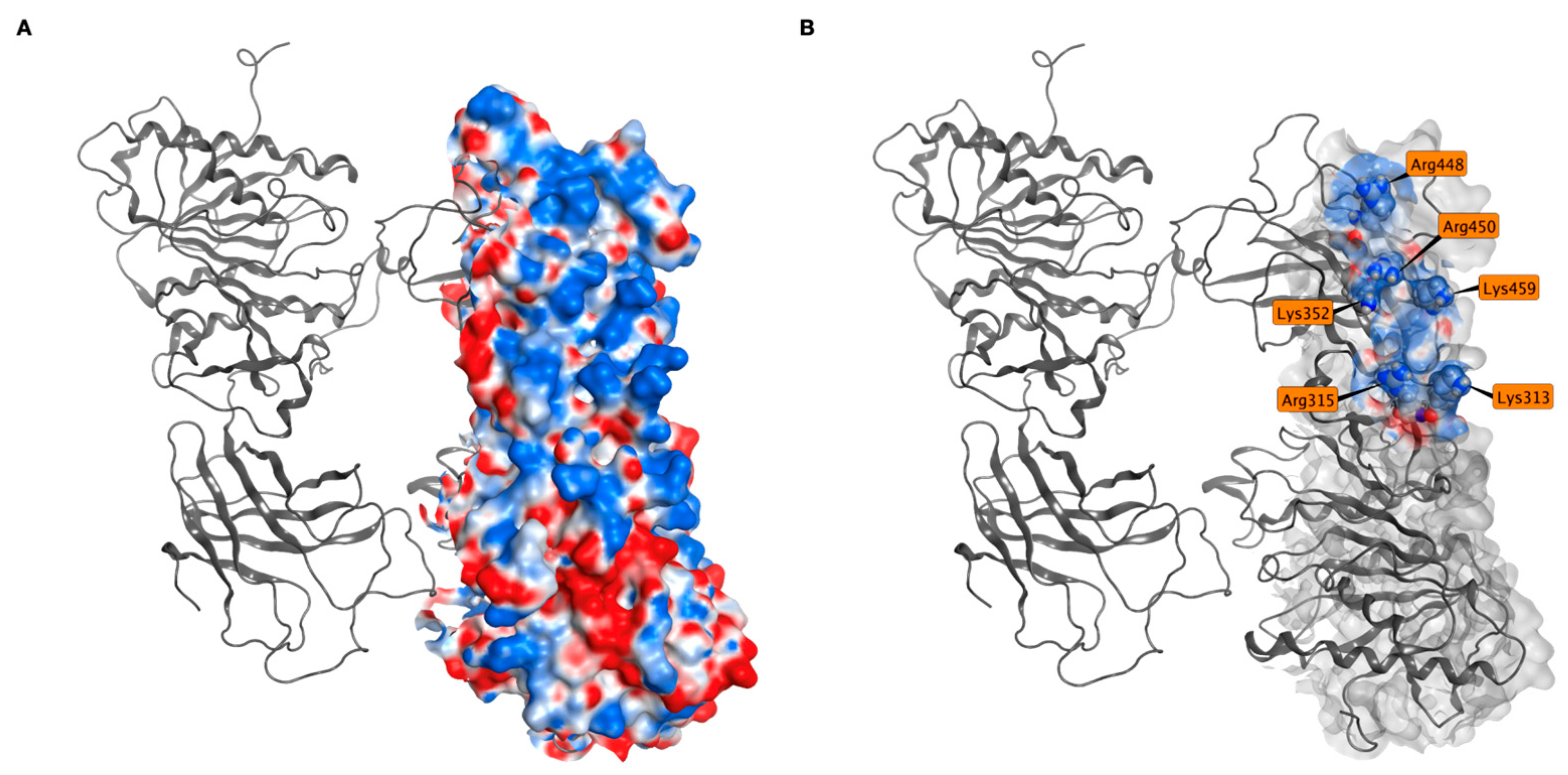
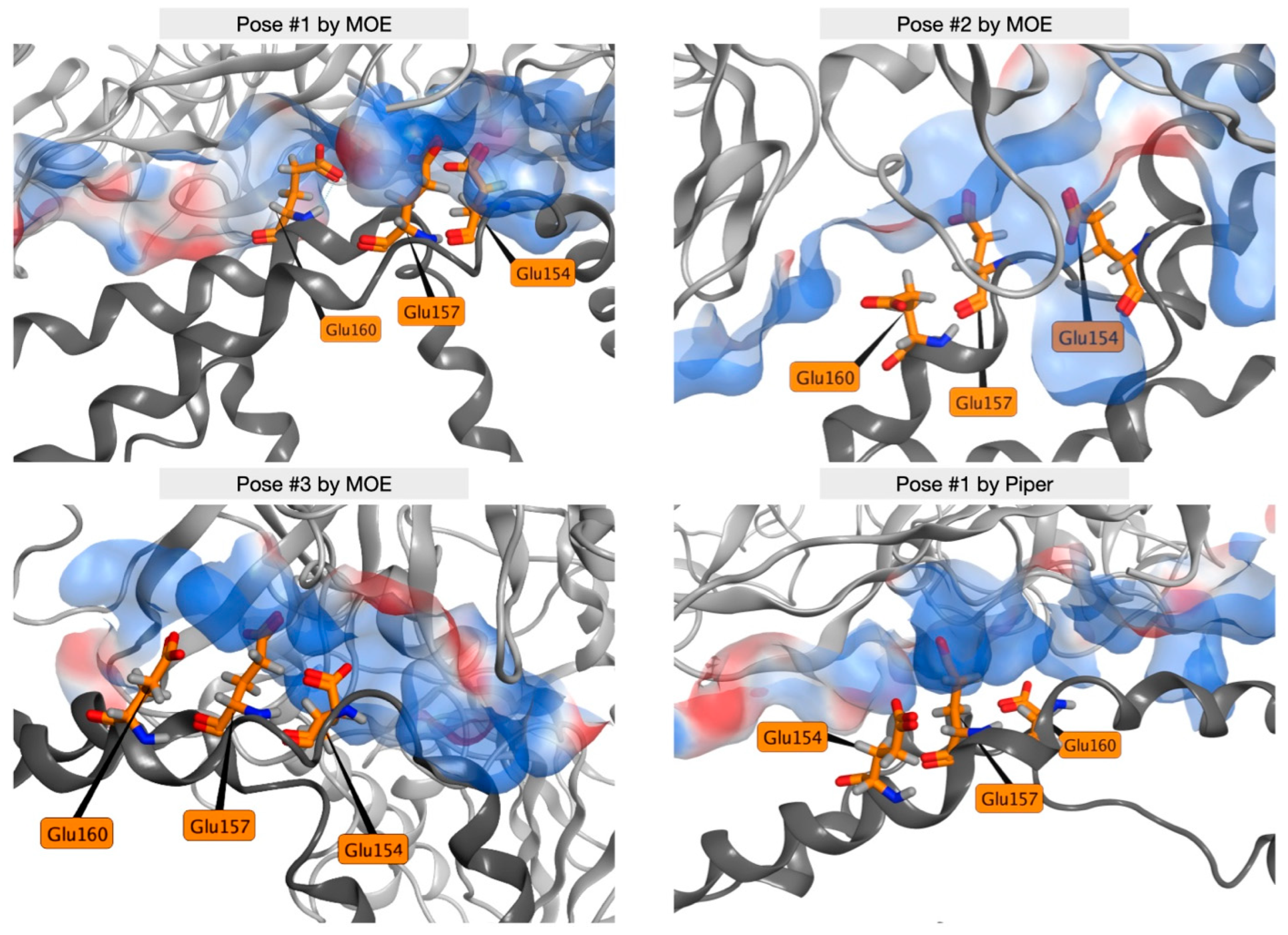
2.4. MD Simulations of Docking Poses and Interactions Analysis
3. Discussion
4. Materials and Methods
4.1. De Novo Modeling of the N-Terminal ANGPTL3 Trimer
4.2. Homology Modeling of the Endothelial Lipase Dimer
4.3. MD Simulations
- NPT ensemble, 12 ps at 10 K 10 K and restraints of 50 kcal/mol/Å on protein atoms;
- NVT ensemble, 12 ps at 10 K and restraints of 50 kcal/mol/Å on protein heavy atoms;
- NPT ensemble, 12 ps at 10 K and restraints of 50 kcal/mol/Å on protein heavy atoms;
- NPT ensemble, 12 ps at 300 K and restraints of 50 kcal/mol/Å on protein heavy atoms;
- NPT ensemble, 24 ps at 300 K without restraints.
4.4. MD Analysis
4.5. Protein–Protein Docking
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lu, X. Structure and Function of Angiopoietin-like Protein 3 (ANGPTL3) in Atherosclerosis. Curr. Med. Chem. 2020, 27, 5159–5174. [Google Scholar] [CrossRef] [PubMed]
- Kämpfer, H.; Pfeilschifter, J.; Frank, S. Expressional regulation of angiopoietin-1 and -2 and the tie-1 and -2 receptor tyrosine kinases during cutaneous wound healing: A comparative study of normal and impaired repair. Lab. Investig. 2001, 81, 361–373. [Google Scholar] [CrossRef] [PubMed]
- Ono, M.; Shimizugawa, T.; Shimamura, M.; Yoshida, K.; Noji-Sakikawa, C.; Ando, Y.; Koishi, R.; Furukawa, H. Protein region important for regulation of lipid metabolism in angiopoietin-like 3 (ANGPTL3): ANGPTL3 is cleaved and activated in vivo. J. Biol. Chem. 2003, 278, 41804–41809. [Google Scholar] [CrossRef]
- Mohamed, F.; Mansfield, B.S.; Raal, F.J. ANGPTL3 as a Drug Target in Hyperlipidemia and Atherosclerosis. Curr. Atheroscler. Rep. 2022, 24, 959–967. [Google Scholar] [CrossRef] [PubMed]
- Gunn, K.H.; Gutgsell, A.R.; Xu, Y.; Johnson, C.V.; Liu, J.; Neher, S.B. Comparison of angiopoietin-like protein 3 and 4 reveals structural and mechanistic similarities. J. Biol. Chem. 2021, 296, 100312. [Google Scholar] [CrossRef]
- Li, X.; Zhang, Y.; Zhang, M.; Wang, Y. GALNT2 regulates ANGPTL3 cleavage in cells and in vivo of mice. Sci. Rep. 2020, 10, 16168. [Google Scholar] [CrossRef]
- Biterova, E.; Esmaeeli, M.; Alanen, H.I.; Saaranen, M.; Ruddock, L.W. Structures of Angptl3 and Angptl4, modulators of triglyceride levels and coronary artery disease. Sci. Rep. 2018, 8, 6752. [Google Scholar] [CrossRef]
- Koishi, R.; Ando, Y.; Ono, M.; Shimamura, M.; Yasumo, H.; Fujiwara, T.; Horikoshi, H.; Furukawa, H. Angptl3 regulates lipid metabolism in mice. Nat. Genet. 2002, 30, 151–157. [Google Scholar] [CrossRef]
- Adam, R.C.; Mintah, I.J.; Alexa-Braun, C.A.; Shihanian, L.M.; Lee, J.S.; Banerjee, P.; Hamon, S.C.; Kim, H.I.; Cohen, J.C.; Hobbs, H.H.; et al. Angiopoietin-like protein 3 governs LDL-cholesterol levels through endothelial lipase-dependent VLDL clearance. J. Lipid Res. 2020, 61, 1271–1286. [Google Scholar] [CrossRef] [PubMed]
- Pennisi, G.; Maurotti, S.; Ciociola, E.; Jamialahmadi, O.; Bertolazzi, G.; Mirarchi, A.; Bergh, P.-O.; Scionti, F.; Mancina, R.M.; Spagnuolo, R.; et al. ANGPTL3 Downregulation Increases Intracellular Lipids by Reducing Energy Utilization. Arter. Thromb. Vasc. Biol. 2024. Online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Shan, L.; Yu, X.-C.; Liu, Z.; Hu, Y.; Sturgis, L.T.; Miranda, M.L.; Liu, Q. The angiopoietin-like proteins ANGPTL3 and ANGPTL4 inhibit lipoprotein lipase activity through distinct mechanisms. J. Biol. Chem. 2009, 284, 1419–1424. [Google Scholar] [CrossRef] [PubMed]
- Sylvers-Davie, K.L.; Segura-Roman, A.; Salvi, A.M.; Schache, K.J.; Davies, B.S. Angiopoietin-like 3 inhibition of endothelial lipase is not modulated by angiopoietin-like 8. J. Lipid Res. 2021, 62, 100112. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Afroza, H.; Rader, D.J.; Jin, W. Angiopoietin-like protein 3 inhibits lipoprotein lipase activity through enhancing its cleavage by proprotein convertases. J. Biol. Chem. 2010, 285, 27561–27570. [Google Scholar] [CrossRef] [PubMed]
- Minicocci, I.; Santini, S.; Cantisani, V.; Stitziel, N.; Kathiresan, S.; Arroyo, J.A.; Martí, G.; Pisciotta, L.; Noto, D.; Cefalù, A.B.; et al. Clinical characteristics and plasma lipids in subjects with familial combined hypolipidemia: A pooled analysis. J. Lipid Res. 2013, 54, 3481–3490. [Google Scholar] [CrossRef] [PubMed]
- Minicocci, I.; Montali, A.; Robciuc, M.R.; Quagliarini, F.; Censi, V.; Labbadia, G.; Gabiati, C.; Pigna, G.; Sepe, M.L.; Pannozzo, F.; et al. Mutations in the ANGPTL3 gene and familial combined hypolipidemia: A clinical and biochemical characterization. J. Clin. Endocrinol. Metab. 2012, 97, E1266–E1275. [Google Scholar] [CrossRef] [PubMed]
- Raal, F.J.; Rosenson, R.S.; Reeskamp, L.F.; Hovingh, G.K.; Kastelein, J.J.; Rubba, P.; Ali, S.; Banerjee, P.; Chan, K.-C.; Gipe, D.A.; et al. Evinacumab for Homozygous Familial Hypercholesterolemia. N. Engl. J. Med. 2020, 383, 711–720. [Google Scholar] [CrossRef] [PubMed]
- Griffon, N.; Jin, W.; Petty, T.J.; Millar, J.; Badellino, K.O.; Saven, J.G.; Marchadier, D.H.; Kempner, E.S.; Billheimer, J.; Glick, J.M.; et al. Identification of the Active Form of Endothelial Lipase, a Homodimer in a Head-to-Tail Conformation. J. Biol. Chem. 2009, 284, 23322–23330. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- The UniProt Consortium; Bateman, A.; Martin, M.-J.; Orchard, S.; Magrane, M.; Ahmad, S.; Alpi, E.; Bowler-Barnett, E.H.; Britto, R.; Bye-A-Jee, H.; et al. UniProt: The Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 2022, 51, D523–D531. [Google Scholar] [CrossRef]
- McGuffin, L.J.; Bryson, K.; Jones, D.T.J.B. The PSIPRED protein structure prediction server. Bioinformatics 2000, 16, 404–405. [Google Scholar] [CrossRef]
- Wood, C.W.; Woolfson, D.N. CCBuilder 2.0: Powerful and accessible coiled-coil modeling. Protein Sci. 2017, 27, 103–111. [Google Scholar] [CrossRef]
- Arora, R.; Nimonkar, A.V.; Baird, D.; Wang, C.; Chiu, C.-H.; Horton, P.A.; Hanrahan, S.; Cubbon, R.; Weldon, S.; Tschantz, W.R.; et al. Structure of lipoprotein lipase in complex with GPIHBP1. Proc. Natl. Acad. Sci. USA 2019, 116, 10360–10365. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Desmond Molecular Dynamics System. David Elliot Shaw Research, New York, NY, 2021. Maestro-Desmond Interoperability Tools; Schrödinger: New York, NY, USA, 2021. [Google Scholar]
- Palazzolo, L.; Paravicini, C.; Laurenzi, T.; Adobati, S.; Saporiti, S.; Guerrini, U.; Gianazza, E.; Indiveri, C.; Anderson, C.M.H.; Thwaites, D.T.; et al. SLC6A14, a Pivotal Actor on Cancer Stage: When Function Meets Structure. SLAS Discov. Adv. Sci. Drug Discov. 2019, 24, 928–938. [Google Scholar] [CrossRef]
- Laurenzi, T.; Palazzolo, L.; Taiana, E.; Saporiti, S.; Ben Mariem, O.; Guerrini, U.; Neri, A.; Eberini, I. Molecular Modelling of NONO and SFPQ Dimerization Process and RNA Recognition Mechanism. Int. J. Mol. Sci. 2022, 23, 7626. [Google Scholar] [CrossRef] [PubMed]
- Bowers, K.J.; Chow, D.E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D.; et al. Scalable algorithms for molecular dynamics simulations on commodity clusters. In Proceedings of the ACM/IEEE Conference on Supercomputing (SC06), Tampa, FL, USA, 11–17 November 2006. [Google Scholar] [CrossRef]
- Lu, C.; Wu, C.; Ghoreishi, D.; Chen, W.; Wang, L.; Damm, W.; Ross, G.A.; Dahlgren, M.K.; Russell, E.; Von Bargen, C.D.; et al. OPLS4: Improving Force Field Accuracy on Challenging Regimes of Chemical Space. J. Chem. Theory Comput. 2021, 17, 4291–4300. [Google Scholar] [CrossRef] [PubMed]
- Daura, X.; Gademann, K.; Jaun, B.; Seebach, D.; van Gunsteren, W.F.; Mark, A.E.; Rigault, A.; Siegel, J.; Harrowfield, J.; Chevrier, B.; et al. Peptide Folding: When Simulation Meets Experiment, Angew. Chem. Int. Ed. Engl. 1998, 31, 1387–1404. [Google Scholar] [CrossRef]
- Palazzolo, L.; Parravicini, C.; Laurenzi, T.; Guerrini, U.; Indiveri, C.; Gianazza, E.; Eberini, I. In silico Description of LAT1 Transport Mechanism at an Atomistic Level. Front. Chem. 2018, 6, 350. [Google Scholar] [CrossRef] [PubMed]
- Hogues, H.; Gaudreault, F.; Corbeil, C.R.; Deprez, C.; Sulea, T.; Purisima, E.O. ProPOSE: Direct Exhaustive Protein–Protein Docking with Side Chain Flexibility. J. Chem. Theory Comput. 2018, 14, 4938–4947. [Google Scholar] [CrossRef] [PubMed]
- Chemical Computing Group ULC. MOE: Molecular Operating Environment (MOE), 2020.02; Chemical Computing Group ULC: Montreal, QC, Canada, 2021. [Google Scholar]
- Kozakov, D.; Brenke, R.; Comeau, S.R.; Vajda, S. PIPER: An FFT-based protein docking program with pairwise potentials. Proteins 2006, 65, 392–406. [Google Scholar] [CrossRef] [PubMed]
- Laurenzi, T.; Parravicini, C.; Palazzolo, L.; Guerrini, U.; Gianazza, E.; Calabresi, L.; Eberini, I. rHDL modeling and the anchoring mechanism of LCAT activation. J. Lipid Res. 2021, 62, 100006. [Google Scholar] [CrossRef] [PubMed]
- Roe, D.R.; Cheatham, T.E. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pose Rank | Cluster Size (PIPER) | S Score [kcal/mol] (MOE) |
|---|---|---|
| 1 | 41 | −79.3 |
| 2 | 35 | −76.3 |
| 3 | 33 | −75.9 |
| 4 # | 26 | −75.4 |
| 5 * | 26 | −71.5 |
| 6 | 25 | −70.1 |
| 7 | 24 | −68.4 |
| 8 | 24 | −68.3 |
| 9 | 23 | −68.1 |
| 10 # | 21 | −67.8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Montavoci, L.; Ben Mariem, O.; Saporiti, S.; Laurenzi, T.; Palazzolo, L.; Ossoli, A.F.; Guerrini, U.; Calabresi, L.; Eberini, I. In Silico Description of the Direct Inhibition Mechanism of Endothelial Lipase by ANGPTL3. Int. J. Mol. Sci. 2024, 25, 3555. https://doi.org/10.3390/ijms25063555
Montavoci L, Ben Mariem O, Saporiti S, Laurenzi T, Palazzolo L, Ossoli AF, Guerrini U, Calabresi L, Eberini I. In Silico Description of the Direct Inhibition Mechanism of Endothelial Lipase by ANGPTL3. International Journal of Molecular Sciences. 2024; 25(6):3555. https://doi.org/10.3390/ijms25063555
Chicago/Turabian StyleMontavoci, Linda, Omar Ben Mariem, Simona Saporiti, Tommaso Laurenzi, Luca Palazzolo, Alice Federica Ossoli, Uliano Guerrini, Laura Calabresi, and Ivano Eberini. 2024. "In Silico Description of the Direct Inhibition Mechanism of Endothelial Lipase by ANGPTL3" International Journal of Molecular Sciences 25, no. 6: 3555. https://doi.org/10.3390/ijms25063555