
Co-Expression Network Analysis Unveiled lncRNA-mRNA Links Correlated to Epidermal Growth Factor Receptor-Tyrosine Kinase Inhibitor Resistance and/or Intermediate Epithelial-to-Mesenchymal Transition Phenotypes in a Human Non-Small Cell Lung Cancer Cellular Model System

Abstract
:1. Introduction
2. Results
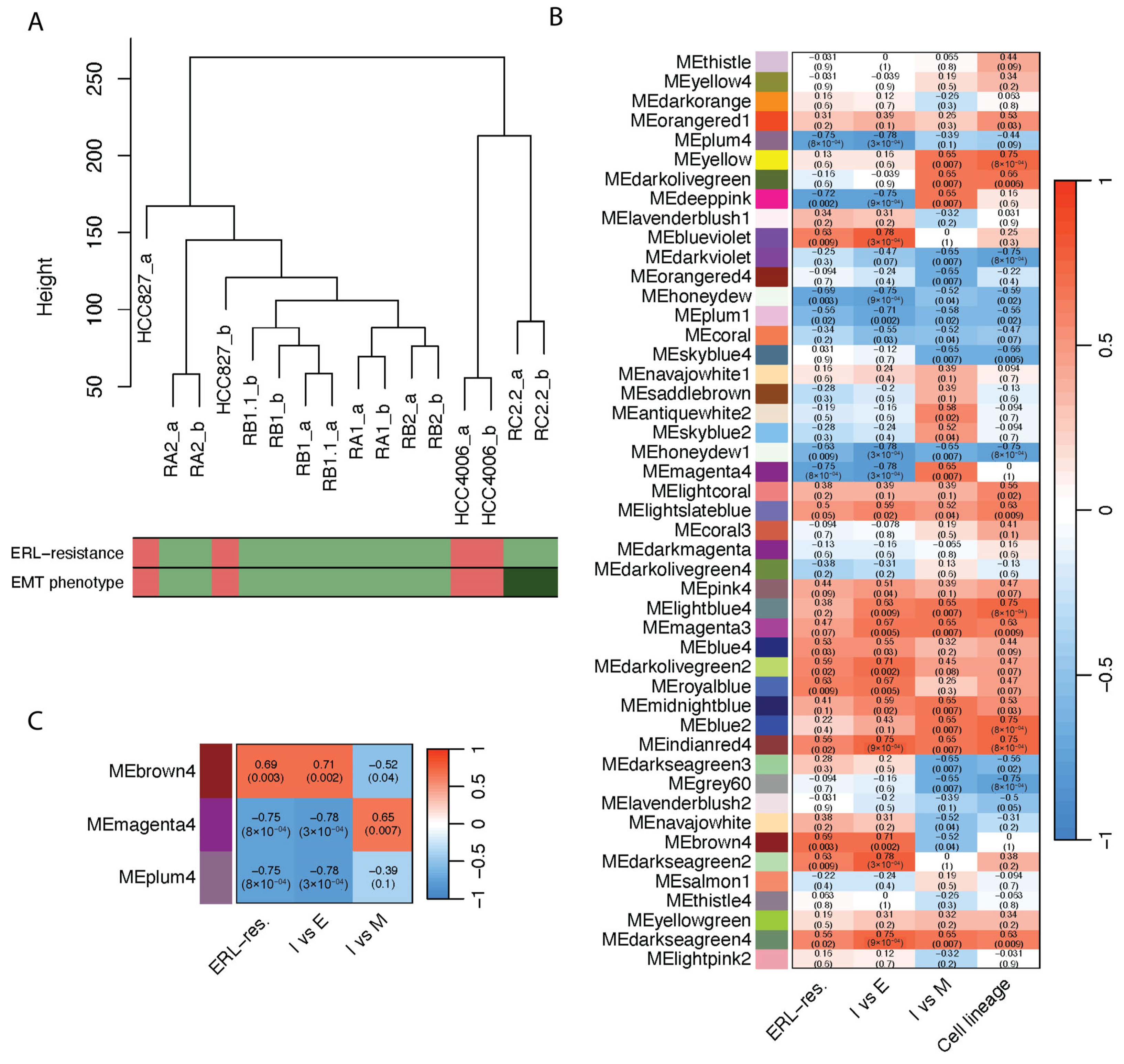
2.1. Construction of a Weighted Gene Co-Expression Network
2.2. Definition of Module–Trait Relationships and Detection of Key Modules
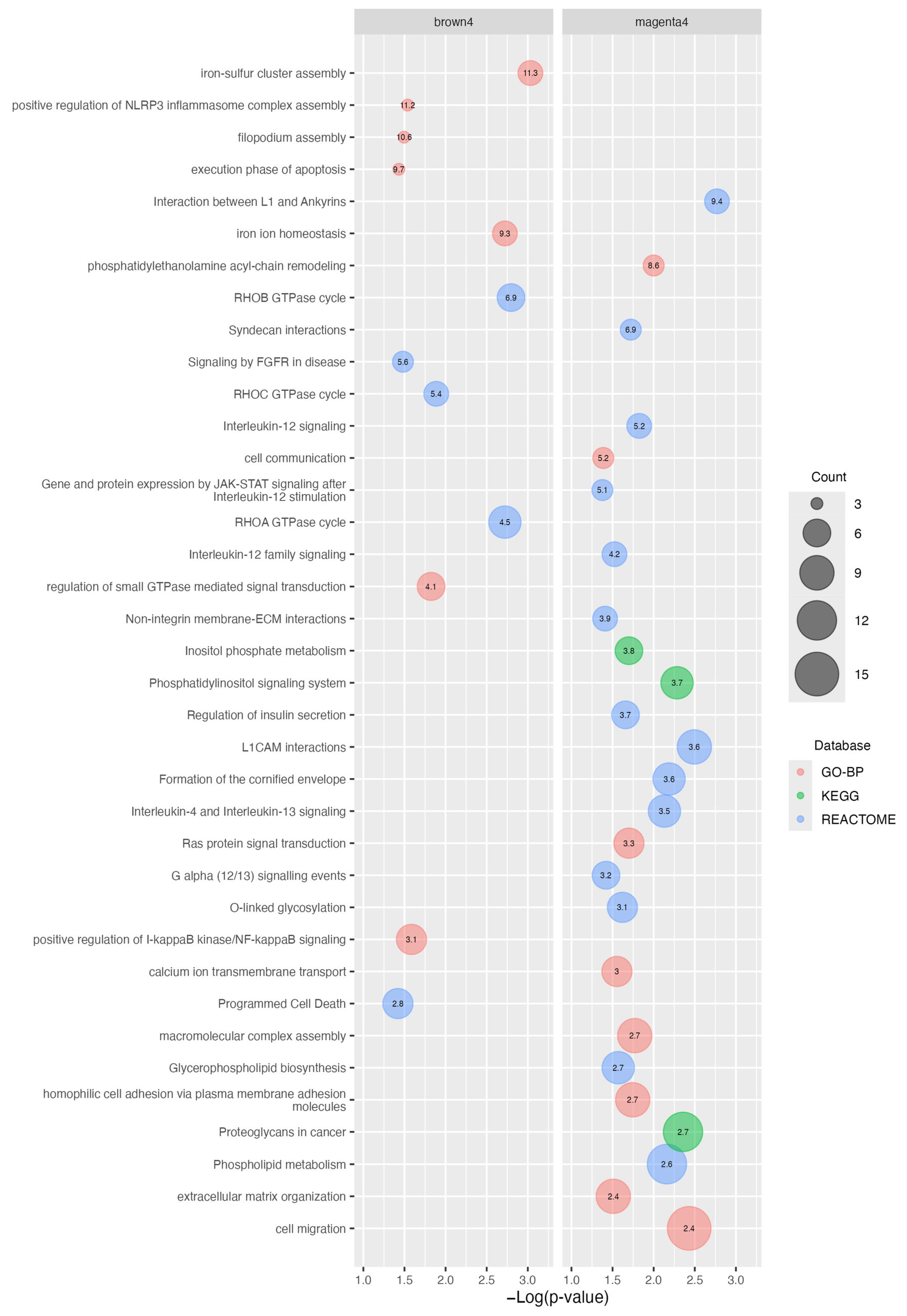
2.3. Functional Enrichment Analysis of the Brown4 and Magenta4 Modules
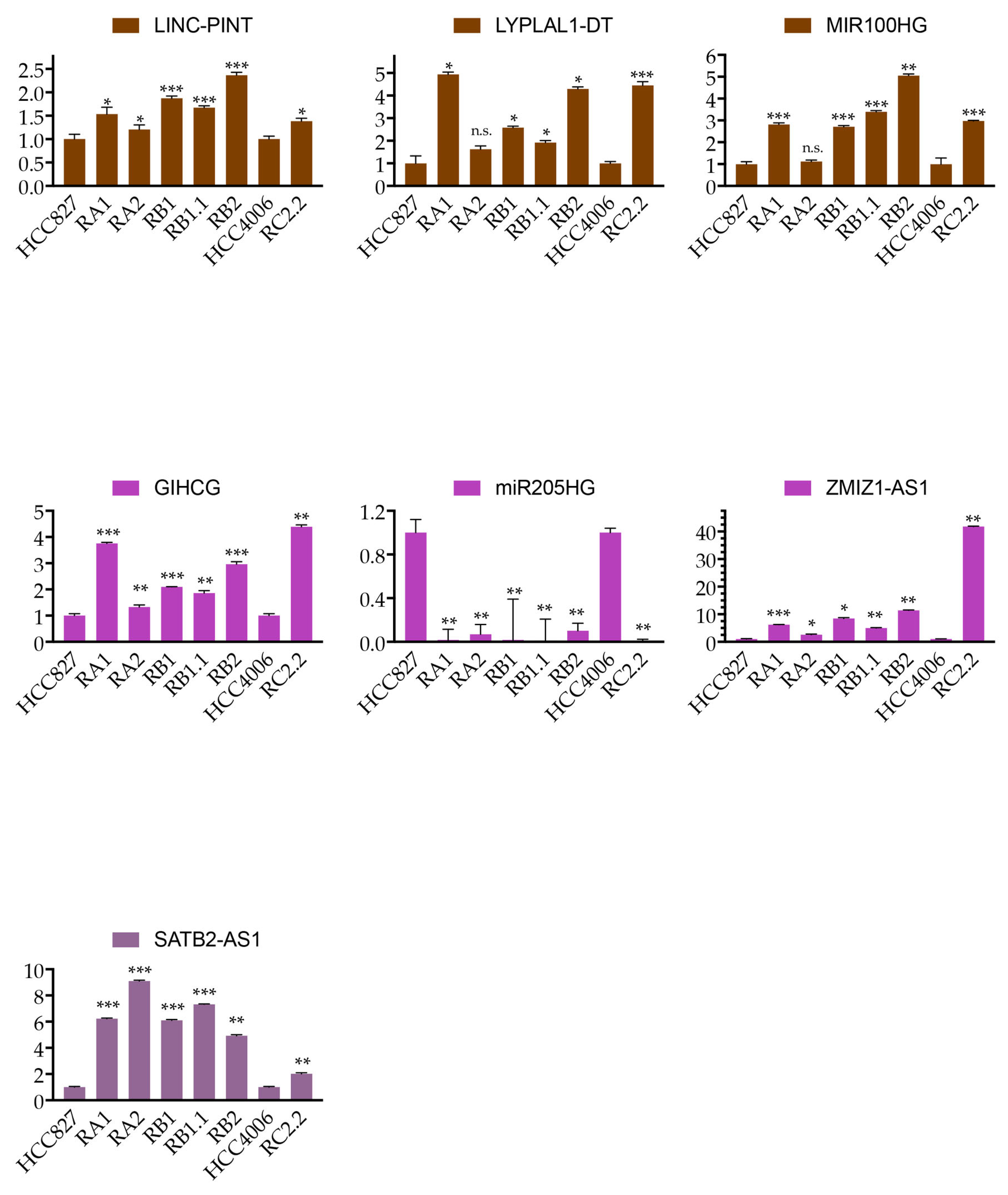
2.4. Selection of the Most Relevant LncRNAs
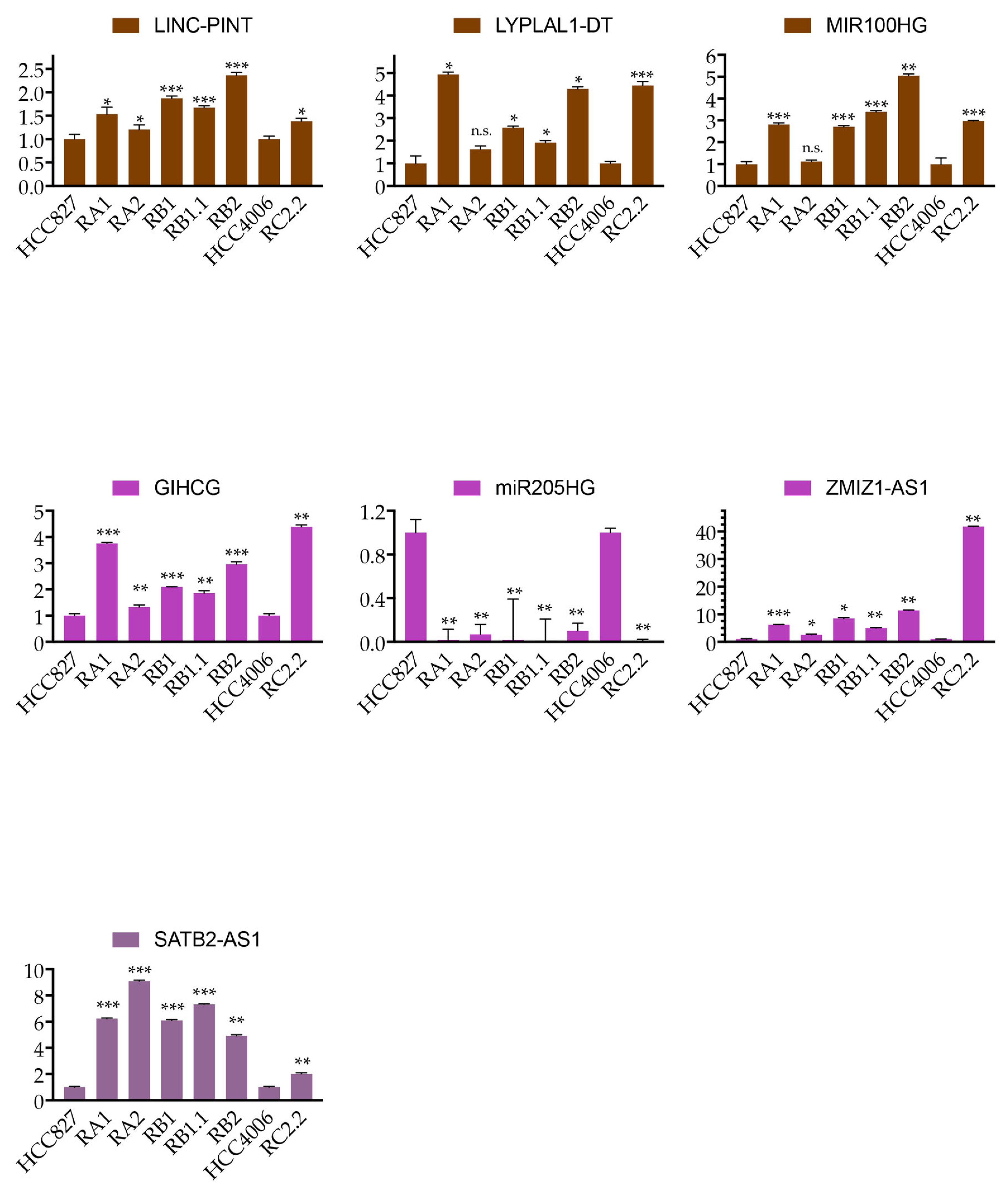
2.5. Expression Validation of Selected LncRNAs
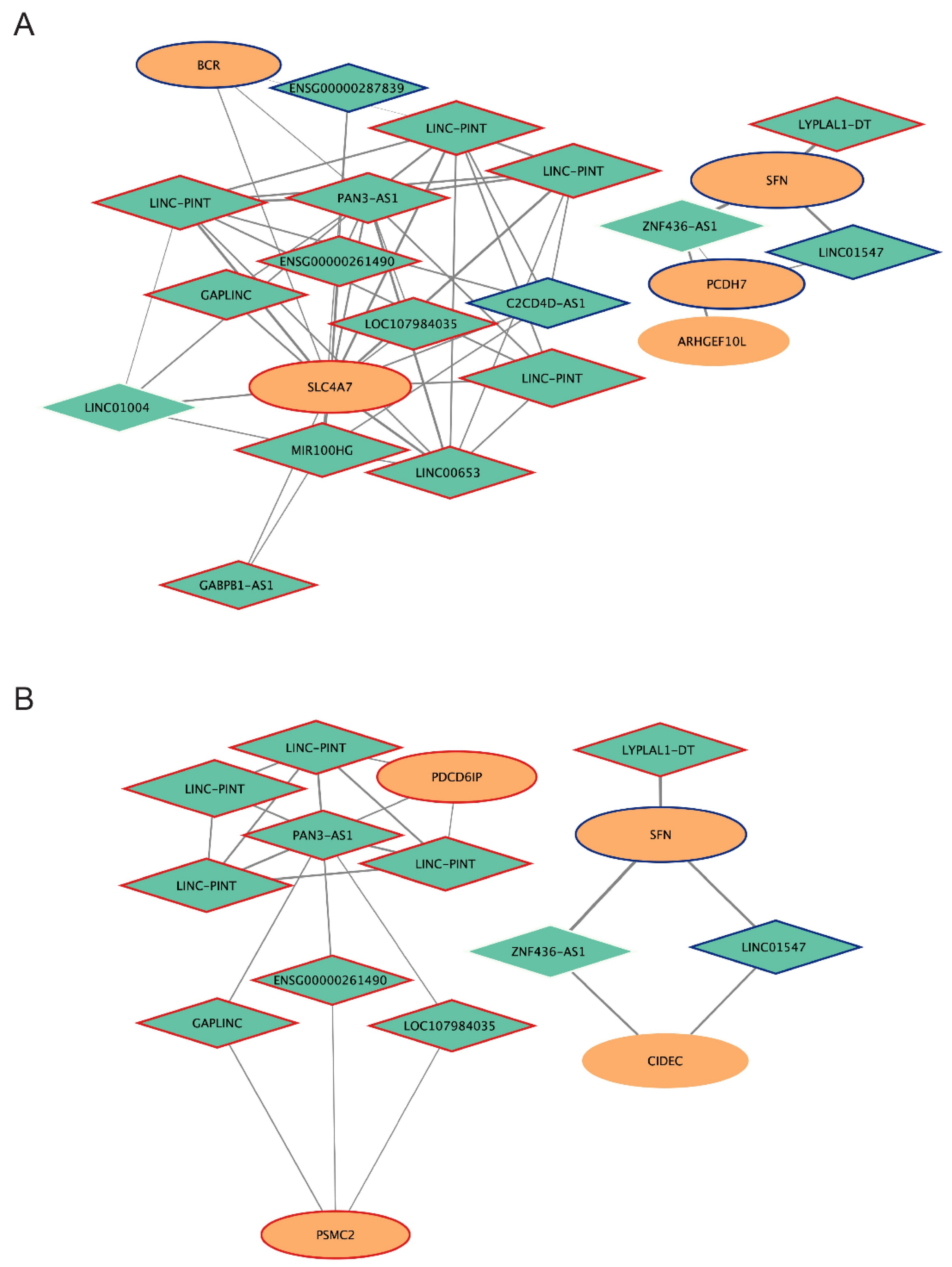
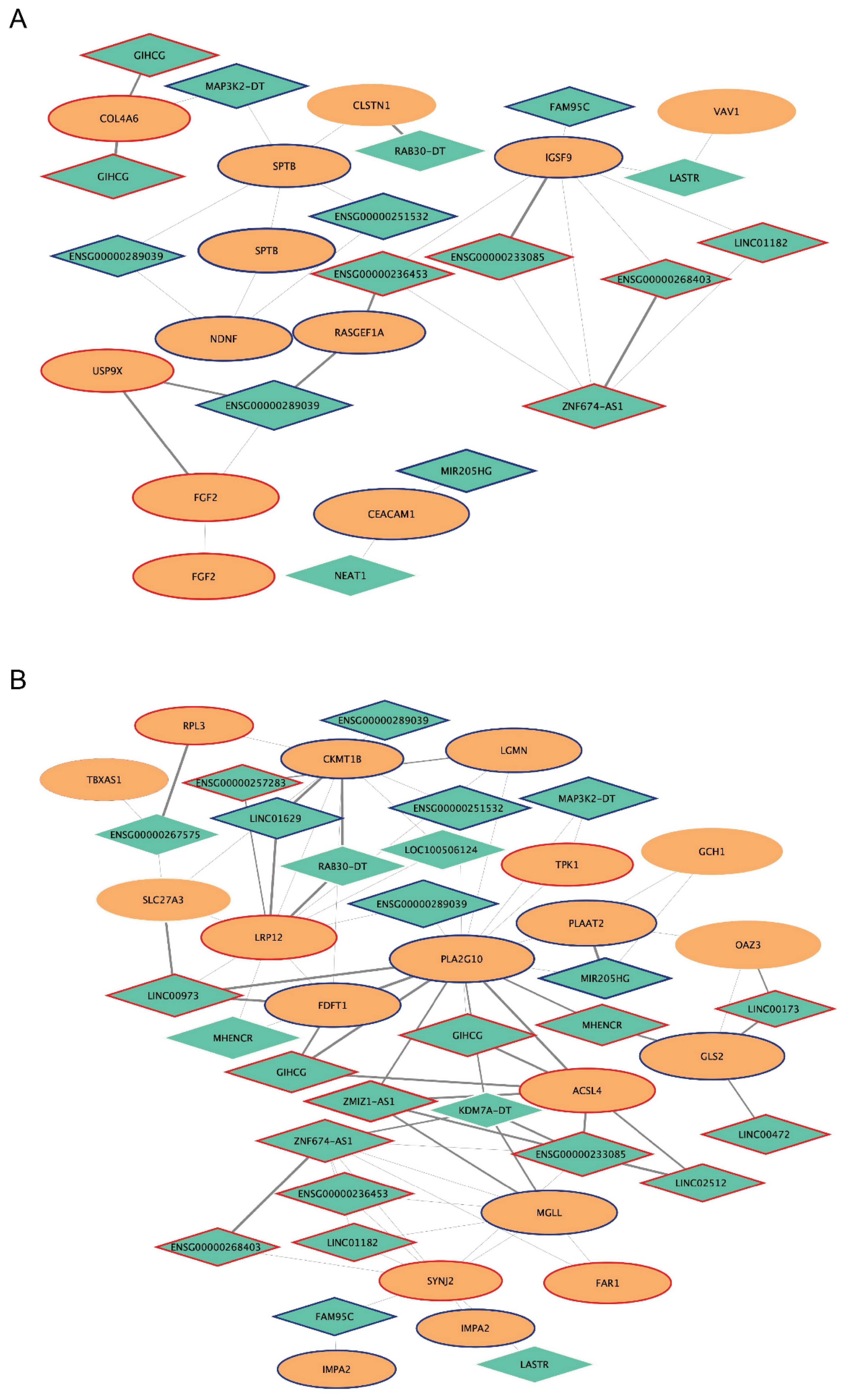
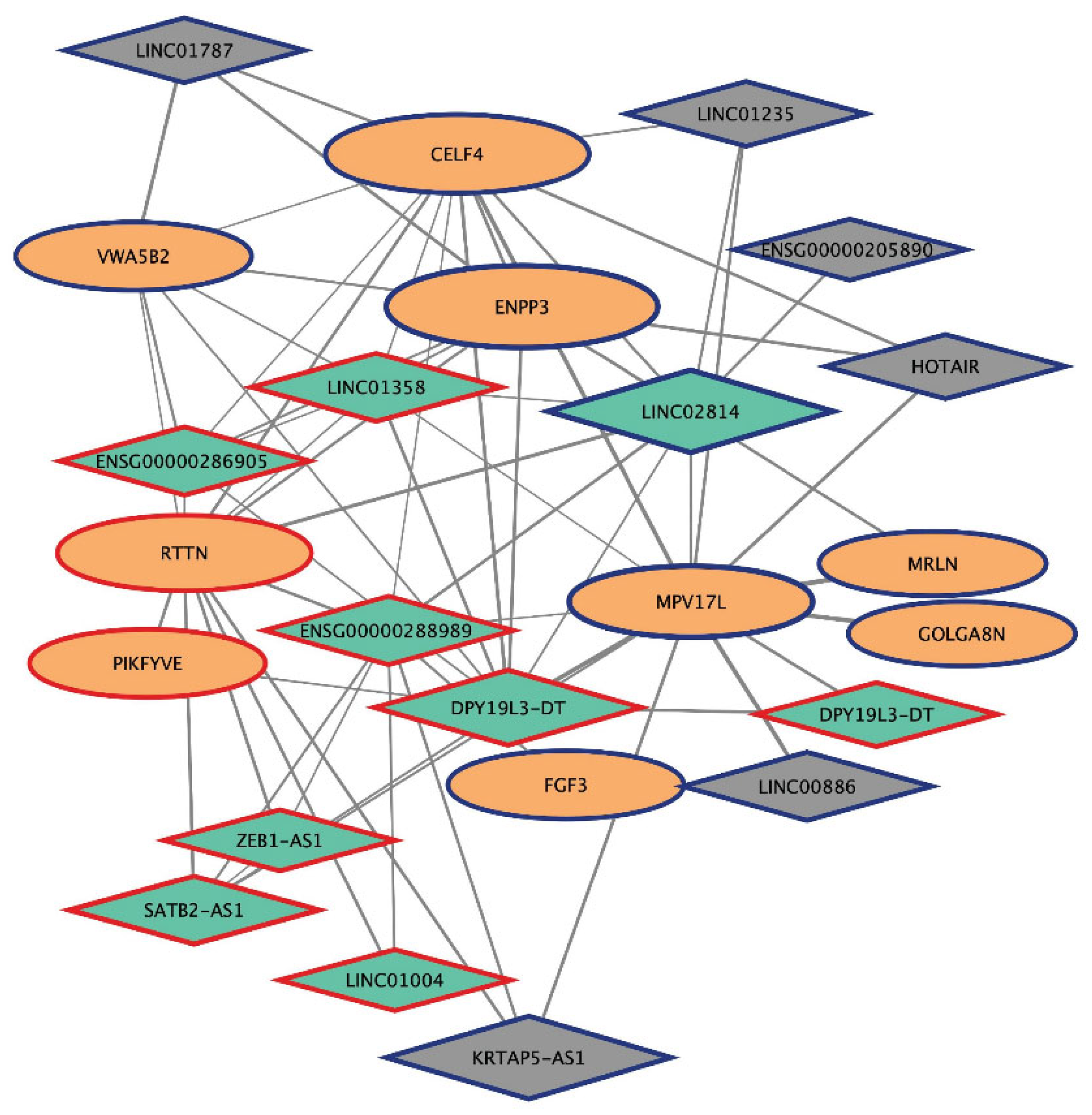
2.6. Analysis of mRNA-lncRNA Sub-Networks
3. Discussion
4. Material and Methods
4.1. Reagents
4.2. Cell Culture
4.3. RNA Extraction and Real-Time qPCR Analysis
4.4. Weighted Gene Co-Expression Network Analysis (WGCNA) and Module Identification
4.5. Modules and Probes Selection
4.6. Functional Annotations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef]
- Mattick, J.S.; Amaral, P.P.; Carninci, P.; Carpenter, S.; Chang, H.Y.; Chen, L.L.; Chen, R.; Dean, C.; Dinger, M.E.; Fitzgerald, K.A.; et al. Long non-coding RNAs: Definitions, functions, challenges and recommendations. Nat. Rev. Mol. Cell Biol. 2023, 24, 430–447. [Google Scholar] [CrossRef] [PubMed]
- Statello, L.; Guo, C.J.; Chen, L.L.; Huarte, M. Gene regulation by long non-coding RNAs and its biological functions. Nat. Rev. Mol. Cell Biol. 2021, 22, 96–118. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Lin, Z.; Li, X.; An, J.; Zheng, Q.; Yang, Y.; Lu, D. HULC cooperates with MALAT1 to aggravate liver cancer stem cells growth through telomere repeat-binding factor 2. Sci. Rep. 2016, 6, 36045. [Google Scholar] [CrossRef] [PubMed]
- Akerman, I.; Tu, Z.; Beucher, A.; Rolando, D.M.Y.; Sauty-Colace, C.; Benazra, M.; Nakic, N.; Yang, J.; Wang, H.; Pasquali, L. Human pancreatic β cell lncRNAs control cell-specific regulatory networks. Cell Metab. 2017, 25, 400–411. [Google Scholar] [CrossRef] [PubMed]
- Shao, T.; Xie, Y.; Shi, J.; Yang, C.; Zou, H.; Li, Y.; Xu, J.; Li, X. Surveying lncRNA-lncRNA cooperations reveals a dominant effect on tumor immunity cross cancers. Commun. Biol. 2022, 5, 1324. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Ma, H.; Chen, H.; Zhang, Z.; Xue, Q. LncRNA in tumorigenesis of non-small-cell lung cancer: From bench to bedside. Cell Death Discov. 2022, 8, 359. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.C.; Ni, J.J.; Cui, W.Y.; Wang, B.Y.; Zhuo, W. Emerging roles of lncRNA in cancer and therapeutic opportunities. Am. J. Cancer Res. 2019, 9, 1354–1366. [Google Scholar] [PubMed]
- Carlevaro-Fita, J.; Lanzós, A.; Feuerbach, L.; Hong, C.; Mas-Ponte, D.; Pedersen, J.S.; Abascal, F.; Amin, S.B.; Bader, G.D.; Barenboim, J.; et al. Cancer LncRNA Census reveals evidence for deep functional conservation of long noncoding RNAs in tumorigenesis. Commun. Biol. 2020, 3, 56. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Wang, R.; Chang, S.; Li, K.; Sun, R.; Wang, M.; Li, Z. Long Non-Coding RNA in Drug Resistance of Non-Small Cell Lung Cancer: A Mini Review. Front. Pharmacol. 2019, 10, 1457. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Li, H.; Hou, S.; Hu, B.; Liu, J.; Wang, J. The Noncoding RNA Expression Profile and the Effect of lncRNA AK126698 on Cisplatin Resistance in Non-Small-Cell Lung Cancer Cell. PLoS ONE 2013, 8, e65309. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Zhang, H.; Zhang, B.; Zhao, J.; Li, T.; Zhao, Y. Microarray Expression Profile of Long Non-Coding RNAs in Paclitaxel-Resistant Human Lung Adenocarcinoma Cells. Oncol. Rep. 2017, 38, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Cheng, N.; Li, X.; Zhao, C.; Ren, S.; Chen, X.; Cai, W.; Zhao, M.; Zhang, Y.; Li, J.; Wang, Q.; et al. Microarray Expression Profile of Long Non-Coding RNAs in EGFR-TKIs Resistance of Human Non-Small Cell Lung Cancer. Oncol. Rep. 2015, 33, 833–839. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Yu, D.-D.; Hu, Y.; Yan, D.; Chen, X.; Cao, H.-X.; Yu, S.-R.; Wang, Z.; Feng, J.-F. Genome-Wide Profiling of Long Non-Coding RNA Expression Patterns in the EGFR-TKI Resistance of Lung Adenocarcinoma by Microarray. Oncol. Rep. 2016, 35, 3371–3386. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Boggon, T.J.; Dayaram, T.; Jänne, P.A.; Kocher, O.; Meyerson, M.; Johnson, B.E.; Eck, M.J.; Tenen, D.G.; Halmos, B. EGFR Mutation and Resistance of Non-Small-Cell Lung Cancer to Gefitinib. N. Engl. J. Med. 2005, 352, 786–792. [Google Scholar] [CrossRef] [PubMed]
- Kosaka, T.; Yatabe, Y.; Endoh, H.; Yoshida, K.; Hida, T.; Tsuboi, M.; Tada, H.; Kuwano, H.; Mitsudomi, T. Analysis of Epidermal Growth Factor Receptor Gene Mutation in Patients with Non-Small Cell Lung Cancer and Acquired Resistance to Gefitinib. Clin. Cancer Res. 2006, 12, 5764–5769. [Google Scholar] [CrossRef] [PubMed]
- Passaro, A.; Mok, T.; Peters, S.; Popat, S.; Ahn, M.J.; de Marinis, F. Recent Advances on the Role of EGFR Tyrosine Kinase Inhibitors in the Management of NSCLC with Uncommon, Non Exon 20 Insertions, EGFR Mutations. J. Thorac. Oncol. 2021, 16, 764–773. [Google Scholar] [CrossRef] [PubMed]
- Jakobsen, K.R.; Demuth, C.; Sorensen, B.S.; Nielsen, A.L. The Role of Epithelial to Mesenchymal Transition in Resistance to Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors in Non-Small Cell Lung Cancer. Transl. Lung Cancer Res. 2016, 5, 172–182. [Google Scholar] [CrossRef] [PubMed]
- Dongre, A.; Weinberg, R.A. New Insights into the Mechanisms of Epithelial–Mesenchymal Transition and Implications for Cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The Basics of Epithelial-Mesenchymal Transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A. Epithelial Plasticity: A Common Theme in Embryonic and Cancer Cells. Science 2013, 342, 1234850. [Google Scholar] [CrossRef] [PubMed]
- Jolly, M.K.; Boareto, M.; Huang, B.; Jia, D.; Lu, M.; Ben-Jacob, E.; Onuchic, J.N.; Levine, H. Implications of the Hybrid Epithelial/Mesenchymal Phenotype in Metastasis. Front. Oncol. 2015, 5, 155. [Google Scholar] [CrossRef] [PubMed]
- Brabletz, S.; Schuhwerk, H.; Brabletz, T.; Stemmler, M.P. Dynamic EMT: A Multi-Tool for Tumor Progression. EMBO J. 2021, 40, e108647. [Google Scholar] [CrossRef] [PubMed]
- Jolly, M.K.; Mani, S.A.; Levine, H. Hybrid Epithelial/Mesenchymal Phenotype(s): The ‘Fittest’ for Metastasis? Biochim. Biophys. Acta-Rev. Cancer 2018, 1870, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Pastushenko, I.; Brisebarre, A.; Sifrim, A.; Fioramonti, M.; Revenco, T.; Boumahdi, S.; Van Keymeulen, A.; Brown, D.; Moers, V.; Lemaire, S.; et al. Identification of the Tumour Transition States Occurring during EMT. Nature 2018, 556, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, A.P.; Vasaikar, S.V.; Tomczak, K.; Tripathi, S.; den Hollander, P.; Arslan, E.; Chakraborty, P.; Soundararajan, R.; Jolly, M.K.; Rai, K.; et al. Identification of EMT Signaling Cross-Talk and Gene Regulatory Networks by Single-Cell RNA Sequencing. Proc. Natl. Acad. Sci. USA 2021, 118, e2102050118. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Guidelines and Definitions for Research on Epithelial–Mesenchymal Transition. Nat. Rev. Mol. Cell Biol. 2020, 21, 341–352. [Google Scholar] [CrossRef]
- Canciello, A.; Cerveró-Varona, A.; Peserico, A.; Mauro, A.; Russo, V.; Morrione, A.; Giordano, A.; Barboni, B. “In Medio Stat Virtus”: Insights into Hybrid E/M Phenotype Attitudes. Front. Cell Dev. Biol. 2022, 10, 1038841. [Google Scholar] [CrossRef] [PubMed]
- Fustaino, V.; Presutti, D.; Colombo, T.; Cardinali, B.; Papoff, G.; Brandi, R.; Bertolazzi, P.; Felici, G.; Ruberti, G. Characterization of Epithelial-Mesenchymal Transition Intermediate/Hybrid Phenotypes Associated to Resistance to EGFR Inhibitors in Non-Small Cell Lung Cancer Cell Lines. Oncotarget 2017, 8, 103340–103363. [Google Scholar] [CrossRef] [PubMed]
- Malagoli Tagliazucchi, G.; Wiecek, A.J.; Withnell, E.; Secrier, M. Genomic and Microenvironmental Heterogeneity Shaping Epithelial-to-Mesenchymal Trajectories in Cancer. Nat. Commun. 2023, 14, 789. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, S.; Xing, J.; Levine, H.; Jolly, M.K. Mathematical Modeling of Plasticity and Heterogeneity in EMT. Methods Mol. Biol. 2021, 2179, 385–413. [Google Scholar] [CrossRef] [PubMed]
- Khanbabaei, H.; Ebrahimi, S.; García-Rodríguez, J.L.; Ghasemi, Z.; Pourghadamyari, H.; Mohammadi, M.; Kristensen, L.S. Non-Coding RNAs and Epithelial Mesenchymal Transition in Cancer: Molecular Mechanisms and Clinical Implications. J. Exp. Clin. Cancer Res. 2022, 41, 278. [Google Scholar] [CrossRef] [PubMed]
- Gugnoni, M.; Ciarrocchi, A. Long Noncoding RNA and Epithelial Mesenchymal Transition in Cancer. Int. J. Mol. Sci. 2019, 20, 1924. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R Package for Weighted Correlation Network Analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Derrien, T.; Johnson, R.; Bussotti, G.; Tanzer, A.; Djebali, S.; Tilgner, H.; Guernec, G.; Martin, D.; Merkel, A.; Knowles, D.G.; et al. The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Res. 2012, 22, 1775–1789. [Google Scholar] [CrossRef] [PubMed]
- Presutti, D.; Santini, S.; Cardinali, B.; Papoff, G.; Lalli, C.; Samperna, S.; Fustaino, V.; Giannini, G.; Ruberti, G. MET Gene Amplification and MET Receptor Activation Are not Sufficient to Predict Efficacy of Combined MET and EGFR Inhibitors in EGFR TKI-Resistant NSCLC Cells. PLoS ONE 2015, 10, e0143333. [Google Scholar] [CrossRef] [PubMed]
- Svensmark, J.H.; Brakebusch, C. Rho GTPases in Cancer: Friend or Foe? Oncogene 2019, 38, 7447–7456. [Google Scholar] [CrossRef] [PubMed]
- Phuyal, S.; Farhan, H. Multifaceted Rho GTPase Signaling at the Endomembranes. Front. Cell Dev. Biol. 2019, 7, 127. [Google Scholar] [CrossRef] [PubMed]
- Ghafouri-Fard, S.; Noroozi, R.; Abak, A.; Taheri, M.; Salimi, A. Emerging Role of lncRNAs in the Regulation of Rho GTPase Pathway. Biomed. Pharmacother. 2021, 140, 111731. [Google Scholar] [CrossRef] [PubMed]
- Saliani, M.; Mirzaiebadizi, A.; Mosaddeghzadeh, N.; Ahmadian, M.R. RHO GTPase-Related Long Noncoding RNAs in Human Cancers. Cancers 2021, 13, 5386. [Google Scholar] [CrossRef] [PubMed]
- Cardone, R.A.; Casavola, V.; Reshkin, S.J. The Role of Disturbed pH Dynamics and the Na+/H+ Exchanger in Metastasis. Nat. Rev. Cancer 2005, 5, 786–795. [Google Scholar] [CrossRef] [PubMed]
- Webb, B.A.; Chimenti, M.; Jacobson, M.P.; Barber, D.L. Dysregulated pH: A Perfect Storm for Cancer Progression. Nat. Rev. Cancer 2011, 11, 671–677. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Zhou, Y.; Skaro, M.F.; Wu, Y.; Qu, Z.; Mao, F.; Zhao, S.; Xu, Y. Metabolic Reprogramming in Cancer Is Induced to Increase Proton Production. Cancer Res. 2020, 80, 1143–1155. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Chang, W.; Lu, X.; Wang, J.; Zhang, C.; Xu, Y. Acid-Base Homeostasis and Implications to the Phenotypic Behaviors of Cancer. Genom. Proteom. Bioinform. 2022. ahead of print. [Google Scholar] [CrossRef]
- Liu, Y.; White, K.A.; Barber, D.L. Intracellular pH Regulates Cancer and Stem Cell Behaviors: A Protein Dynamics Perspective. Front. Oncol. 2020, 10, 1401. [Google Scholar] [CrossRef] [PubMed]
- Gorbatenko, A.; Olesen, C.; Boedtkjer, E.; Pedersen, S. Regulation and Roles of Bicarbonate Transport in Cancer. Front. Physiol. 2014, 5, 130. [Google Scholar] [CrossRef] [PubMed]
- Ali, E.S.; Lipońska, A.; O’Hara, B.P.; Amici, D.R.; Torno, M.D.; Gao, P.; Asara, J.M.; Yap, M.N.F.; Mendillo, M.L.; Ben-Sahra, I. The mTORC1-SLC4A7 Axis Stimulates Bicarbonate Import to Enhance de Novo Nucleotide Synthesis. Mol. Cell 2022, 82, 3284–3298.e7. [Google Scholar] [CrossRef] [PubMed]
- Koe, J.C.; Hewton, K.G.; Parker, S.J. SLC4A7 and mTORC1 Raise Nucleotide Synthesis with Bicarbonate. Mol. Cell 2022, 82, 3121–3123. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Li, G.; Liu, Z.; Ma, H.; Yuan, W.; Lu, Z.; Zhang, D.; Ling, H.; Zhang, F.; Liu, Y.; et al. Bicarbonate transporter SLC4A7 promotes EMT and metastasis of HNSCC by activating the PI3K/AKT/mTOR signaling pathway. Mol. Carcinog. 2023, 62, 628–640. [Google Scholar] [CrossRef]
- Toft, N.J.; Axelsen, T.V.; Pedersen, H.L.; Mele, M.; Burton, M.; Balling, E.; Johansen, T.; Thomassen, M.; Christiansen, P.M.; Boedtkjer, E. Acid-Base Transporters and pH Dynamics in Human Breast Carcinomas Predict Proliferative Activity, Metastasis, and Survival. eLife 2021, 10, e68447. [Google Scholar] [CrossRef] [PubMed]
- Chuang, T.H.; Xu, X.; Kaartinen, V.; Heisterkamp, N.; Groffen, J.; Bokoch, G.M. Abr and Bcr are multifunctional regulators of the Rho GTP-binding protein family. Proc. Natl. Acad. Sci. USA 1995, 92, 10282–10286. [Google Scholar] [CrossRef] [PubMed]
- Diekmann, D.; Brill, S.; Garrett, M.D.; Totty, N.; Hsuan, J.; Monfries, C.; Hall, C.; Lim, L.; Hall, A. Bcr encodes a GTPase-activating protein for p21rac. Nature 1991, 351, 400–402. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.J.; Cunnick, J.M.; Yi, S.J.; Kaartinen, V.; Groffen, J.; Heisterkamp, N. Abr and Bcr, two homologous Rac GTPase-activating proteins control multiple cellular functions of murine macrophages. Mol. Cell. Biol. 2007, 27, 899–911. [Google Scholar] [CrossRef] [PubMed]
- Dubash, A.D.; Koetsier, J.L.; Amargo, E.V.; Najor, N.A.; Harmon, R.M.; Green, K.J. The GEF Bcr activates RhoA/MAL signaling to promote keratinocyte differentiation via desmoglein-1. J. Cell Biol. 2013, 202, 653–666. [Google Scholar] [CrossRef] [PubMed]
- Olson, M.F.; Sahai, E. The actin cytoskeleton in cancer cell motility. Clin. Exp. Metastasis 2009, 26, 273–287. [Google Scholar] [CrossRef] [PubMed]
- Lawson, C.D.; Ridley, A.J. Rho GTPase signaling complexes in cell migration and invasion. J. Cell Biol. 2018, 217, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Radu, M.; Semenova, G.; Kosoff, R.; Chernoff, J. PAK signalling during the development and progression of cancer. Nat. Rev. Cancer 2014, 14, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Bustelo, X.R. RHO GTPases in cancer: Known facts, open questions, and therapeutic challenges. Biochem. Soc. Trans. 2018, 46, 741–760. [Google Scholar] [CrossRef] [PubMed]
- Cooke, M.; Kreider-Letterman, G.; Baker, M.J.; Zhang, S.; Sullivan, N.T.; Eruslanov, E.; Abba, M.C.; Goicoechea, S.M.; García-Mata, R.; Kazanietz, M.G. FARP1, ARHGEF39, and TIAM2 are essential receptor tyrosine kinase effectors for Rac1-dependent cell motility in human lung adenocarcinoma. Cell Rep. 2021, 37, 109905. [Google Scholar] [CrossRef]
- Bukhari, I.; Khan, M.R.; Li, F.; Swiatczak, B.; Thorne, R.F.; Zheng, P.; Mi, Y. Clinical Implications of lncRNA LINC-PINT in Cancer. Front. Mol. Biosci. 2023, 10, 1097694. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Lv, J.; Zhang, X.; Zhang, Q.; Li, Z.; Lu, J.; Huo, X.; Guo, M.; Liu, X.; Gao, R.; et al. ELAVL4 Promotes the Tumorigenesis of Small Cell Lung Cancer by Stabilizing LncRNA LYPLAL1-DT and Enhancing Profilin 2 Activation. FASEB J. 2023, 37, e23170. [Google Scholar] [CrossRef] [PubMed]
- Ghafouri-Fard, S.; Harsij, A.; Farahzadi, H.; Hussen, B.M.; Taheri, M.; Mokhtari, M. A concise review on the role of MIR100HG in human disorders. J. Cell. Mol. Med. 2023, 27, 2278–2289. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Wang, Z.; Yu, S.; Liu, D.; Sun, L. LncmiRHG-MIR100HG: A New Budding Star in Cancer. Front. Oncol. 2022, 12, 997532. [Google Scholar] [CrossRef] [PubMed]
- Sui, C.J.; Zhou, Y.M.; Shen, W.F.; Dai, B.H.; Lu, J.J.; Zhang, M.F.; Yang, J.M. Long noncoding RNA GIHCG promotes hepatocellular carcinoma progression through epigenetically regulating miR-200b/a/429. J. Mol. Med. 2016, 94, 1281–1296. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Mao, L.; Li, L.; He, Z.; Wang, N.; Song, Y. Long noncoding RNA GIHCG functions as an oncogene and serves as a serum diagnostic biomarker for cervical cancer. J. Cancer 2019, 10, 672–681. [Google Scholar] [CrossRef] [PubMed]
- He, Z.H.; Qin, X.H.; Zhang, X.L.; Yi, J.W.; Han, J.Y. Long noncoding RNA GIHCG is a potential diagnostic and prognostic biomarker and therapeutic target for renal cell carcinoma. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Nath, A.; Lau, E.Y.T.; Lee, A.M.; Geeleher, P.; Cho, W.C.S.; Huang, R.S. Discovering long noncoding RNA predictors of anticancer drug sensitivity beyond protein-coding genes. Proc. Natl. Acad. Sci. USA 2019, 116, 22020–22029. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, Y.; Yang, B.; Cao, H.; Yang, C.X.; Ouyang, W.; Zhang, S.M.; Yang, G.F.; Zhou, F.X.; Zhou, Y.F.; et al. Elevated expression of USP9X correlates with poor prognosis in human non-small cell lung cancer. J. Thorac. Dis. 2015, 7, 672–679. [Google Scholar] [CrossRef] [PubMed]
- Pardali, K.; Moustakas, A. Actions of TGF-β as tumor suppressor and pro-metastatic factor in human cancer. Biochim. Biophys. Acta 2007, 1775, 21–62. [Google Scholar] [CrossRef] [PubMed]
- Berger, W.; Setinek, U.; Mohr, T.; Kindas-Mugge, I.; Vetterlein, M.; Dekan, G.; Eckersberger, F.; Caldas, C.; Micksche, M. Evidence for a role of FGF-2 and FGF receptors in the proliferation of non–small cell lung cancer cells. Int. J. Cancer 1999, 83, 415–423. [Google Scholar] [CrossRef]
- Marek, L.; Ware, K.E.; Fritzsche, A.; Hercule, P.; Helton, W.R.; Smith, J.E.; McDermott, L.A.; Coldren, C.D.; Nemenoff, R.A.; Merrick, D.T.; et al. Fibroblast growth factor (FGF) and FGF receptor-mediated autocrine signaling in non–small-cell lung cancer cells. Mol. Pharmacol. 2009, 75, 196–207. [Google Scholar] [CrossRef] [PubMed]
- Terai, H.; Soejima, K.; Yasuda, H.; Nakayama, S.; Hamamoto, J.; Arai, D.; Ishioka, K.; Ohgino, K.; Ikemura, S.; Sato, T.; et al. Activation of the FGF2-FGFR1 Autocrine Pathway: A Novel Mechanism of Acquired Resistance to Gefitinib in NSCLC. Mol. Cancer Res. 2013, 11, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Jing, C.; Cao, H.; Wu, J.; Ma, R.; Zhang, J.; Liu, S.; Wang, Z. Long noncoding RNA ZMIZ1-AS1 promotes gefitinib resistance via binding to hnRNPA1. Precis. Med. Sci. 2021, 10, 133–137. [Google Scholar] [CrossRef]
- Martínez-Reyes, I.; Chandel, N.S. Cancer metabolism: Looking forward. Nat. Rev. Cancer 2021, 21, 669–680. [Google Scholar] [CrossRef] [PubMed]
- Sciacovelli, M.; Frezza, C. Metabolic reprogramming and epithelial-to-mesenchymal transition in cancer. FEBS J. 2017, 284, 3132–3144. [Google Scholar] [CrossRef] [PubMed]
- Fedele, M.; Sgarra, R.; Battista, S.; Cerchia, L.; Manfioletti, G. The Epithelial-Mesenchymal Transition at the Crossroads between Metabolism and Tumor Progression. Int. J. Mol. Sci. 2022, 23, 800. [Google Scholar] [CrossRef] [PubMed]
- Eltayeb, K.; La Monica, S.; Tiseo, M.; Alfieri, R.; Fumarola, C. Reprogramming of Lipid Metabolism in Lung Cancer: An Overview with Focus on EGFR-Mutated Non-Small Cell Lung Cancer. Cells 2022, 11, 413. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Qiu, M.; Xing, X.; Zhou, J.; Yao, H.; Li, M.; Yin, R.; Hou, Y.; Li, Y.; Pan, S.; et al. Lung cancer scRNA-seq and lipidomics reveal aberrant lipid metabolism for early-stage diagnosis. Sci. Transl. Med. 2022, 14, eabk2756. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhu, T.; Wang, X.; Xiong, F.; Hu, Z.; Qiao, X.; Yuan, X.; Wang, D. ACSL3 and ACSL4, Distinct Roles in Ferroptosis and Cancers. Cancers 2022, 14, 5896. [Google Scholar] [CrossRef] [PubMed]
- Ha, N.T.; Lee, C.H. Roles of Farnesyl-Diphosphate Farnesyltransferase 1 in Tumour and Tumour Microenvironments. Cells 2020, 9, 2352. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Li, W. Monoacylglycerol lipase inhibitors: Modulators for lipid metabolism in cancer malignancy, neurological and metabolic disorders. Acta Pharm. Sin. B 2020, 10, 582–602. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Wang, X.; Curtiss, C.; Landas, S.; Rong, R.; Sheikh, M.S.; Huang, Y. Monoglyceride lipase gene knockout in mice leads to increased incidence of lung adenocarcinoma. Cell Death Dis. 2018, 9, 36. [Google Scholar] [CrossRef]
- Buczkowska, J.; Szeliga, M. Two Faces of Glutaminase GLS2 in Carcinogenesis. Cancers 2023, 15, 5566. [Google Scholar] [CrossRef]
- Ramirez-Peña, E.; Arnold, J.; Shivakumar, V.; Joseph, R.; Vidhya Vijay, G.; den Hollander, P.; Bhangre, N.; Allegakoen, P.; Prasad, R.; Conley, Z.; et al. The Epithelial to Mesenchymal Transition Promotes Glutamine Independence by Suppressing GLS2 Expression. Cancers 2019, 11, 1610. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.H.; Zhu, J.W.; Xu, H.H.; Zhang, G.Q.; Wang, Y.; Liu, Y.M.; Liang, J.B.; Wang, Y.X.; Wu, Y.; Guo, Q.F. A novel antisense long non-coding RNA SATB2-AS1 overexpresses in osteosarcoma and increases cell proliferation and growth. Mol. Cell. Biochem. 2017, 430, 47–56. [Google Scholar] [CrossRef]
- Wu, G.; Wang, Q.; Zhu, T.; Fu, L.; Li, Z.; Wu, Y.; Zhang, C. Identification and Validation of Immune-Related LncRNA Prognostic Signature for Lung Adenocarcinoma. Front. Genet. 2021, 12, 681277. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Xu, X.; Pan, B.; Chen, X.; Lin, K.; Zeng, K.; Liu, X.; Xu, T.; Sun, L.; Qin, J.; et al. LncRNA SATB2-AS1 inhibits tumor metastasis and affects the tumor immune cell microenvironment in colorectal cancer by regulating SATB2. Mol. Cancer 2019, 18, 135. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. Fast R Functions for Robust Correlations and Hierarchical Clustering. J. Stat. Softw. 2012, 46, i11. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Horvath, S. A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol. 2005, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Huang, d.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Biotype | Cytoband Location | Module |
|---|---|---|---|
| LINC-PINT | lincRNA | 7q32.3 | brown4 |
| MIOS-DT | divergent lncRNA | 7p21.3 | |
| LYPLAL1-DT | divergent lncRNA | 1q41 | |
| LINC01547 | lincRNA | 21q22.3 | |
| MIR100HG | miRNA host gene | 11q24.1 | |
| ZNF436-AS1 | antisense lncRNA | p36.12 | |
| LOC107984035 | uncharacterized | 9p11.2 | |
| LINC00653 | lincRNA | 20p11.23 | |
| LHX1-DT | divergent lncRNA | 17q12 | |
| GAPLINC | lincRNA | 18p11.31 | |
| PAN3-AS1 | antisense lncRNA | 13q12.2 | |
| LINC01004 | lincRNA | 7q22.3 | |
| ENSG00000261490 | uncharacterized | 4p16.1 | |
| ENSG00000232850 | antisense lncRNA | 9q34.11 | |
| C2CD4D-AS1 | antisense lncRNA | 1q21.3 | |
| NFE2L1-DT | divergent lncRNA | 17q21.32 | |
| MIR4435-2HG | miRNA host gene | 2q13 | |
| ENSG00000234141 | lncRNA | 7p21.3 | |
| ENSG00000287839 | uncharacterized | 1q22 | |
| GABPB1-AS1 | antisense lncRNA | 15q21.2 | |
| ENSG00000289039 | uncharacterized | 10q26.3 | magenta4 |
| MHENCR | lincRNA | 20q13.33 | |
| ENSG00000233085 | lincRNA | 6q27 | |
| ENSG00000251532 | lincRNA | 5p15.33 | |
| ENSG00000236453 | lincRNA | 7q21.3 | |
| SPART-AS1 | antisense lncRNA | 13q13.3 | |
| LINC00973 | lincRNA | 3q12.1 | |
| MAP3K2-DT | divergent lncRNA | 2q14.3 | |
| ZNF674-AS1 | antisense lncRNA | Xp11.3 | |
| GIHCG | lncRNA | 12q14.1 | |
| MIR205HG | miRNA host gene | 1q32.2 | |
| ZMIZ1-AS1 | antisense lncRNA | 10q22.3 | |
| NEAT1 | lincRNA | 11q13.1 | |
| RAB30-DT | divergent lncRNA | 11q14.1 | |
| LINC01629 | lincRNA | 14q24.3 | |
| LOC100506124 | lncRNA | 2q24.3 | |
| ENSG00000267575 | lincRNA | 19q11 | |
| LINC00472 | lincRNA | 6q13 | |
| FAM95C | lncRNA | 9p13.1 | |
| LINC02512 | lincRNA | 4q33 | |
| LINC01182 | lincRNA | 4p15.33 | |
| LASTR | lincRNA | 10p15.1 | |
| ENSG00000268403 | antisense lncRNA | 11p15.4 | |
| ENSG00000257283 | antisense lncRNA | 12q22 | |
| KDM7A-DT | divergent LncRNA | 7q34 | |
| LINC00173 | lincRNA | 12q24.22 |
| Gene Name | Biotype | Cytoband Location | Module |
|---|---|---|---|
| LINC02814 | lincRNA | 1q42.13 | plum4 |
| DPY19L3-DT | divergent lncRNA | 19q13.11 | |
| ENSG00000288989 | uncharacterized | 9q34.13 | |
| SATB2-AS1 | antisense lncRNA | 2q33.1 | |
| LINC01358 | lincRNA | 1p32.1 | |
| ENSG00000286905 | uncharacterized | 2p25.3 | |
| ZEB1-AS1 | antisense lncRNA | 10p11.22 | |
| LINC01004 | lincRNA | 7q22.3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fustaino, V.; Papoff, G.; Ruberti, F.; Ruberti, G. Co-Expression Network Analysis Unveiled lncRNA-mRNA Links Correlated to Epidermal Growth Factor Receptor-Tyrosine Kinase Inhibitor Resistance and/or Intermediate Epithelial-to-Mesenchymal Transition Phenotypes in a Human Non-Small Cell Lung Cancer Cellular Model System. Int. J. Mol. Sci. 2024, 25, 3863. https://doi.org/10.3390/ijms25073863
Fustaino V, Papoff G, Ruberti F, Ruberti G. Co-Expression Network Analysis Unveiled lncRNA-mRNA Links Correlated to Epidermal Growth Factor Receptor-Tyrosine Kinase Inhibitor Resistance and/or Intermediate Epithelial-to-Mesenchymal Transition Phenotypes in a Human Non-Small Cell Lung Cancer Cellular Model System. International Journal of Molecular Sciences. 2024; 25(7):3863. https://doi.org/10.3390/ijms25073863
Chicago/Turabian StyleFustaino, Valentina, Giuliana Papoff, Francesca Ruberti, and Giovina Ruberti. 2024. "Co-Expression Network Analysis Unveiled lncRNA-mRNA Links Correlated to Epidermal Growth Factor Receptor-Tyrosine Kinase Inhibitor Resistance and/or Intermediate Epithelial-to-Mesenchymal Transition Phenotypes in a Human Non-Small Cell Lung Cancer Cellular Model System" International Journal of Molecular Sciences 25, no. 7: 3863. https://doi.org/10.3390/ijms25073863