
Therapeutic Potential of Regorafenib in Cisplatin-Resistant Bladder Cancer with High Epithelial–Mesenchymal Transition and Stemness Properties
 , and
, and
Abstract
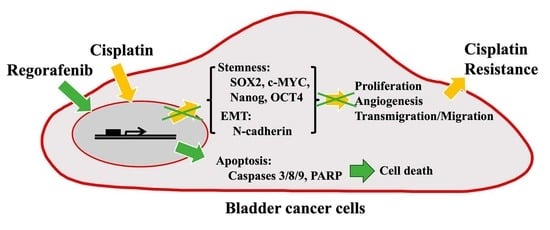
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
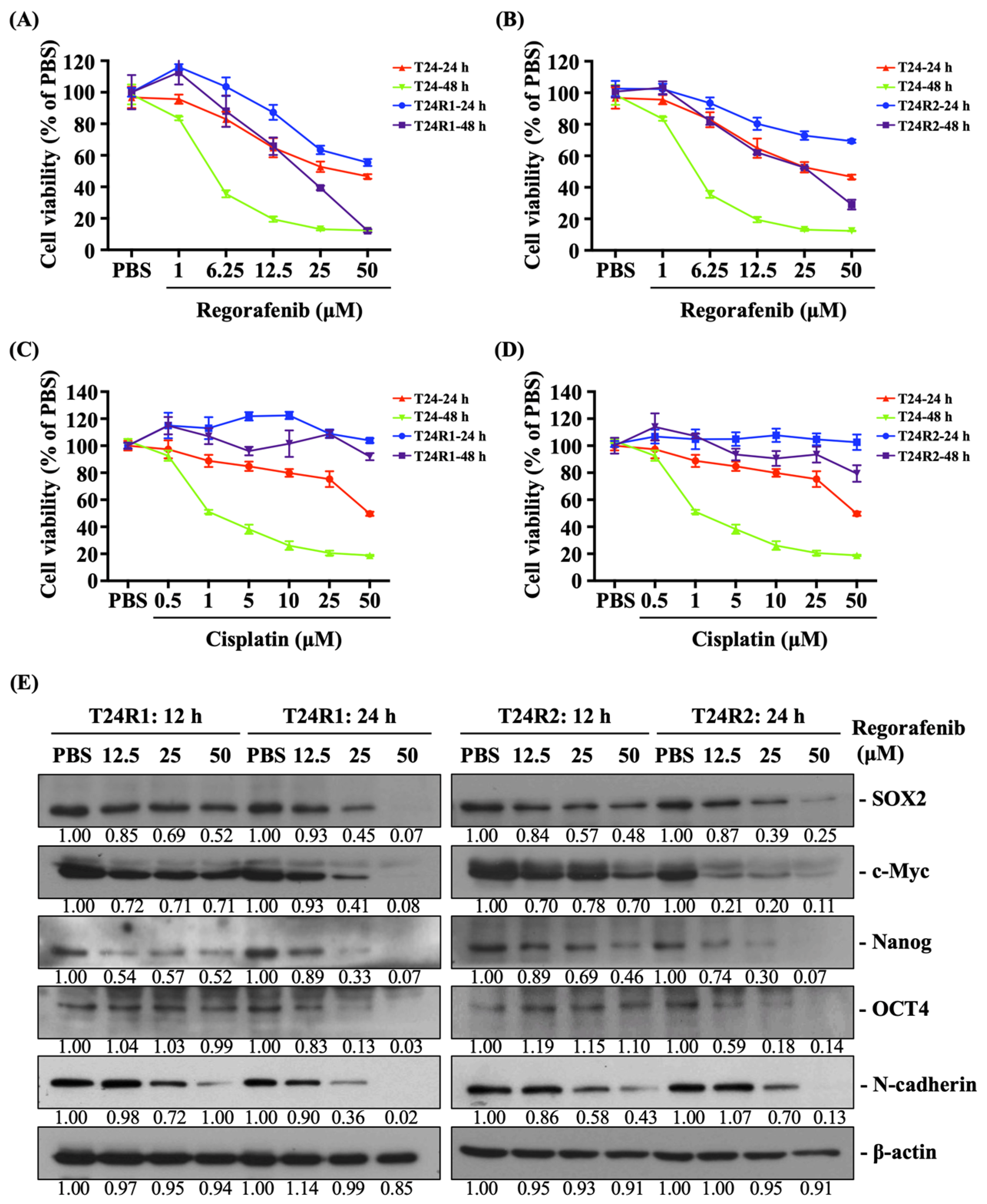
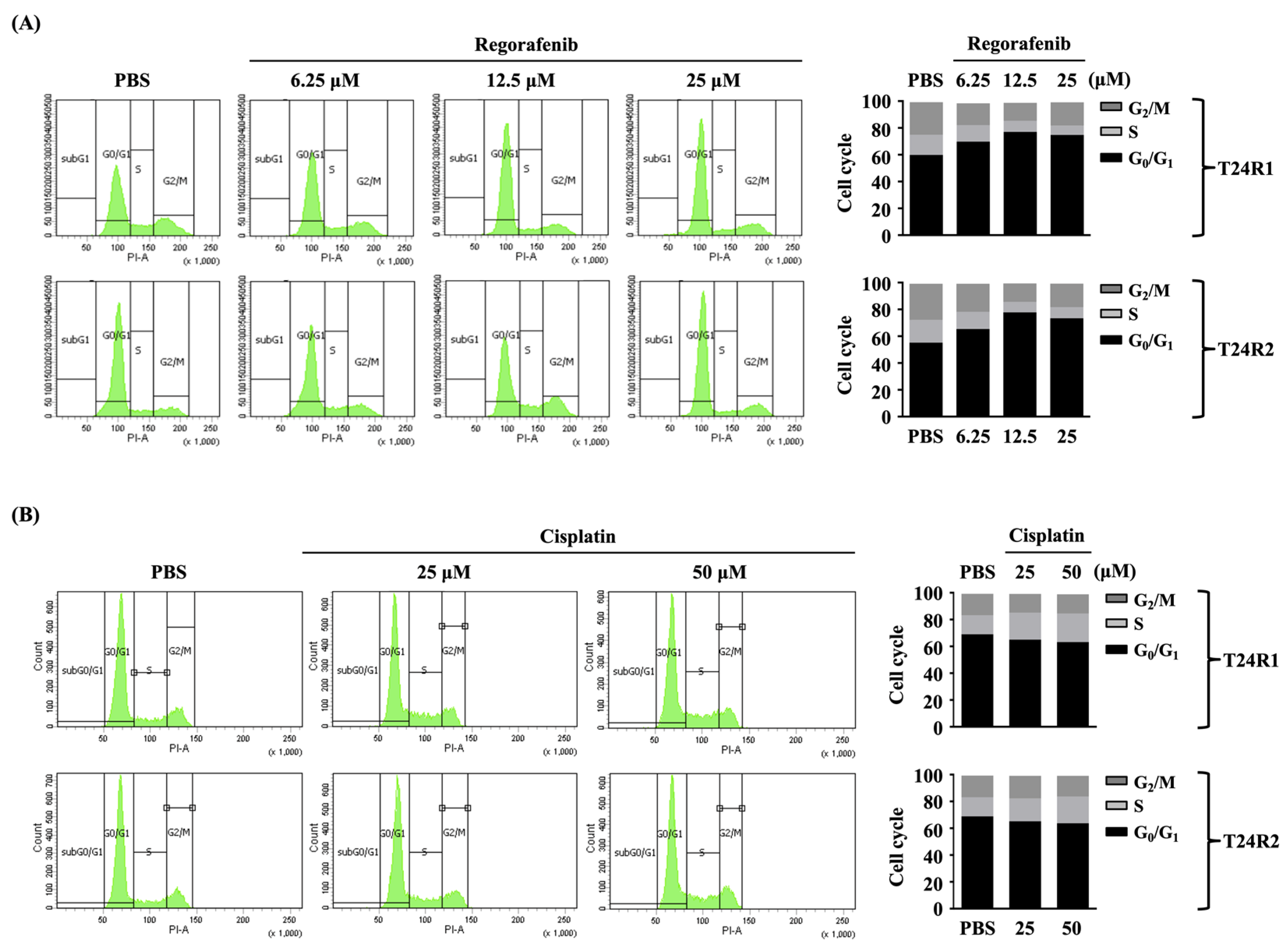
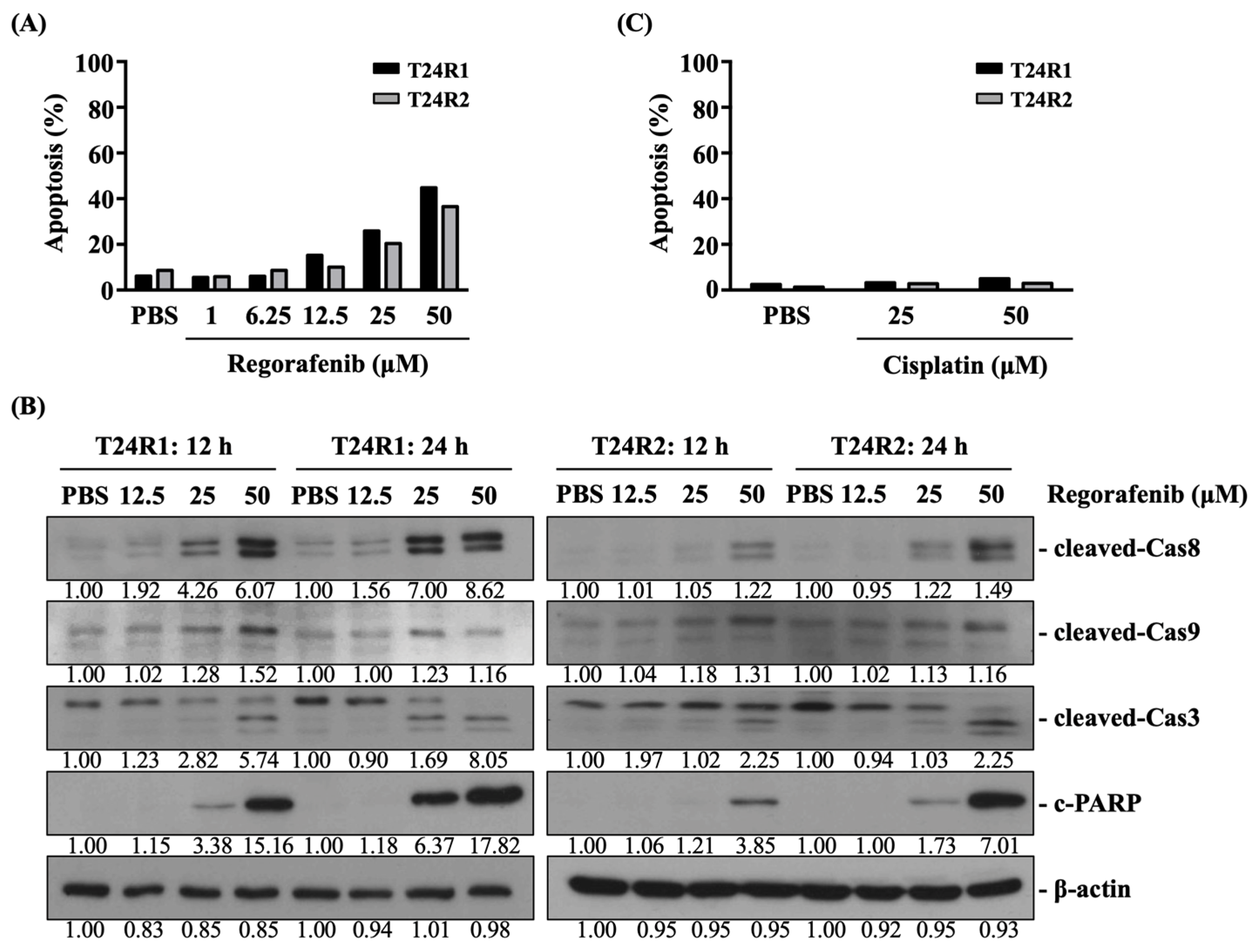
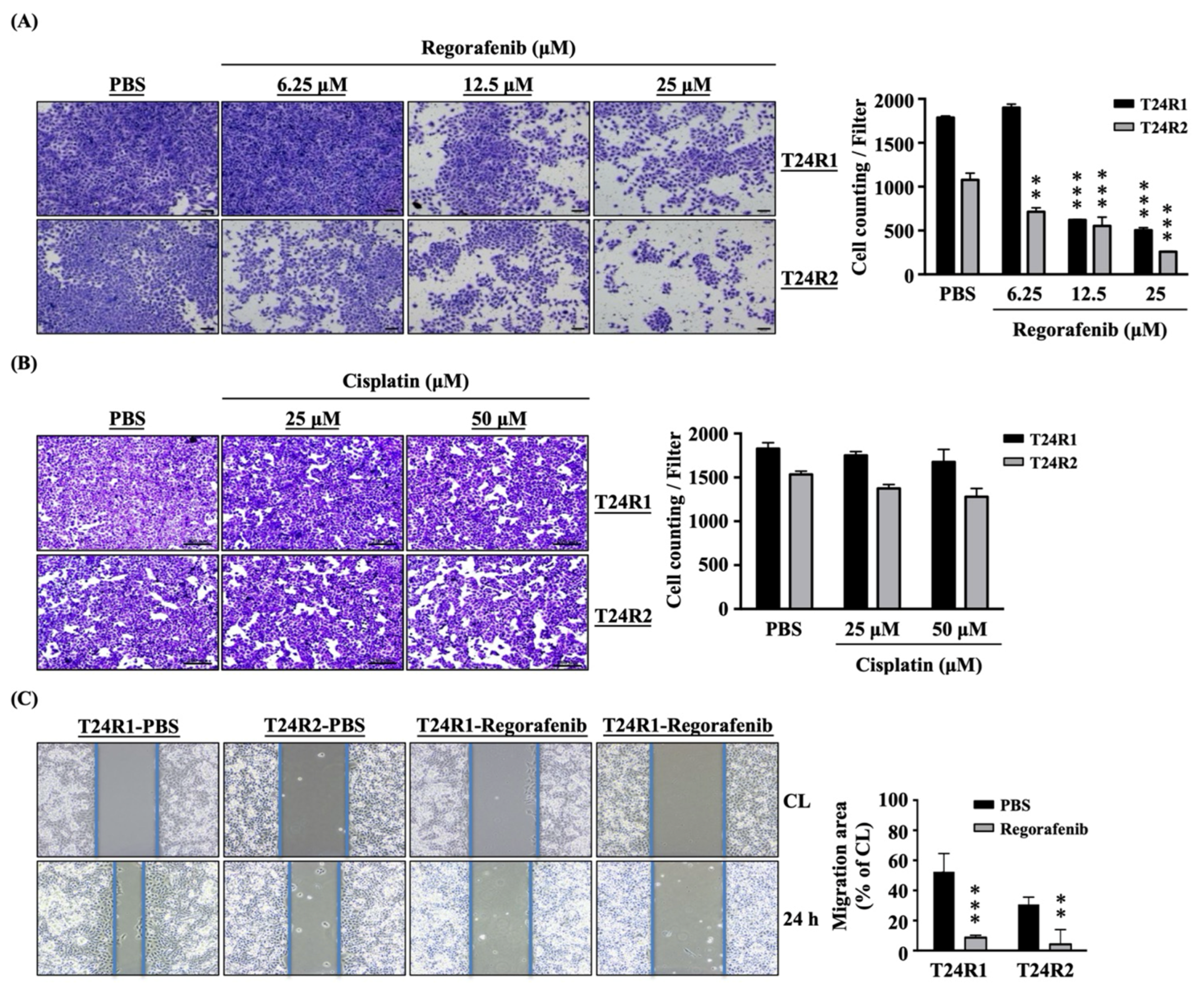
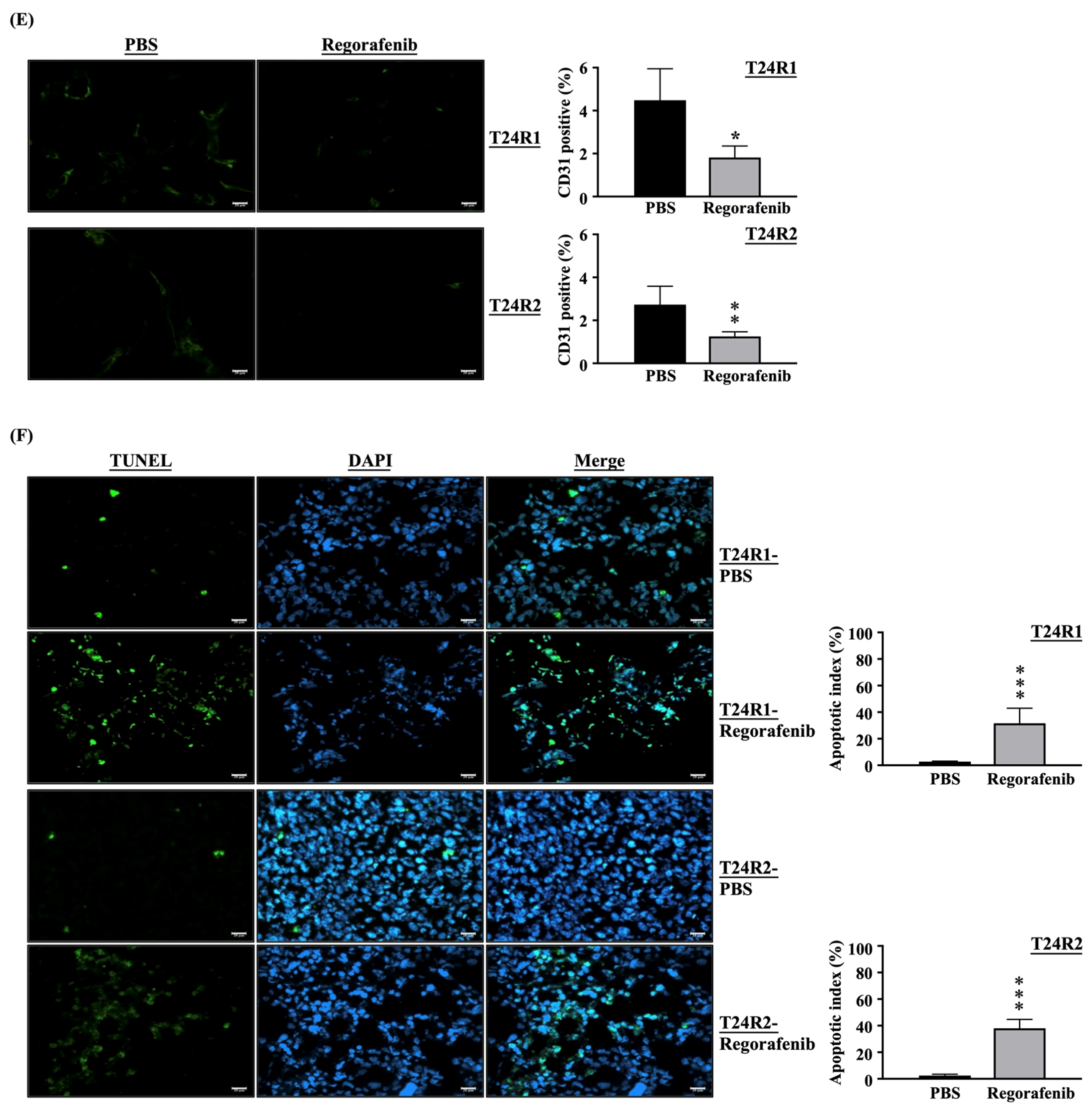
2. Results
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. Cell Viability
4.4. Apoptosis
4.5. Western Blot
4.6. Cell Cycle Distribution Analysis
4.7. Transmigration Assay (Transwell)
4.8. Migration Assay (Wound Healing)
4.9. Animal Model
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- García-Caballero, M.; Torres-Vargas, J.A.; Marrero, A.D.; Martínez-Poveda, B.; Medina, M.; Quesada, A.R. Angioprevention of Urologic Cancers by Plant-Derived Foods. Pharmaceutics 2022, 14, 256. [Google Scholar] [CrossRef] [PubMed]
- Tse, R.T.; Zhao, H.; Wong, C.Y.; Kong, A.W.; Chan, R.C.; To, K.F.; Ng, C.F.; Teoh, J.Y. In vitro assessment of intra-operative and post-operative environment in reducing bladder cancer recurrence. Sci. Rep. 2022, 12, 22. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Jia, B.; Shen, Z. Metformin and bladder cancer: Drug repurposing as a potential tool for novel therapy: A review. Medicine 2022, 101, e31635. [Google Scholar] [CrossRef] [PubMed]
- Stein, J.P.; Lieskovsky, G.; Cote, R.; Groshen, S.; Feng, A.C.; Boyd, S.; Skinner, E.; Bochner, B.; Thangathurai, D.; Mikhail, M.; et al. Radical cystectomy in the treatment of invasive bladder cancer: Long-term results in 1054 patients. J. Clin. Oncol. 2001, 19, 666–675. [Google Scholar] [CrossRef] [PubMed]
- Yin, M.; Joshi, M.; Meijer, R.P.; Glantz, M.; Holder, S.; Harvey, H.A.; Kaag, M.; Fransen van de Putte, E.E.; Horenblas, S.; Drabick, J.J. Neoadjuvant chemotherapy for muscle-invasive bladder cancer: A systematic review and two-step meta-analysis. Oncologist 2016, 21, 708–715. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Ho, J.N.; Jin, H.; Lee, S.C.; Lee, S.E.; Hong, S.K.; Lee, J.W.; Lee, E.S.; Byun, S.S. Upregulated expression of BCL2, MCM7, and CCNE1 indicate cisplatin-resistance in the set of two human bladder cancer cell lines: T24 cisplatin sensitive and T24R2 cisplatin resistant bladder cancer cell lines. Investig. Clin. Urol. 2016, 57, 63–72. [Google Scholar] [CrossRef]
- Tchounwou, P.B.; Dasari, S.; Noubissi, F.K.; Ray, P.; Kumar, S. Advances in our understanding of the molecular mechanisms of action of csplatin in cancer therapy. J. Exp. Pharmacol. 2021, 13, 303–328. [Google Scholar] [CrossRef]
- Kim, K.H.; Lee, H.W.; Ha, H.K.; Seo, H.K. Perioperative systemic therapy in muscle invasive bladder cancer: Current standard method, biomarkers and emerging strategies. Investig. Clin. Urol. 2023, 64, 202–218. [Google Scholar] [CrossRef]
- Jabir, M.S.; Abood, N.A.; Jawad, M.H.; Öztürk, K.; Kadhim, H.; Albukhaty, S.; Al-Shammari, A.; AlMalki, F.A.; Albaqami, J.; Sulaiman, G.M. Gold nanoparticles loaded TNF-α and CALNN peptide as a drug delivery system and promising therapeutic agent for breast cancer cells. Mater. Technol. 2022, 37, 3152–3166. [Google Scholar] [CrossRef]
- Ibrahim, A.A.; Kareem, M.M.; Al-Noor, T.H.; Al-Muhimeed, T.; AlObaid, A.A.; Albukhaty, S.; Sulaiman, G.M.; Jabir, M.; Taqi, Z.J.; Sahib, U.I. Pt(II)-thiocarbohydrazone complex as cytotoxic agent and apoptosis inducer in caov-3 and HT-29 cells through the p53 and caspase-8 pathways. Pharmaceuticals 2021, 14, 509. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Xu, C.; Gao, X.; Yao, Q. Platinum-based drugs for cancer therapy and anti-tumor strategies. Theranostics 2022, 12, 2115–2132. [Google Scholar] [CrossRef] [PubMed]
- Williams, E.D.; Gao, D.; Redfern, A.; Thompson, E.W. Controversies around epithelial-mesenchymal plasticity in cancer metastasis. Nat. Rev. Cancer 2019, 19, 716–732. [Google Scholar] [CrossRef] [PubMed]
- Chin, V.L.; Lim, C.L. Epithelial-mesenchymal plasticity-engaging stemness in an interplay of phenotypes. Stem Cell Investig. 2019, 6, 25. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Dai, X.; Zhang, Q.; Dai, Z. Gene expression of OCT4, SOX2, KLF4 and MYC (OSKM) induced pluripotent stem cells: Identification for potential mechanisms. Diagn. Pathol. 2015, 10, 35. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Wang, Y.; Sun, L.; Xu, S.; Li, C.; Jin, X. The origin and evolution of bladder cancer stem cells. Front. Cell Dev. Biol. 2022, 10, 950241. [Google Scholar] [CrossRef] [PubMed]
- Migita, T.; Ueda, A.; Ohishi, T.; Hatano, M.; Seimiya, H.; Horiguchi, S.I.; Koga, F.; Shibasaki, F. Epithelial-mesenchymal transition promotes SOX2 and NANOG expression in bladder cancer. Lab. Invest. 2017, 97, 567–576. [Google Scholar] [CrossRef]
- Garg, M. Epithelial plasticity in urothelial carcinoma: Current advancements and future challenges. World J. Stem Cells 2016, 8, 260–267. [Google Scholar] [CrossRef]
- Li, Y.; Lin, K.; Yang, Z.; Han, N.; Quan, X.; Guo, X.; Li, C. Bladder cancer stem cells: Clonal origin and therapeutic perspectives. Oncotarget 2017, 8, 66668–66679. [Google Scholar] [CrossRef]
- Chen, G.; Chen, Y.; Xu, R.; Zhang, G.; Zou, X.; Wu, G. Impact of SOX2 function and regulation on therapy resistance in bladder cancer. Front. Oncol. 2022, 12, 1020675. [Google Scholar] [CrossRef]
- Cohen, P.; Cross, D.; Jänne, P.A. Kinase drug discovery 20 years after imatinib: Progress and future directions. Nat. Rev. Drug Discov. 2021, 20, 551–569. [Google Scholar] [CrossRef] [PubMed]
- Bhullar, K.S.; Lagarón, N.O.; McGowan, E.M.; Parmar, I.; Jha, A.; Hubbard, B.P.; Rupasinghe, H.P.V. Kinase-targeted cancer therapies: Progress, challenges and future directions. Mol. Cancer 2018, 17, 48. [Google Scholar] [CrossRef] [PubMed]
- Grothey, A.; Blay, J.Y.; Pavlakis, N.; Yoshino, T.; Bruix, J. Evolving role of regorafenib for the treatment of advanced cancers. Cancer Treat. Rev. 2020, 86, 101993. [Google Scholar] [CrossRef] [PubMed]
- Hofheinz, R.D.; Bruix, J.; Demetri, G.D.; Grothey, A.; Marian, M.; Bartsch, J.; Odom, D. Effect of regorafenib in delaying definitive deterioration in health-related quality of life in patients with advanced cancer of three different tumor types. Cancer Manag. Res. 2021, 13, 5523–5533. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.Y.; Cheng, T.S.; Chuang, S.C.; Chang, W.T.; Liang, P.C.; Hsu, C.T.; Wei, Y.J.; Jang, T.Y.; Yeh, M.L.; Huang, C.I.; et al. Regorafenib for Taiwanese patients with unresectable hepatocellular carcinoma after sorafenib failure: Impact of alpha-fetoprotein levels. Cancer Med. 2022, 11, 104–116. [Google Scholar] [CrossRef] [PubMed]
- Agulnik, M.; Schulte, B.; Robinson, S.; Hirbe, A.C.; Kozak, K.; Chawla, S.P.; Attia, S.; Rademaker, A.; Zhang, H.; Abbinanti, S.; et al. An open-label single-arm phase II study of regorafenib for the treatment of angiosarcoma. Eur. J. Cancer 2021, 154, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.D.; Sanoff, H.K.; Poklepovic, A.S.; Soares, H.; Kim, J.; Lyu, J.; Liu, Y.; Nixon, A.B.; Kim, D.W. A multi-institutional phase 2 trial of regorafenib in refractory advanced biliary tract cancer. Cancer 2020, 126, 3464–3470. [Google Scholar] [CrossRef]
- Bozzarelli, S.; Rimassa, L.; Giordano, L.; Sala, S.; Tronconi, M.C.; Pressiani, T.; Smiroldo, V.; Prete, M.G.; Spaggiari, P.; Personeni, N.; et al. Regorafenib in patients with refractory metastatic pancreatic cancer: A Phase II study (RESOUND). Future Oncol. 2019, 15, 4009–4017. [Google Scholar] [CrossRef]
- Hsu, F.T.; Sun, C.C.; Wu, C.H.; Lee, Y.J.; Chiang, C.H.; Wang, W.S. Regorafenib induces apoptosis and inhibits metastatic potential of human bladder carcinoma cells. Anticancer. Res. 2017, 37, 4919–4926. [Google Scholar]
- Chiang, C.H.; Chung, J.G.; Hsu, F.T. Regorefenib induces extrinsic/intrinsic apoptosis and inhibits MAPK/NF-κB-modulated tumor progression in bladder cancer in vitro and in vivo. Environ. Toxicol. 2019, 34, 679–688. [Google Scholar] [CrossRef]
- Zangouei, A.S.; Barjasteh, A.H.; Rahimi, H.R.; Mojarrad, M.; Moghbeli, M. Role of tyrosine kinases in bladder cancer progression: An overview. Cell Commun. Signal 2020, 18, 127. [Google Scholar] [CrossRef] [PubMed]
- Osawa, H. Response to regorafenib at an initial dose of 120 mg as salvage therapy for metastatic colorectal cancer. Mol. Clin. Oncol. 2017, 6, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Hirano, G.; Makiyama, A.; Makiyama, C.; Esaki, T.; Oda, H.; Uchino, K.; Komoda, M.; Tanaka, R.; Matsushita, Y.; Mitsugi, K.; et al. Reduced dose of salvage-line regorafenib monotherapy for metastatic colorectal cancer in Japan. Anticancer. Res. 2015, 35, 371–377. [Google Scholar] [PubMed]
- Fukuoka, S.; Hara, H.; Takahashi, N.; Kojima, T.; Kawazoe, A.; Asayama, M.; Yoshii, T.; Kotani, D.; Tamura, H.; Mikamoto, Y.; et al. Regorafenib plus nivolumab in patients with advanced gastric or colorectal cancer: An open-label, dose-escalation, and dose-expansion phase Ib trial (REGONIVO, EPOC1603). J. Clin. Oncol. 2020, 38, 2053–2061. [Google Scholar] [CrossRef] [PubMed]
- Fondevila, F.; Méndez-Blanco, C.; Fernández-Palanca, P.; González-Gallego, J.; Mauriz, J.L. Anti-tumoral activity of single and combined regorafenib treatments in preclinical models of liver and gastrointestinal cancers. Exp. Mol. Med. 2019, 51, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Kodama, H.; Yoshida, M.; Goto, M.; Nakamura, M.; Hasegawa, H.; Ikeda, A.; Tokunaga, Y.; Tanigawa, K.; Terazawa, T.; Sakai, D.; et al. [Regorafenib versus S-1 plus bevacizumab for metastatic colorectal cancer as salvage line-A phase II study (OGSG1301)]. Gan To Kagaku Ryoho 2021, 48, 1241–1246. [Google Scholar]
- Roviello, G.; Catalano, M.; Santi, R.; Santoni, M.; Galli, I.C.; Amorosi, A.; Polom, W.; De Giorgi, U.; Nesi, G. Neoadjuvant treatment in muscle-invasive bladder cancer: From the beginning to the latest developments. Front. Oncol. 2022, 12, 912699. [Google Scholar] [CrossRef]
- Xiao, Y.; Lin, F.T.; Lin, W.C. ACTL6A promotes repair of cisplatin-induced DNA damage, a new mechanism of platinum resistance in cancer. Proc. Natl. Acad. Sci. USA 2021, 118, e2015808118. [Google Scholar] [CrossRef]
- Kiss, R.C.; Xia, F.; Acklin, S. Targeting DNA damage response and repair to enhance therapeutic index in cisplatin-based cancer treatment. Int. J. Mol. Sci. 2021, 22, 8199. [Google Scholar] [CrossRef]
- Elfadadny, A.; El-Husseiny, H.M.; Abugomaa, A.; Ragab, R.F.; Mady, E.A.; Aboubakr, M.; Samir, H.; Mandour, A.S.; El-Mleeh, A.; El-Far, A.H.; et al. Role of multidrug resistance-associated proteins in cancer therapeutics: Past, present, and future perspectives. Environ. Sci. Pollut. Res. Int. 2021, 28, 49447–49466. [Google Scholar] [CrossRef]
- Runyan, R.B.; Savagner, P. Epithelial-mesenchymal transition and plasticity in the developmental basis of cancer and fibrosis. Dev. Dyn. 2018, 247, 330–331. [Google Scholar] [CrossRef]
- Dzobo, K.; Senthebane, D.A.; Ganz, C.; Thomford, N.E.; Wonkam, A.; Dandara, C. Advances in therapeutic targeting of cancer stem cells within the tumor microenvironment: An updated review. Cells 2020, 9, 1896. [Google Scholar] [CrossRef]
- Abugomaa, A.; Elbadawy, M.; Yamawaki, H.; Usui, T.; Sasaki, K. Emerging roles of cancer stem cells in bladder cancer progression, tumorigenesis, and resistance to chemotherapy: A potential therapeutic target for bladder cancer. Cells 2020, 9, 235. [Google Scholar] [CrossRef]
- Taoka, Y.; Matsumoto, K.; Ohashi, K.; Minamida, S.; Hagiwara, M.; Nagi, S.; Saito, T.; Kodera, Y.; Iwamura, M. Protein expression profile related to cisplatin resistance in bladder cancer cell lines detected by two-dimensional gel electrophoresis. Biomed. Res. 2015, 36, 253–261. [Google Scholar] [CrossRef]
- Rieger, A.M.; Nelson, K.L.; Konowalchuk, J.D.; Barreda, D.R. Modified annexin V/propidium iodide apoptosis assay for accurate assessment of cell death. J. Vis. Exp. 2011, 50, 2597. [Google Scholar]
- Wu, M.H.; Hsu, W.B.; Chen, M.H.; Shi, C.S. Inhibition of neddylation suppresses osteoclast differentiation and function in vitro and alleviates osteoporosis in vivo. Biomedicines 2022, 10, 2355. [Google Scholar] [CrossRef]
- Wang, Z.; Yang, B.; Zhang, M.; Guo, W.; Wu, Z.; Wang, Y.; Jia, L.; Li, S.; Xie, W.; Yang, D. lncRNA epigenetic landscape analysis identifies EPIC1 as an oncogenic incRNA that interacts with MYC and promotes cell-cycle progression in cancer. Cancer Cell 2018, 33, 706–720. [Google Scholar] [CrossRef]
- Lee, S.R.; Roh, Y.G.; Kim, S.K.; Lee, J.S.; Seol, S.Y.; Lee, H.H.; Kim, W.T.; Kim, W.J.; Heo, J.; Cha, H.J.; et al. Activation of EZH2 and SUZ12 regulated by E2F1 predicts the disease progression and aggressive characteristics of bladder cancer. Clin. Cancer Res. 2015, 21, 5391–5403. [Google Scholar] [CrossRef]
- Lu, C.C.; Chu, P.Y.; Hsia, S.M.; Wu, C.H.; Tung, Y.T.; Yen, G.C. Insulin induction instigates cell proliferation and metastasis in human colorectal cancer cells. Int. J. Oncol. 2017, 50, 736–744. [Google Scholar] [CrossRef] [PubMed]
- Cai, F.; Chen, M.; Zha, D.; Zhang, P.; Zhang, X.; Cao, N.; Wang, J.; He, Y.; Fan, X.; Zhang, W.; et al. Curcumol potentiates celecoxib-induced growth inhibition and apoptosis in human non-small cell lung cancer. Oncotarget 2017, 8, 115526–115545. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuan, F.-C.; Li, J.-M.; Huang, Y.-C.; Chang, S.-F.; Shi, C.-S. Therapeutic Potential of Regorafenib in Cisplatin-Resistant Bladder Cancer with High Epithelial–Mesenchymal Transition and Stemness Properties. Int. J. Mol. Sci. 2023, 24, 17610. https://doi.org/10.3390/ijms242417610
Kuan F-C, Li J-M, Huang Y-C, Chang S-F, Shi C-S. Therapeutic Potential of Regorafenib in Cisplatin-Resistant Bladder Cancer with High Epithelial–Mesenchymal Transition and Stemness Properties. International Journal of Molecular Sciences. 2023; 24(24):17610. https://doi.org/10.3390/ijms242417610
Chicago/Turabian StyleKuan, Feng-Che, Jhy-Ming Li, Yun-Ching Huang, Shun-Fu Chang, and Chung-Sheng Shi. 2023. "Therapeutic Potential of Regorafenib in Cisplatin-Resistant Bladder Cancer with High Epithelial–Mesenchymal Transition and Stemness Properties" International Journal of Molecular Sciences 24, no. 24: 17610. https://doi.org/10.3390/ijms242417610