
The Microglial Transcriptome of Age-Associated Deep Subcortical White Matter Lesions Suggests a Neuroprotective Response to Blood–Brain Barrier Dysfunction
 , , , , ,
, , , , ,
Abstract
:1. Introduction
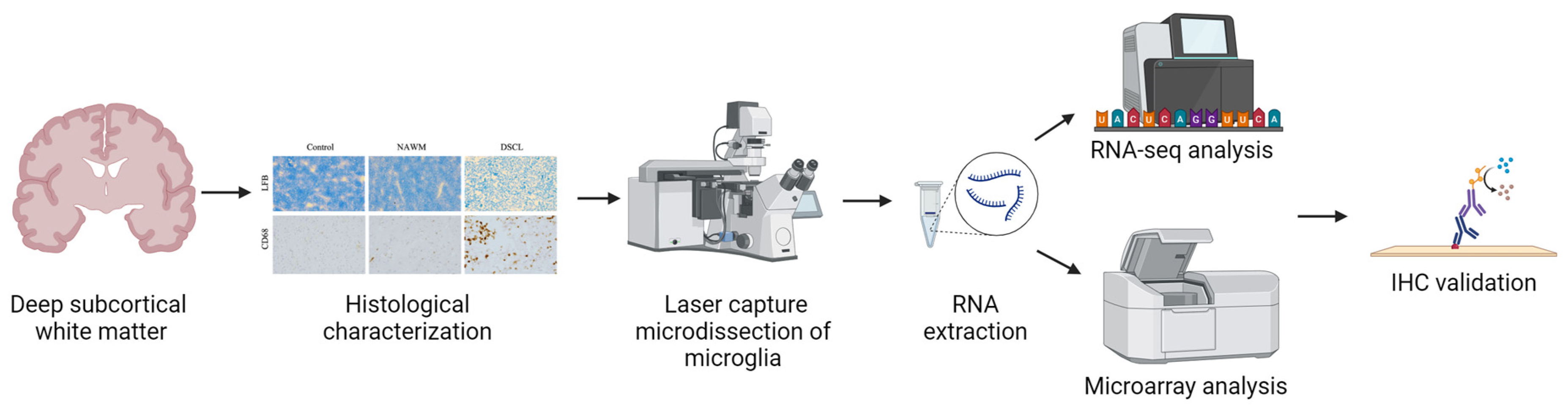
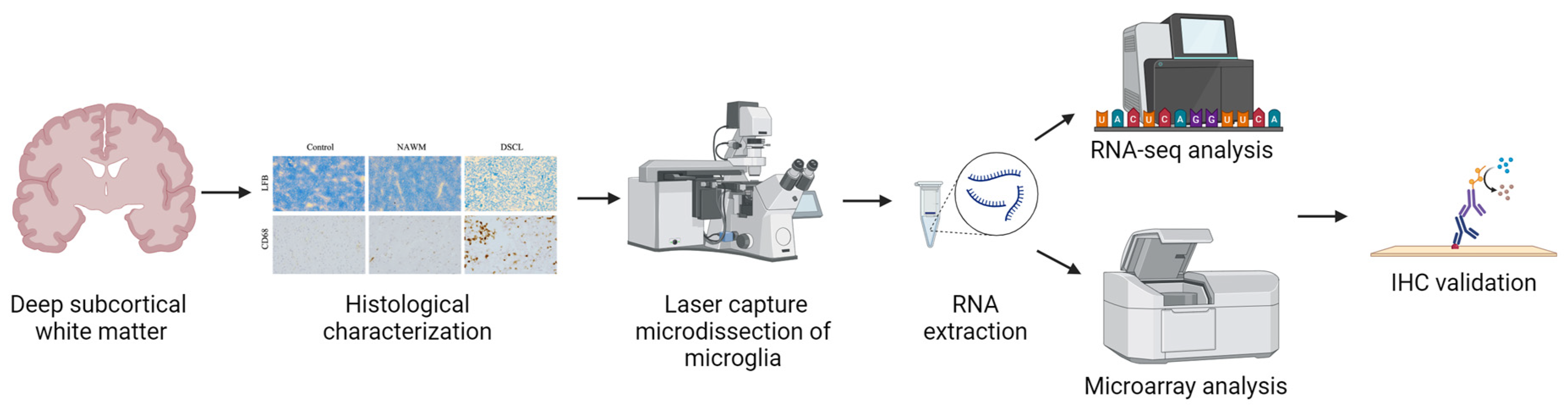
2. Results
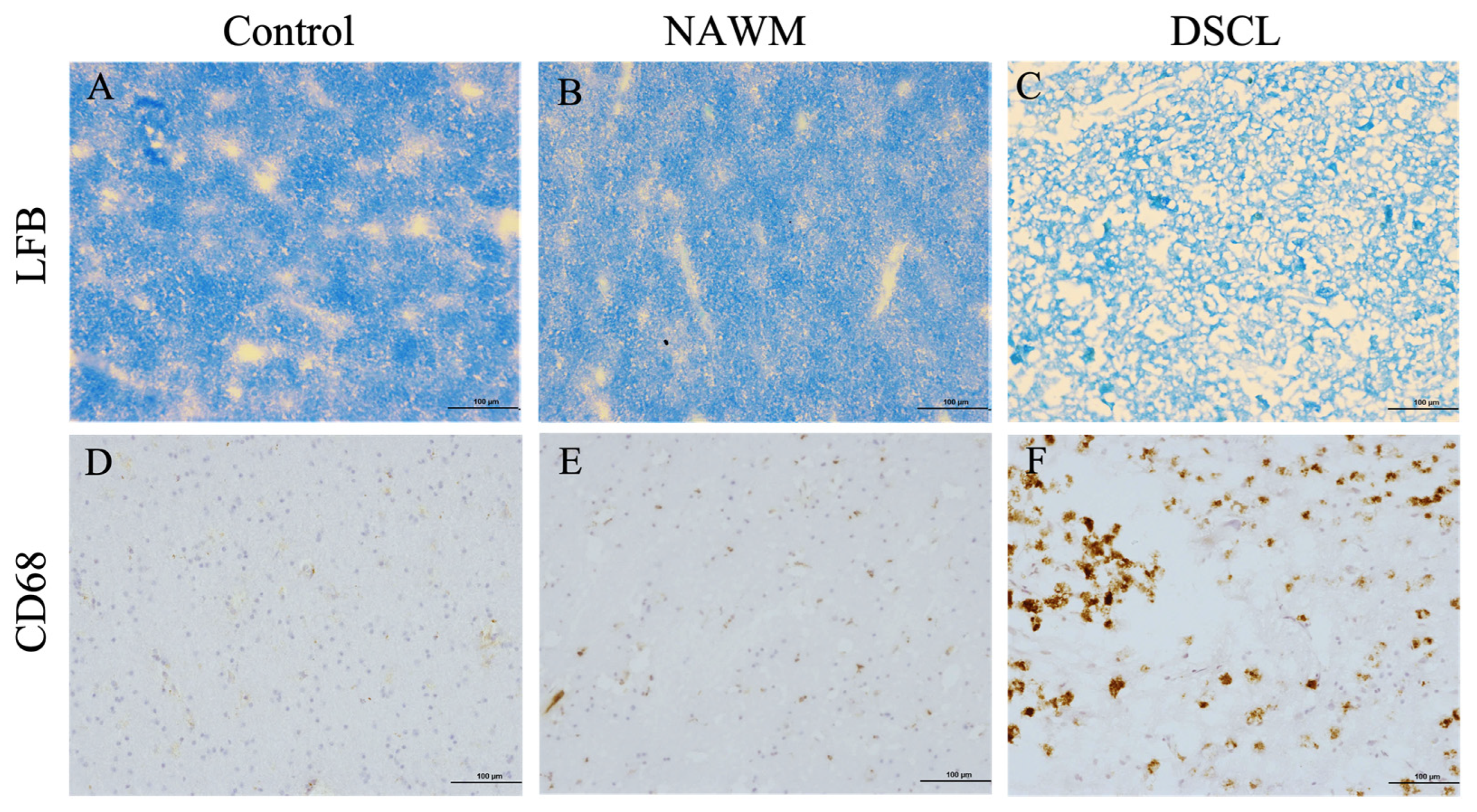
2.1. Histological Characterization Confirms the Classification of Sampled WM
2.2. RNA-Sequencing Is Not Compatible with Immuno-LCM-ed Post-Mortem Tissue in the CFAS Cohort
2.3. Microarray Transcriptomic Profiling Is Compatible with Immuno-LCM in the CFAS Cohort
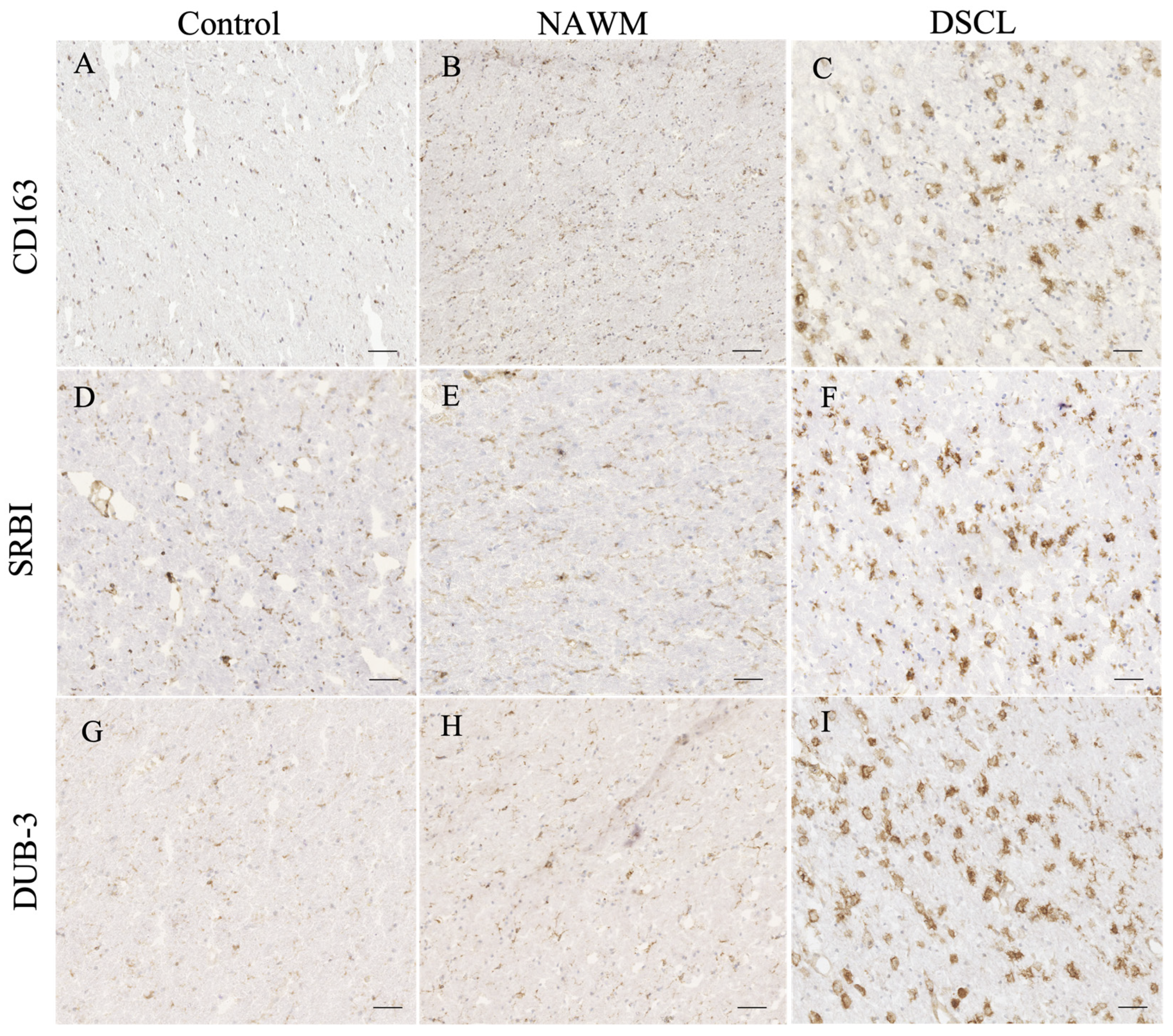
2.4. Validation of Candidate Gene Expression
3. Discussion
4. Materials and Methods
4.1. Case Selection and Histological Characterization
4.2. Laser Capture Microdissection (LCM) of Microglia
4.3. RNA-Sequencing
RNA-Seq Data Analysis
4.4. Microarray
Microarray Data Analysis
4.5. Immunohistochemical Validation of Protein Changes
Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Prins, N.D.; Scheltens, P. White matter hyperintensities, cognitive impairment and dementia: An update. Nat. Rev. Neurol. 2015, 11, 157. [Google Scholar] [CrossRef] [PubMed]
- Brayne, C.; McCracken, C.; Matthews, F.E. Cohort profile: The Medical Research Council cognitive function and ageing study (CFAS). Int. J. Epidemiol. 2006, 35, 1140–1145. [Google Scholar] [CrossRef] [PubMed]
- Wharton, S.B.; Brayne, C.; Savva, G.M.; Matthews, F.E.; Forster, G.; Simpson, J.; Lace, G.; Ince, P.G. Epidemiological neuropathology: The MRC cognitive function and aging study experience. J. Alzheimers Dis. 2011, 25, 359–372. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Viqar, F.; Zimmerman, M.E.; Narkhede, A.; Tosto, G.; Benzinger, T.L.; Marcus, D.S.; Fagan, A.M.; Goate, A.; Fox, N.C.; et al. White matter hyperintensities are a core feature of Alzheimer’s disease: Evidence from the dominantly inherited Alzheimer network. Ann. Neurol. 2016, 79, 929–939. [Google Scholar] [CrossRef] [PubMed]
- Hanyu, H.; Koyama, Y.; Umekida, K.; Momose, T.; Watanabe, S.; Sato, T. Factors and brain imaging features associated with cognition in oldest-old patients with Alzheimer-type dementia. J. Neurol. Sci. 2024, 10, 122929. [Google Scholar] [CrossRef]
- De Groot, J.C.; De Leeuw, F.E.; Oudkerk, M.; Van Gijn, J.; Hofman, A.; Jolles, J.; Breteler, M.M. Periventricular cerebral white matter lesions predict rate of cognitive decline. Ann. Neurol. 2002, 52, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.-Y.; Ou, Y.-N.; Shen, X.-N.; Qu, Y.; Ma, Y.-H.; Wang, Z.-T.; Dong, Q.; Tan, L.; Yu, J.-T. White matter hyperintensities and risks of cognitive impairment and dementia: A systematic review and meta-analysis of 36 prospective studies. Neurosci. Biobehav. Rev. 2021, 120, 16–27. [Google Scholar] [CrossRef]
- Freeze, W.M.; Jacobs, H.I.; De Jong, J.J.; Verheggen, I.C.; Gronenschild, E.H.; Palm, W.M.; Hoff, E.I.; Wardlaw, J.M.; Jansen, J.F.A.; Verhey, F.R.; et al. White matter hyperintensities mediate the association between blood-brain barrier leakage and information processing speed. Neurobiol. Aging 2020, 85, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Prinz, M.; Priller, J. Microglia and brain macrophages in the molecular age: From origin to neuropsychiatric disease. Nat. Rev. Neurosci. 2014, 15, 300–312. [Google Scholar] [CrossRef]
- Wharton, S.B.; Simpson, J.E.; Brayne, C.; Ince, P.G. Age-Associated White Matter Lesions: The MRC Cognitive Function and Ageing Study. Brain Pathol. 2015, 25, 35–43. [Google Scholar] [CrossRef]
- Fadul, M.M.; Heath, P.R.; Cooper-Knock, J.; Kurz, J.M.; Al-Azzawi, H.A.; Ali, Z.; Smith, T.; Matthews, F.M.; Brayne, C.; Wharton, S.B.; et al. Transcriptomic analysis of age-associated periventricular lesions reveals dysregulation of the immune response. Int. J. Mol. Sci. 2020, 21, 7924. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Kalpouzos, G.; Bäckman, L.; Qiu, C.; Laukka, E.J. Association of white matter hyperintensity accumulation with domain-specific cognitive decline: A population-based cohort study. Neurobiol. Aging. 2023, 132, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Ozsolak, F.; Milos, P.M. RNA sequencing: Advances, challenges and opportunities. Nat. Rev. Genet. 2011, 12, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Lonergan, W.; Whistler, T.; Vernon, S.D. Comparison of target labeling methods for use with Affymetrix GeneChips. BMC Biotechnol. 2007, 7, 24. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Hu, C.; Huang, C.; Wei, Y.; Li, X.; Hu, M.; Li, H.; Wu, J.; Czajkowsky, D.M.; Guo, Y.; et al. Robust Acquisition of Spatial Transcriptional Programs in Tissues with Immunofluorescence-Guided Laser Capture Microdissection. Front. Cell Dev. Biol. 2022, 10, 853188. [Google Scholar] [CrossRef]
- Almeida, D.; Turecki, G. Profiling cell-type specific gene expression in post-mortem human brain samples through laser capture microdissection. Methods 2022, 207, 3–10. [Google Scholar] [CrossRef]
- Mauney, S.A.; Pietersen, C.Y.; Sonntag, K.-C.; Woo, T.-U.W. Differentiation of oligodendrocyte precursors is impaired in the prefrontal cortex in schizophrenia. Schizophr. Res. 2015, 169, 374–380. [Google Scholar] [CrossRef] [PubMed]
- Sadrzadeh, S.M.; Bozorgmehr, J. Haptoglobin phenotypes in health and disorders. Am. J. Clin. Pathol. 2004, 121, S97–S104. [Google Scholar] [CrossRef]
- Bulters, D.; Gaastra, B.; Zolnourian, A.; Alexander, S.; Ren, D.; Blackburn, S.L.; Borsody, M.; Doré, S.; Galea, J.; Iihara, K.; et al. Haemoglobin scavenging in intracranial bleeding: Biology and clinical implications. Nat. Rev. Neurol. 2018, 14, 416–432. [Google Scholar] [CrossRef]
- Brown, N.; Alkhayer, K.; Clements, R.; Singhal, N.; Gregory, R.; Azzam, S.; Li, S.; Freeman, E.; McDonough, J. Neuronal Hemoglobin Expression and Its Relevance to Multiple Sclerosis Neuropathology. J. Mol. Neurosci. 2016, 59, 1–17. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Hua, Y.; Keep, R.F.; Liu, W.; Wang, M.M.; Xi, G. Hemoglobin Expression in Neurons and Glia After Intracerebral Hemorrhage. In Intracerebral Hemorrhage Research; Acta Neurochirurgica Supplementum; Zhang, J., Colohan, A., Eds.; Springer: Vienna, Austria, 2011; Volume 111, pp. 133–137. [Google Scholar]
- Borda, J.T.; Alvarez, X.; Mohan, M.; Hasegawa, A.; Bernardino, A.; Jean, S.; Aye, P.; Lackner, A.A. CD163, a marker of perivascular macrophages, is up-regulated by microglia in simian immunodeficiency virus encephalitis after haptoglobin-hemoglobin complex stimulation and is suggestive of breakdown of the blood-brain barrier. Am. J. Pathol. 2008, 172, 725–737. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Lu, P.; Wu, S.; Yang, J.; Liu, W.; Zhang, Z.; Xu, Q. CD163-mediated small-vessel injury in Alzheimer’s Disease: An exploration from neuroimaging to transcriptomics. Int. J. Mol. Sci. 2024, 25, 2293. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.-F.; Zhang, X.; Su, Y.-G.; Zhao, R.; Wang, Y.-Y. The role of the deubiquitinating enzyme DUB3/USP17 in cancer: A narrative review. Cancer Cell Int. 2021, 21, 455. [Google Scholar] [CrossRef] [PubMed]
- Ruan, J.; Schlüter, D.; Wang, X. Deubiquitinating enzymes (DUBs): DoUBle-edged swords in CNS autoimmunity. J. Neuroinflamm. 2020, 17, 102. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Ramakrishna, S.; Kim, K.-S. Critical roles of deubiquitinating enzymes in the nervous system and neurodegenerative disorders. Mol. Cells 2020, 43, 203. [Google Scholar] [PubMed]
- Snyder, N.A.; Silva, G.M. Deubiquitinating enzymes (DUBs): Regulation, homeostasis, and oxidative stress response. J. Biol. Chem. 2021, 297, 101077. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Ruan, J.; Chen, M.; Li, Z.; Manjengwa, G.; Schlüter, D.; Song, W.; Wang, X. Deubiquitinating enzymes (DUBs): Decipher underlying basis of neurodegenerative diseases. Mol. Psychiatry 2022, 27, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Weidauer, S.; Wagner, M.; Hattingen, E. White Matter Lesions in Adults—A Differential Diagnostic Approach. Fortschr. Röntgenstr. 2020, 192, 1154–1173. [Google Scholar] [CrossRef]
- Rao, M.S.; Van Vleet, T.R.; Ciurlionis, R.; Buck, W.R.; Mittelstadt, S.W.; Blomme, E.A.G.; Liguori, M.J. Comparison of RNA-Seq and Microarray Gene Expression Platforms for the Toxicogenomic Evaluation of Liver from Short-Term Rat Toxicity Studies. Front. Genet. 2019, 9, 636. [Google Scholar] [CrossRef]
- Schurch, N.J.; Schofield, P.; Gierliński, M.; Cole, C.; Sherstnev, A.; Singh, V.; Wrobel, N.; Gharbi, K.; Simpson, G.G.; Owen-Hughes, T.; et al. How many biological replicates are needed in an RNA-seq experiment and which differential expression tool should you use? RNA 2016, 22, 839–851. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Sherman, B.; Lempicki, R. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | Length | M Seq (×106) | % Fail | % GC | % Dup | % Over-Representative |
|---|---|---|---|---|---|---|
| Sample 1_fastq | 101 bp | 3.1 | 45% | 87% | 76.5% | 23.5% |
| Sample 2_fastq | 101 bp | 6 | 36% | 87% | 77.7% | 23.5% |
| Sample 3_fastq | 101 bp | 6.9 | 36% | 90% | 82.3% | 25.0% |
| Sample 4_fastq | 101 bp | 7.8 | 36% | 88% | 80.0% | 25.0% |
| Sample 5_fastq | 101 bp | 16.5 | 45% | 83% | 67.3% | 21.5% |
| Sample 6_fastq | 101 bp | 6.3 | 36% | 89% | 80.9% | 25.0% |
| Sample 7_fastq | 101 bp | 6.1 | 45% | 90% | 81.7% | 23.5% |
| Sample 8_fastq | 101 bp | 9.5 | 45% | 80% | 66.8% | 19.7% |
| Sample 9_fastq | 101 bp | 16.8 | 45% | 83% | 78.8% | 21.5% |
| Sample 10_fastq | 101 bp | 8.4 | 36% | 89% | 80.1% | 19.7% |
| Sample 11_fastq | 101 bp | 9.6 | 36% | 89% | 81.2% | 25.0% |
| Sample 12_fastq | 101 bp | 8.9 | 45% | 90% | 80.8% | 19.7% |
| Annotated Cluster | Enrichment Score | Gene Count | Benjamini p-Value | Directional Change | Differentially Expressed Genes |
|---|---|---|---|---|---|
| Haptoglobin binding | 2.5 | 3 | 1.7 × 10−2 | Up | Haemoglobin-α1 (HBA1) Haemoglobin-α2 (HBA2) Haemoglobin-β (HBB) |
| Haptoglobin–haemoglobin complex | 3 | 2.4 × 10−2 | |||
| TBC | 2.66 | 7 | 6.3 × 10−6 | Down | TBC1 domain family member: (TBC1D3B, TBC1D3E, TBC1D3F, TBC1D3G, TBC1D3H, TBC1D3I, TBC1D3L) |
| Rab-GTPase-TBC domain | 4.3 × 10−3 |
| Annotated Cluster | Enrichment Score | Gene Count | Benjamini p-Value | Directional Change | Differentially Expressed Genes |
|---|---|---|---|---|---|
| Protein deubiquitination | 5.14 | 9 | 1.3 × 10−4 | Down | Ubiquitin specific peptidase 17-like family member: USP17L5, USP17L19, USP17L24, USP17L25, USP17L26, USP17L27, USP17L28, USP17L29, USP17L30, Ring finger protein 8 (RNF8) Toll interaction protein (TOLLIP) |
| Ubiquitin-dependent protein catabolic process | 11 | 1.3 × 10−4 |
| WM Group | Control (n = 4) | NAWM (n = 4) | DSCLs (n = 4) |
|---|---|---|---|
| CD163 | 0.47 (0.29–0.85) | 1.41 (0.43–3.24) | 4.07 (2.12–6.41) |
| SRBI | 0.46 (0.22–0.70) | 0.83 (0.74–0.93) | 7.02 (1.85–15.09) * |
| Dub3 | 0.25 (0.20–0.28) | 0.44 (0.30–0.66) | 4.44 (1.09–7.89) * |
| Primary Antibody | Species | Clonality | Isotype | Dilution | Supplier |
|---|---|---|---|---|---|
| CD163 | Rabbit | Monoclonal | IgG | 1:500 | Abcam, UK |
| Dub3 | Rabbit | Polyclonal | IgG | 1:100 | Novusbio, UK |
| SRBI | Rabbit | Monoclonal | IgG | 1:500 | Abcam, UK |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Almansouri, T.; Waller, R.; Wharton, S.B.; Heath, P.R.; Matthews, F.E.; Brayne, C.; van Eeden, F.; Simpson, J.E. The Microglial Transcriptome of Age-Associated Deep Subcortical White Matter Lesions Suggests a Neuroprotective Response to Blood–Brain Barrier Dysfunction. Int. J. Mol. Sci. 2024, 25, 4445. https://doi.org/10.3390/ijms25084445
Almansouri T, Waller R, Wharton SB, Heath PR, Matthews FE, Brayne C, van Eeden F, Simpson JE. The Microglial Transcriptome of Age-Associated Deep Subcortical White Matter Lesions Suggests a Neuroprotective Response to Blood–Brain Barrier Dysfunction. International Journal of Molecular Sciences. 2024; 25(8):4445. https://doi.org/10.3390/ijms25084445
Chicago/Turabian StyleAlmansouri, Taghreed, Rachel Waller, Stephen B. Wharton, Paul R. Heath, Fiona E. Matthews, Carol Brayne, Fredericus van Eeden, and Julie E. Simpson. 2024. "The Microglial Transcriptome of Age-Associated Deep Subcortical White Matter Lesions Suggests a Neuroprotective Response to Blood–Brain Barrier Dysfunction" International Journal of Molecular Sciences 25, no. 8: 4445. https://doi.org/10.3390/ijms25084445