
MicroRNA-Mediated Suppression of Glial Cell Line-Derived Neurotrophic Factor Expression Is Modulated by a Schizophrenia-Associated Non-Coding Polymorphism
 ,
,
Abstract
:1. Introduction
2. Results
2.1. Case–Control Association Analysis
2.2. Functional Analysis
2.2.1. In Silico Prediction of microRNA Binding
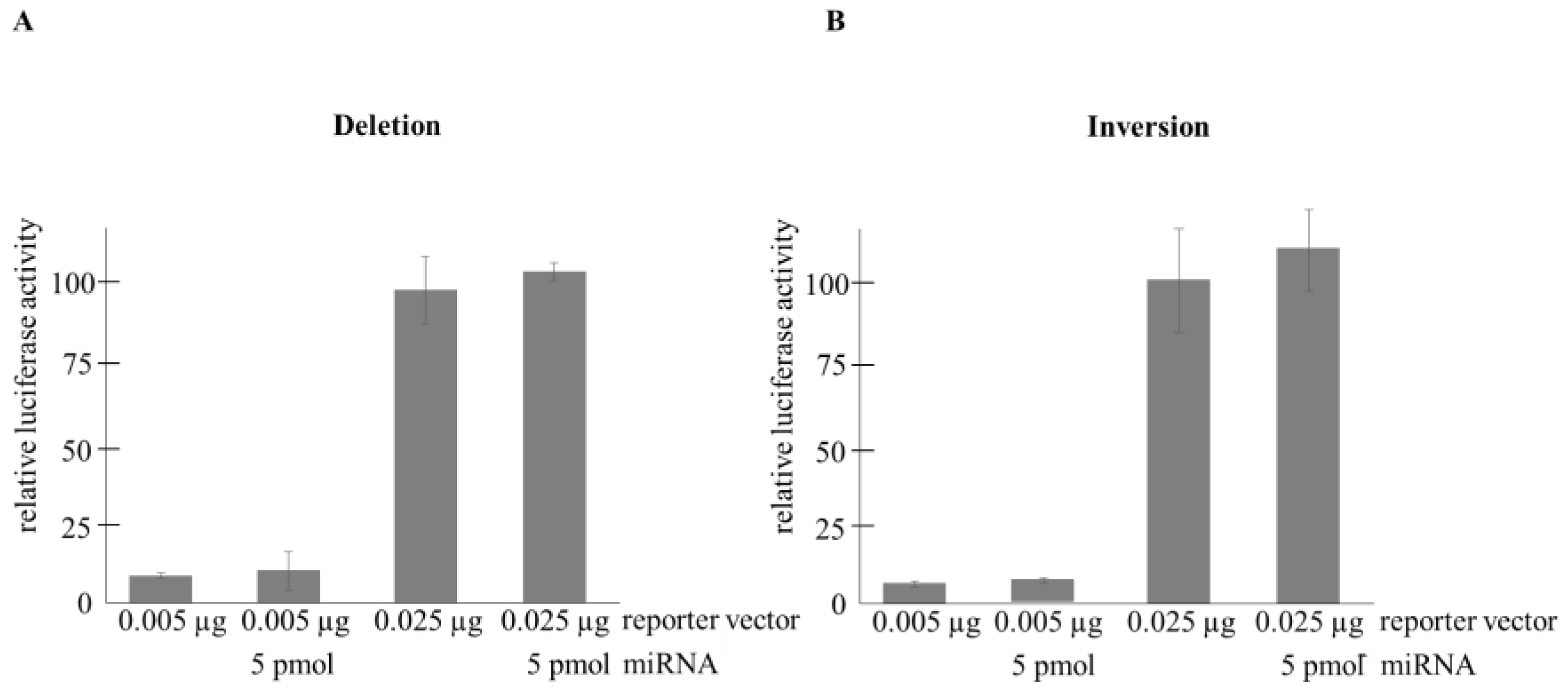
2.2.2. Transient Transfection-Based Reporter Assays
3. Discussion
4. Materials and Methods
4.1. Participants
4.2. Principles of SNP Selection
4.3. DNA Sampling and Purification
4.4. SNP Genotyping
4.5. In Silico Tools
4.6. Construction of Reporter Vectors
4.7. Cell cultures and Transfection
4.8. In Vitro Reporter Assays
4.9. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- McGrath, J.; Saha, S.; Chant, D.; Welham, J. Schizophrenia: A concise overview of incidence, prevalence, and mortality. Epidemiol. Rev. 2008, 30, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Davies, C.; Segre, G.; Estrade, A.; Radua, J.; De Micheli, A.; Provenzani, U.; Oliver, D.; Salazar de Pablo, G.; Ramella-Cravaro, V.; Besozzi, M.; et al. Prenatal and perinatal risk and protective factors for psychosis: A systematic review and meta-analysis. Lancet Psychiatry 2020, 7, 399–410. [Google Scholar] [CrossRef] [PubMed]
- Trubetskoy, V.; Pardinas, A.F.; Qi, T.; Panagiotaropoulou, G.; Awasthi, S.; Bigdeli, T.B.; Bryois, J.; Chen, C.Y.; Dennison, C.A.; Hall, L.S.; et al. Mapping genomic loci implicates genes and synaptic biology in schizophrenia. Nature 2022, 604, 502–508. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.S.; Chu, C.L.; Wu, C.C.; Lu, T. Serum nerve growth factor beta, brain- and glial-derived neurotrophic factor levels and psychopathology in unmedicated patients with schizophrenia. J. Chin. Med. Assoc. 2018, 81, 577–581. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Zhou, C.; Gao, J.; Duan, W.; Yu, M.; Xiao, W.; Zhang, X.; Dong, H.; Wang, X.; Zhang, X. Serum BDNF and GDNF in Chinese male patients with deficit schizophrenia and their relationships with neurocognitive dysfunction. BMC Psychiatry 2019, 19, 254. [Google Scholar] [CrossRef] [PubMed]
- Dremencov, E.; Jezova, D.; Barak, S.; Gaburjakova, J.; Gaburjakova, M.; Kutna, V.; Ovsepian, S.V. Trophic factors as potential therapies for treatment of major mental disorders. Neurosci. Lett. 2021, 764, 136194. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Kim, D.J. GFRA1: A Novel Molecular Target for the Prevention of Osteosarcoma Chemoresistance. Int. J. Mol. Sci. 2018, 19, 1078. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Braun, R.E. Cyclical expression of GDNF is required for spermatogonial stem cell homeostasis. Development 2018, 145, dev151555. [Google Scholar] [CrossRef] [PubMed]
- Costantini, F.; Shakya, R. GDNF/Ret signaling and the development of the kidney. Bioessays 2006, 28, 117–127. [Google Scholar] [CrossRef]
- Tao, L.; Ma, W.; Wu, L.; Xu, M.; Yang, Y.; Zhang, W.; Sha, W.; Li, H.; Xu, J.; Feng, R.; et al. Glial cell line-derived neurotrophic factor (GDNF) mediates hepatic stellate cell activation via ALK5/Smad signalling. Gut 2019, 68, 2214–2227. [Google Scholar] [CrossRef]
- Brisch, R.; Saniotis, A.; Wolf, R.; Bielau, H.; Bernstein, H.G.; Steiner, J.; Bogerts, B.; Braun, K.; Jankowski, Z.; Kumaratilake, J.; et al. The role of dopamine in schizophrenia from a neurobiological and evolutionary perspective: Old fashioned, but still in vogue. Front. Psychiatry 2014, 5, 47. [Google Scholar] [CrossRef] [PubMed]
- Matlik, K.; Voikar, V.; Vilenius, C.; Kulesskaya, N.; Andressoo, J.O. Two-fold elevation of endogenous GDNF levels in mice improves motor coordination without causing side-effects. Sci. Rep. 2018, 8, 11861. [Google Scholar] [CrossRef] [PubMed]
- Tunca, Z.; Kivircik Akdede, B.; Ozerdem, A.; Alkin, T.; Polat, S.; Ceylan, D.; Bayin, M.; Cengizcetin Kocuk, N.; Simsek, S.; Resmi, H.; et al. Diverse glial cell line-derived neurotrophic factor (GDNF) support between mania and schizophrenia: A comparative study in four major psychiatric disorders. Eur. Psychiatry 2015, 30, 198–204. [Google Scholar] [CrossRef] [PubMed]
- Turkmen, B.A.; Yazici, E.; Erdogan, D.G.; Suda, M.A.; Yazici, A.B. BDNF, GDNF, NGF and Klotho levels and neurocognitive functions in acute term of schizophrenia. BMC Psychiatry 2021, 21, 562. [Google Scholar] [CrossRef] [PubMed]
- Xiao, W.; Ye, F.; Ma, L.; Tang, X.; Li, J.; Dong, H.; Sha, W.; Zhang, X. Atypical antipsychotic treatment increases glial cell line-derived neurotrophic factor serum levels in drug-free schizophrenic patients along with improvement of psychotic symptoms and therapeutic effects. Psychiatry Res. 2016, 246, 617–622. [Google Scholar] [CrossRef] [PubMed]
- Yuan, B.; Yuan, M. Changes of Mental State and Serum Prolactin Levels in Patients with Schizophrenia and Depression after Receiving the Combination Therapy of Amisulpride and Chloroprothixol Tablets. Comput. Math. Methods Med. 2022, 2022, 6580030. [Google Scholar] [CrossRef] [PubMed]
- Skibinska, M.; Kapelski, P.; Pawlak, J.; Rajewska-Rager, A.; Dmitrzak-Weglarz, M.; Szczepankiewicz, A.; Czerski, P.; Twarowska-Hauser, J. Glial Cell Line-Derived Neurotrophic Factor (GDNF) serum level in women with schizophrenia and depression, correlation with clinical and metabolic parameters. Psychiatry Res. 2017, 256, 396–402. [Google Scholar] [CrossRef]
- Niitsu, T.; Shirayama, Y.; Matsuzawa, D.; Shimizu, E.; Hashimoto, K.; Iyo, M. Association between serum levels of glial cell-line derived neurotrophic factor and attention deficits in schizophrenia. Neurosci. Lett. 2014, 575, 37–41. [Google Scholar] [CrossRef]
- Hidese, S.; Hattori, K.; Sasayama, D.; Tsumagari, T.; Miyakawa, T.; Matsumura, R.; Yokota, Y.; Ishida, I.; Matsuo, J.; Yoshida, S.; et al. Cerebrospinal fluid neuroplasticity-associated protein levels in patients with psychiatric disorders: A multiplex immunoassay study. Transl. Psychiatry 2020, 10, 161. [Google Scholar] [CrossRef]
- Matlik, K.; Garton, D.R.; Montano-Rodriguez, A.R.; Olfat, S.; Eren, F.; Casserly, L.; Damdimopoulos, A.; Panhelainen, A.; Porokuokka, L.L.; Kopra, J.J.; et al. Elevated endogenous GDNF induces altered dopamine signalling in mice and correlates with clinical severity in schizophrenia. Mol. Psychiatry 2022, 27, 3247–3261. [Google Scholar] [CrossRef]
- Shiwaku, H.; Katayama, S.; Kondo, K.; Nakano, Y.; Tanaka, H.; Yoshioka, Y.; Fujita, K.; Tamaki, H.; Takebayashi, H.; Terasaki, O.; et al. Autoantibodies against NCAM1 from patients with schizophrenia cause schizophrenia-related behavior and changes in synapses in mice. Cell Rep. Med. 2022, 3, 100597. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Kunugi, H.; Nanko, S. Glial cell line-derived neurotrophic factor (GDNF) gene and schizophrenia: Polymorphism screening and association analysis. Psychiatry Res. 2001, 104, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Michelato, A.; Bonvicini, C.; Ventriglia, M.; Scassellati, C.; Randazzo, R.; Bignotti, S.; Beneduce, R.; Riva, M.A.; Gennarelli, M. 3′ UTR (AGG)n repeat of glial cell line-derived neurotrophic factor (GDNF) gene polymorphism in schizophrenia. Neurosci. Lett. 2004, 357, 235–237. [Google Scholar] [CrossRef] [PubMed]
- Williams, H.J.; Norton, N.; Peirce, T.; Dwyer, S.; Williams, N.M.; Moskvina, V.; Owen, M.J.; O’Donovan, M.C. Association analysis of the glial cell line-derived neurotrophic factor (GDNF) gene in schizophrenia. Schizophr. Res. 2007, 97, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.C.; Chen, C.; Zhu, F.; Jia, W.; Gao, C.G. Association of the GDNF gene with depression and heroin dependence, but not schizophrenia, in a Chinese population. Psychiatry Res. 2013, 210, 1296–1298. [Google Scholar] [CrossRef] [PubMed]
- Souza, R.P.; Romano-Silva, M.A.; Lieberman, J.A.; Meltzer, H.Y.; MacNeil, L.T.; Culotti, J.G.; Kennedy, J.L.; Wong, A.H. Genetic association of the GDNF alpha-receptor genes with schizophrenia and clozapine response. J. Psychiatr. Res. 2010, 44, 700–706. [Google Scholar] [CrossRef]
- Safari, R.; Tunca, Z.; Ozerdem, A.; Ceylan, D.; Yalcin, Y.; Sakizli, M. Glial cell-derived neurotrophic factor gene polymorpisms affect severity and functionality of bipolar disorder. J. Integr. Neurosci. 2017, 16, 471–481. [Google Scholar] [CrossRef]
- Kotyuk, E.; Keszler, G.; Nemeth, N.; Ronai, Z.; Sasvari-Szekely, M.; Szekely, A. Glial cell line-derived neurotrophic factor (GDNF) as a novel candidate gene of anxiety. PLoS ONE 2013, 8, e80613. [Google Scholar] [CrossRef]
- Kotyuk, E.; Nemeth, N.; Halmai, Z.; Faludi, G.; Sasvari-Szekely, M.; Szekely, A. Association between mood characteristics and polymorphisms of glial cell line-derived neurotrophic factor (GNDF) in patients with depression. Neuropsychopharmacol. Hung. 2013, 15, 63–72. [Google Scholar]
- Kotyuk, E.; Nemeth, N.; Ronai, Z.; Demetrovics, Z.; Sasvari-Szekely, M.; Szekely, A. Association between smoking behaviour and genetic variants of glial cell line-derived neurotrophic factor. J. Genet. 2016, 95, 811–818. [Google Scholar] [CrossRef]
- Vereczkei, A.; Barta, C.; Magi, A.; Farkas, J.; Eisinger, A.; Kiraly, O.; Belik, A.; Griffiths, M.D.; Szekely, A.; Sasvari-Szekely, M.; et al. FOXN3 and GDNF Polymorphisms as Common Genetic Factors of Substance Use and Addictive Behaviors. J. Pers. Med. 2022, 12, 690. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Pagliaroli, L.; Vereczkei, A.; Kotyuk, E.; Langstieh, B.; Demetrovics, Z.; Barta, C. Association of GDNF and CNTNAP2 gene variants with gambling. J. Behav. Addict. 2019, 8, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Huertas-Fernandez, I.; Gomez-Garre, P.; Madruga-Garrido, M.; Bernal-Bernal, I.; Bonilla-Toribio, M.; Martin-Rodriguez, J.F.; Caceres-Redondo, M.T.; Vargas-Gonzalez, L.; Carrillo, F.; Pascual, A.; et al. GDNF gene is associated with tourette syndrome in a family study. Mov. Disord. 2015, 30, 1115–1120. [Google Scholar] [CrossRef] [PubMed]
- Rethelyi, J.M.; Bakker, S.C.; Polgar, P.; Czobor, P.; Strengman, E.; Pasztor, P.I.; Kahn, R.S.; Bitter, I. Association study of NRG1, DTNBP1, RGS4, G72/G30, and PIP5K2A with schizophrenia and symptom severity in a Hungarian sample. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2010, 153B, 792–801. [Google Scholar] [CrossRef] [PubMed]
- Keller, A.; Groger, L.; Tschernig, T.; Solomon, J.; Laham, O.; Schaum, N.; Wagner, V.; Kern, F.; Schmartz, G.P.; Li, Y.; et al. miRNATissueAtlas2: An update to the human miRNA tissue atlas. Nucleic Acids Res. 2022, 50, D211–D221. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhao, X.; Wu, Z.; Huang, G.; Lin, R.; Chen, H.; Xu, K.; Sun, K.; Zhou, H.; Shu, J. Identification of Differentially Expressed mRNAs and miRNAs and Related Regulatory Networks in Cumulus Oophorus Complexes Associated with Fertilization. Reprod. Sci. 2024. online, ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, M.; Ghosh, S.; Goswami, S. Investigating the interference of single nucleotide polymorphisms with miRNA mediated gene regulation in pancreatic ductal adenocarcinoma: An in silico approach. Gene 2022, 819, 146259. [Google Scholar] [CrossRef] [PubMed]
- Mirabbasi, R.; Ebrahimi, S.O.; Tavakoli, F.; Reiisi, S. Novel polymorphism rs12402181 in the mature sequence of hsa-miR-3117-3p has a protective effect against breast cancer development by affecting miRNA processing and function. 3 Biotech 2023, 13, 349. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Li, Y.; Zhu, X.; Wang, K.; Zhu, X.; Jiang, Y.; Xu, L.; Li, J. Circular RNA-VPS13A attenuates diabetes-induced enteric glia damage by targeting miR-182/GDNF Axis. Acta Biochim. Biophys. Sin. 2022, 54, 999–1007. [Google Scholar] [CrossRef]
- Ravegnini, G.; Serrano, C.; Simeon, V.; Sammarini, G.; Nannini, M.; Roversi, E.; Urbini, M.; Ferre, F.; Ricci, R.; Tarantino, G.; et al. The rs17084733 variant in the KIT 3′ UTR disrupts a miR-221/222 binding site in gastrointestinal stromal tumour: A sponge-like mechanism conferring disease susceptibility. Epigenetics 2019, 14, 545–557. [Google Scholar] [CrossRef]
- Konovalova, J.; Gerasymchuk, D.; Arroyo, S.N.; Kluske, S.; Mastroianni, F.; Pereyra, A.V.; Domanskyi, A. Human-Specific Regulation of Neurotrophic Factors MANF and CDNF by microRNAs. Int. J. Mol. Sci. 2021, 22, 9691. [Google Scholar] [CrossRef]
- Akkouh, I.A.; Hughes, T.; Steen, V.M.; Glover, J.C.; Andreassen, O.A.; Djurovic, S.; Szabo, A. Transcriptome analysis reveals disparate expression of inflammation-related miRNAs and their gene targets in iPSC-astrocytes from people with schizophrenia. Brain Behav. Immun. 2021, 94, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Gash, D.M.; Gerhardt, G.A.; Bradley, L.H.; Wagner, R.; Slevin, J.T. GDNF clinical trials for Parkinson’s disease: A critical human dimension. Cell Tissue Res. 2020, 382, 65–70. [Google Scholar] [CrossRef]
- Shim, S.H.; Hwangbo, Y.; Yoon, H.J.; Kwon, Y.J.; Lee, H.Y.; Hwang, J.A.; Kim, Y.K. Increased levels of plasma glial-derived neurotrophic factor in children with attention deficit hyperactivity disorder. Nord. J. Psychiatry 2015, 69, 546–551. [Google Scholar] [CrossRef] [PubMed]
- Oh-hashi, K.; Hirata, Y.; Kiuchi, K. Characterization of 3′-untranslated region of the mouse GDNF gene. BMC Mol. Biol. 2012, 13, 2. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Kopra, J.; Varendi, K.; Porokuokka, L.L.; Panhelainen, A.; Kuure, S.; Marshall, P.; Karalija, N.; Harma, M.A.; Vilenius, C.; et al. GDNF Overexpression from the Native Locus Reveals its Role in the Nigrostriatal Dopaminergic System Function. PLoS Genet. 2015, 11, e1005710. [Google Scholar] [CrossRef]
- Matlik, K.; Olfat, S.; Cowlishaw, M.C.; Moreno, E.D.; Ollila, S.; Andressoo, J.O. In vivo modulation of endogenous gene expression via CRISPR/Cas9-mediated 3’UTR editing. Heliyon 2023, 9, e13844. [Google Scholar] [CrossRef]
- Yue, X.H.; Guo, L.; Wang, Z.Y.; Jia, T.H. Inhibition of miR-17-5p promotes mesenchymal stem cells to repair spinal cord injury. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 3899–3907. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, K. Downregulation of MicroRNA-33-5p Protected Bupivacaine-Induced Apoptosis in Murine Dorsal Root Ganglion Neurons Through GDNF. Neurotox. Res. 2019, 35, 860–866. [Google Scholar] [CrossRef]
- Zheng, T.; Zhang, T.; Zhang, W.; Lv, K.; Jia, D.; Yang, F.; Sun, Y.; Lian, J.; Wang, R. Icariside II facilitates the differentiation of ADSCs to schwann cells and restores erectile dysfunction through regulation of miR-33/GDNF axis. Biomed. Pharmacother. 2020, 125, 109888. [Google Scholar] [CrossRef]
- Shen, W.S.; Li, C.F.; Zhou, Z.S.; Zhai, N.N.; Pan, L.P. MicroRNA-204 silencing relieves pain of cervical spondylotic radiculopathy by targeting GDNF. Gene Ther. 2020, 27, 254–265. [Google Scholar] [CrossRef] [PubMed]
- Weng, N.; Sun, J.; Kuang, S.; Lan, H.; He, Q.; Yang, H.; Zhang, L.; Xue, H. MicroRNA-451 Aggravates Kainic Acid-induced Seizure and Neuronal Apoptosis by Targeting GDNF. Curr. Neurovasc. Res. 2020, 17, 50–57. [Google Scholar] [CrossRef]
- Yoong, L.F.; Wan, G.; Too, H.P. Glial cell-line derived neurotrophic factor and neurturin regulate the expressions of distinct miRNA precursors through the activation of GFRα2. J. Neurochem. 2006, 98, 1149–1158. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; Zhu, X.; Sun, Y.; Li, Z.; Li, X.; Ai, L.; He, Y.; Liu, Y.; Jia, N.; Hu, G.; et al. Identification of Peripheral Blood miRNA Biomarkers in First-Episode Drug-Free Schizophrenia Patients Using Bioinformatics Strategy. Mol. Neurobiol. 2022, 59, 4730–4746. [Google Scholar] [CrossRef]
- Usuba, W.; Urabe, F.; Yamamoto, Y.; Matsuzaki, J.; Sasaki, H.; Ichikawa, M.; Takizawa, S.; Aoki, Y.; Niida, S.; Kato, K.; et al. Circulating miRNA panels for specific and early detection in bladder cancer. Cancer Sci. 2019, 110, 408–419. [Google Scholar] [CrossRef]
- Alqurashi, N.; Hashimi, S.M.; Alowaidi, F.; Ivanovski, S.; Farag, A.; Wei, M.Q. miR-496, miR-1185, miR-654, miR-3183 and miR-495 are downregulated in colorectal cancer cells and have putative roles in the mTOR pathway. Oncol. Lett. 2019, 18, 1657–1668. [Google Scholar] [CrossRef]
- Wang, T.W.; Chern, E.; Hsu, C.W.; Tseng, K.C.; Chao, H.M. SIRT1-Mediated Expression of CD24 and Epigenetic Suppression of Novel Tumor Suppressor miR-1185-1 Increases Colorectal Cancer Stemness. Cancer Res. 2020, 80, 5257–5269. [Google Scholar] [CrossRef]
- Garcia-Lacarte, M.; Mansego, M.L.; Zulet, M.A.; Martinez, J.A.; Milagro, F.I. miR-1185-1 and miR-548q Are Biomarkers of Response to Weight Loss and Regulate the Expression of GSK3B. Cells 2019, 8, 1548. [Google Scholar] [CrossRef] [PubMed]
- Delay, C.; Grenier-Boley, B.; Amouyel, P.; Dumont, J.; Lambert, J.C. miRNA-dependent target regulation: Functional characterization of single-nucleotide polymorphisms identified in genome-wide association studies of Alzheimer’s disease. Alzheimers Res. Ther. 2016, 8, 20. [Google Scholar] [CrossRef]
- Kovacs-Nagy, R.; Elek, Z.; Szekely, A.; Nanasi, T.; Sasvari-Szekely, M.; Ronai, Z. Association of aggression with a novel microRNA binding site polymorphism in the wolframin gene. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2013, 162B, 404–412. [Google Scholar] [CrossRef]
- Balázs, J.; Bitter, I.; Hideg, K.; Vitrai, J. The Hungarian version of the M.I.N.I. and the M.I.N.I. Plus. Psychiatr. Hung. 1998, 13, 160–168. (In Hungarian) [Google Scholar]
- Liu, H.; Naismith, J.H. An efficient one-step site-directed deletion, insertion, single and multiple-site plasmid mutagenesis protocol. BMC Biotechnol. 2008, 8, 91. [Google Scholar] [CrossRef] [PubMed]
- Barrett, J.C.; Fry, B.; Maller, J.; Daly, M.J. Haploview: Analysis and visualization of LD and haplotype maps. Bioinformatics 2005, 21, 263–265. [Google Scholar] [CrossRef] [PubMed]
- Menyhart, O.; Weltz, B.; Gyorffy, B. MultipleTesting.com: A tool for life science researchers for multiple hypothesis testing correction. PLoS ONE 2021, 16, e0245824. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
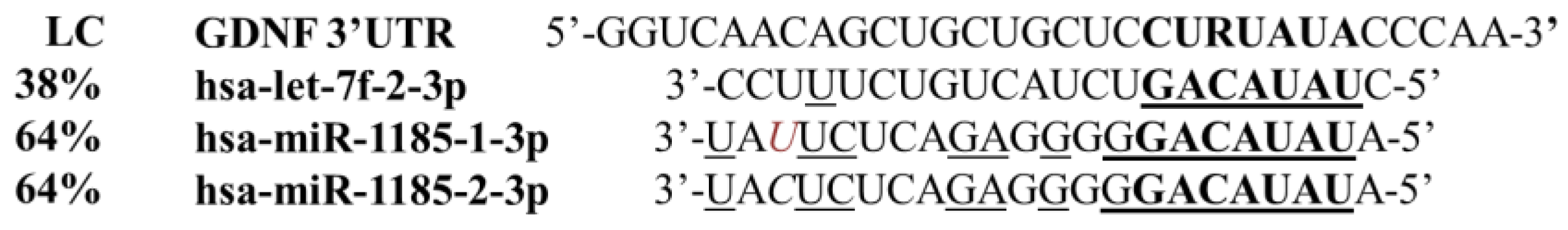{kind=link}
{kind=link}
{kind=link}
| SNP | Allele | Genomic Location (GRCh38) | Intragenic Location | MAF | TaqMan ID | References |
|---|---|---|---|---|---|---|
| rs11111 | A/G | 5:37814000 | 3′ UTR | 0.16 (G) | C___8813050_1_ | [28,30] |
| rs1549250 | G/T | 5:37821119 | intronic | 0.42 (G) | C__11553504_10 | [28,30,31] |
| rs2910702 | A/G | 5:37828202 | intronic | 0.25 (G) | C__15948353_10 | [24,25,28,30] |
| rs3096140 | C/T | 5:37832731 | intronic | 0.30 (C) | C___1395038_20 | [28,30,33] |
| rs3812047 | A/G | 5:37835296 | intronic | 0.13 (A) | C__27492935_10 | [28,29,30] |
| SNP | Genotype | Genotype Frequency (%) | HWE | p Value | Allele Frequency (%) | p Value | |||
|---|---|---|---|---|---|---|---|---|---|
| Control | Patient | Control | Patient | Genotype | Control | Patient | Allele | ||
| rs11111 | AA | 75.9 | 66.2 | 0.132 | 0.44 | 0.006 | 86.7 | 80.9 | |
| AG | 21.7 | 29.5 | 0.001 | ||||||
| GG | 2.4 | 4.3 | 13.3 | 19.1 | |||||
| rs1549250 | TT | 33.5 | 33.3 | 0.806 | 0.799 | 0.988 | 57.7 | 57.3 | |
| TG | 48.4 | 48.1 | 0.957 | ||||||
| GG | 18.1 | 18.6 | 42.3 | 42.7 | |||||
| rs2910702 | AA | 54.8 | 59.9 | 0.667 | 0.557 | 0.360 | 74.3 | 77.0 | |
| AG | 38.9 | 34.1 | 0.157 | ||||||
| GG | 6.3 | 6.0 | 25.7 | 23.0 | |||||
| rs3096140 | CC | 48.0 | 51.1 | 0.700 | 0.132 | 0.329 | 69.5 | 70.0 | |
| CT | 43.0 | 37.8 | 0.424 | ||||||
| TT | 9.0 | 11.1 | 30.5 | 30.0 | |||||
| rs3812047 | AA | 76.6 | 74.3 | 0.781 | 0.785 | 0.755 | 87.4 | 86.4 | |
| AG | 21.8 | 24.0 | 0.494 | ||||||
| GG | 1.7 | 1.7 | 12.6 | 13.6 | |||||
| SNP | Positive Factor | Negative Factor | Hostility Factor | Cognitive Factor | Depression Factor |
|---|---|---|---|---|---|
| rs11111 AG | 0.0320 | 0.2669 | 0.4243 | 0.0405 | 0.2466 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Keszler, G.; Vékony, B.; Elek, Z.; Nemoda, Z.; Angyal, N.; Bánlaki, Z.; Kovács-Nagy, R.; Rónai, Z.; Réthelyi, J.M. MicroRNA-Mediated Suppression of Glial Cell Line-Derived Neurotrophic Factor Expression Is Modulated by a Schizophrenia-Associated Non-Coding Polymorphism. Int. J. Mol. Sci. 2024, 25, 4477. https://doi.org/10.3390/ijms25084477
Keszler G, Vékony B, Elek Z, Nemoda Z, Angyal N, Bánlaki Z, Kovács-Nagy R, Rónai Z, Réthelyi JM. MicroRNA-Mediated Suppression of Glial Cell Line-Derived Neurotrophic Factor Expression Is Modulated by a Schizophrenia-Associated Non-Coding Polymorphism. International Journal of Molecular Sciences. 2024; 25(8):4477. https://doi.org/10.3390/ijms25084477
Chicago/Turabian StyleKeszler, Gergely, Bálint Vékony, Zsuzsanna Elek, Zsófia Nemoda, Nóra Angyal, Zsófia Bánlaki, Réka Kovács-Nagy, Zsolt Rónai, and János M. Réthelyi. 2024. "MicroRNA-Mediated Suppression of Glial Cell Line-Derived Neurotrophic Factor Expression Is Modulated by a Schizophrenia-Associated Non-Coding Polymorphism" International Journal of Molecular Sciences 25, no. 8: 4477. https://doi.org/10.3390/ijms25084477