
Single-Cell Informatics for Tumor Microenvironment and Immunotherapy


Abstract
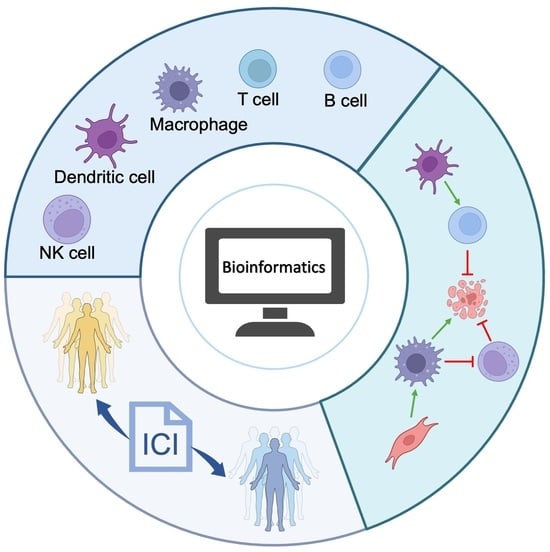
:
1. Introduction
2. Single-Cell Technologies
2.1. Single-Cell Sequencing
2.2. Spatial Single-Cell Technologies
3. Bioinformatics Tools for Single-Cell Data Analysis
3.1. Bioinformatics Dissection of Tumor–Immune Cell Microenvironment
3.1.1. Quality Control
3.1.2. Gene Count Normalization
3.1.3. Confounder Correction
3.1.4. Feature Selection
3.1.5. Dimensionality Reduction
3.1.6. Cell Clustering
3.1.7. Annotation
3.2. Bioinformatics Analysis of Tumor–Immune Cell Communication
3.2.1. Ligand–Receptor Interaction
3.2.2. Intracellular Signaling Communication
3.2.3. Spatial-Based Communication Inference
3.2.4. Experimental Validation of the Inferred CCC
4. Application of Single-Cell Bioinformatics Tools in Understanding Patient Heterogeneity and Treatment Responses
4.1. Utilizing Bioinformatics Analysis to Explore the Tumor Microenvironment (TME)
4.2. Application of Bioinformatics Analysis Regarding Tumor–Immune Cell Communication
5. Challenges and Future Directions
5.1. Challenges Faced by TME Dissection via Single-Cell Technologies
5.2. Challenges Faced by Cell–Cell Interaction Analysis via Single-Cell Technologies
5.3. Challenges of Integrating Diverse Single-Cell Datasets
5.4. Outlook: Translational Insights of Single-Cell Technologies in Clinical Application
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Li, N.; Zhu, Q.; Tian, Y.; Ahn, K.J.; Wang, X.; Cramer, Z.; Jou, J.; Folkert, I.W.; Yu, P.; Adams-Tzivelekidis, S.; et al. Mapping and modeling human colorectal carcinoma interactions with the tumor microenvironment. Nat. Commun. 2023, 14, 7915. [Google Scholar] [CrossRef] [PubMed]
- Nofech-Mozes, I.; Soave, D.; Awadalla, P.; Abelson, S. Pan-cancer classification of single cells in the tumour microenvironment. Nat. Commun. 2023, 14, 1615. [Google Scholar] [CrossRef] [PubMed]
- Sorin, M.; Rezanejad, M.; Karimi, E.; Fiset, B.; Desharnais, L.; Perus, L.J.M.; Milette, S.; Yu, M.W.; Maritan, S.M.; Dore, S.; et al. Single-cell spatial landscapes of the lung tumour immune microenvironment. Nature 2023, 614, 548–554. [Google Scholar] [CrossRef] [PubMed]
- Tran, K.A.; Addala, V.; Johnston, R.L.; Lovell, D.; Bradley, A.; Koufariotis, L.T.; Wood, S.; Wu, S.Z.; Roden, D.; Al-Eryani, G.; et al. Performance of tumour microenvironment deconvolution methods in breast cancer using single-cell simulated bulk mixtures. Nat. Commun. 2023, 14, 5758. [Google Scholar] [CrossRef] [PubMed]
- Xue, R.; Zhang, Q.; Cao, Q.; Kong, R.; Xiang, X.; Liu, H.; Feng, M.; Wang, F.; Cheng, J.; Li, Z.; et al. Liver tumour immune microenvironment subtypes and neutrophil heterogeneity. Nature 2022, 612, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Nolz, J.C. Molecular mechanisms of CD8+ T cell trafficking and localization. Cell. Mol. Life Sci. 2015, 72, 2461–2473. [Google Scholar] [CrossRef] [PubMed]
- Schoenberger, S.P.; Toes, R.E.; van der Voort, E.I.; Offringa, R.; Melief, C.J. T-cell help for cytotoxic T lymphocytes is mediated by CD40-CD40L interactions. Nature 1998, 393, 480–483. [Google Scholar] [CrossRef] [PubMed]
- Bennett, S.R.; Carbone, F.R.; Karamalis, F.; Miller, J.F.; Heath, W.R. Induction of a CD8+ cytotoxic T lymphocyte response by cross-priming requires cognate CD4+ T cell help. J. Exp. Med. 1997, 186, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Sokke Umeshappa, C.; Hebbandi Nanjundappa, R.; Xie, Y.; Freywald, A.; Deng, Y.; Ma, H.; Xiang, J. CD154 and IL-2 signaling of CD4+ T cells play a critical role in multiple phases of CD8+ CTL responses following adenovirus vaccination. PLoS ONE 2012, 7, e47004. [Google Scholar] [CrossRef]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef]
- Veglia, F.; Sanseviero, E.; Gabrilovich, D.I. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat. Rev. Immunol. 2021, 21, 485–498. [Google Scholar] [CrossRef] [PubMed]
- Ghoshdastider, U.; Rohatgi, N.; Mojtabavi Naeini, M.; Baruah, P.; Revkov, E.; Guo, Y.A.; Rizzetto, S.; Wong, A.M.L.; Solai, S.; Nguyen, T.T.; et al. Pan-Cancer Analysis of Ligand-Receptor Cross-talk in the Tumor Microenvironment. Cancer Res. 2021, 81, 1802–1812. [Google Scholar] [CrossRef] [PubMed]
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 2000, 192, 1027–1034. [Google Scholar] [CrossRef] [PubMed]
- Hoekstra, M.E.; Bornes, L.; Dijkgraaf, F.E.; Philips, D.; Pardieck, I.N.; Toebes, M.; Thommen, D.S.; van Rheenen, J.; Schumacher, T.N.M. Long-distance modulation of bystander tumor cells by CD8+ T cell-secreted IFNgamma. Nat. Cancer 2020, 1, 291–301. [Google Scholar] [CrossRef] [PubMed]
- Wing, K.; Onishi, Y.; Prieto-Martin, P.; Yamaguchi, T.; Miyara, M.; Fehervari, Z.; Nomura, T.; Sakaguchi, S. CTLA-4 control over Foxp3+ regulatory T cell function. Science 2008, 322, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef] [PubMed]
- Zou, W.; Wolchok, J.D.; Chen, L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci. Transl. Med. 2016, 8, 328rv324. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhang, Z.; Zheng, X.; Tao, H.; Zhang, S.; Ma, J.; Liu, Z.; Wang, J.; Qian, Y.; Cui, P.; et al. Response Efficacy of PD-1 and PD-L1 Inhibitors in Clinical Trials: A Systematic Review and Meta-Analysis. Front. Oncol. 2021, 11, 562315. [Google Scholar] [CrossRef]
- Valero, C.; Lee, M.; Hoen, D.; Zehir, A.; Berger, M.F.; Seshan, V.E.; Chan, T.A.; Morris, L.G.T. Response Rates to Anti-PD-1 Immunotherapy in Microsatellite-Stable Solid Tumors with 10 or More Mutations per Megabase. JAMA Oncol. 2021, 7, 739–743. [Google Scholar] [CrossRef]
- Lim, B.; Lin, Y.; Navin, N. Advancing Cancer Research and Medicine with Single-Cell Genomics. Cancer Cell 2020, 37, 456–470. [Google Scholar] [CrossRef]
- Guruprasad, P.; Lee, Y.G.; Kim, K.H.; Ruella, M. The current landscape of single-cell transcriptomics for cancer immunotherapy. J. Exp. Med. 2021, 218, e20201574. [Google Scholar] [CrossRef]
- Bennett, H.M.; Stephenson, W.; Rose, C.M.; Darmanis, S. Single-cell proteomics enabled by next-generation sequencing or mass spectrometry. Nat. Methods 2023, 20, 363–374. [Google Scholar] [CrossRef]
- Flynn, E.; Almonte-Loya, A.; Fragiadakis, G.K. Single-Cell Multiomics. Annu. Rev. Biomed. Data Sci. 2023, 6, 313–337. [Google Scholar] [CrossRef]
- Zappia, L.; Theis, F.J. Over 1000 tools reveal trends in the single-cell RNA-seq analysis landscape. Genome Biol. 2021, 22, 301. [Google Scholar] [CrossRef]
- Davis, R.T.; Blake, K.; Ma, D.; Gabra, M.B.I.; Hernandez, G.A.; Phung, A.T.; Yang, Y.; Maurer, D.; Lefebvre, A.; Alshetaiwi, H.; et al. Transcriptional diversity and bioenergetic shift in human breast cancer metastasis revealed by single-cell RNA sequencing. Nat. Cell Biol. 2020, 22, 310–320. [Google Scholar] [CrossRef] [PubMed]
- Lozano, A.X.; Chaudhuri, A.A.; Nene, A.; Bacchiocchi, A.; Earland, N.; Vesely, M.D.; Usmani, A.; Turner, B.E.; Steen, C.B.; Luca, B.A.; et al. T cell characteristics associated with toxicity to immune checkpoint blockade in patients with melanoma. Nat. Med. 2022, 28, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Hirz, T.; Mei, S.; Sarkar, H.; Kfoury, Y.; Wu, S.; Verhoeven, B.M.; Subtelny, A.O.; Zlatev, D.V.; Wszolek, M.W.; Salari, K.; et al. Dissecting the immune suppressive human prostate tumor microenvironment via integrated single-cell and spatial transcriptomic analyses. Nat. Commun. 2023, 14, 663. [Google Scholar] [CrossRef] [PubMed]
- Causer, A.; Tan, X.; Lu, X.; Moseley, P.; Teoh, S.M.; Molotkov, N.; McGrath, M.; Kim, T.; Simpson, P.T.; Perry, C.; et al. Deep spatial-omics analysis of Head & Neck carcinomas provides alternative therapeutic targets and rationale for treatment failure. NPJ Precis Oncol. 2023, 7, 89. [Google Scholar] [CrossRef]
- Sathe, A.; Mason, K.; Grimes, S.M.; Zhou, Z.; Lau, B.T.; Bai, X.; Su, A.; Tan, X.; Lee, H.; Suarez, C.J.; et al. Colorectal Cancer Metastases in the Liver Establish Immunosuppressive Spatial Networking between Tumor-Associated SPP1+ Macrophages and Fibroblasts. Clin. Cancer Res. 2023, 29, 244–260. [Google Scholar] [CrossRef]
- Zhu, J.; Fan, Y.; Xiong, Y.; Wang, W.; Chen, J.; Xia, Y.; Lei, J.; Gong, L.; Sun, S.; Jiang, T. Delineating the dynamic evolution from preneoplasia to invasive lung adenocarcinoma by integrating single-cell RNA sequencing and spatial transcriptomics. Exp. Mol. Med. 2022, 54, 2060–2076. [Google Scholar] [CrossRef]
- Picelli, S.; Bjorklund, A.K.; Faridani, O.R.; Sagasser, S.; Winberg, G.; Sandberg, R. Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat. Methods 2013, 10, 1096–1098. [Google Scholar] [CrossRef] [PubMed]
- Jaitin, D.A.; Kenigsberg, E.; Keren-Shaul, H.; Elefant, N.; Paul, F.; Zaretsky, I.; Mildner, A.; Cohen, N.; Jung, S.; Tanay, A.; et al. Massively parallel single-cell RNA-seq for marker-free decomposition of tissues into cell types. Science 2014, 343, 776–779. [Google Scholar] [CrossRef] [PubMed]
- Bageritz, J.; Raddi, G. Single-Cell RNA Sequencing with Drop-Seq. Methods Mol. Biol. 2019, 1979, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Zilionis, R.; Nainys, J.; Veres, A.; Savova, V.; Zemmour, D.; Klein, A.M.; Mazutis, L. Single-cell barcoding and sequencing using droplet microfluidics. Nat. Protoc. 2017, 12, 44–73. [Google Scholar] [CrossRef] [PubMed]
- Jarosch, S.; Kohlen, J.; Wagner, S.; D’Ippolito, E.; Busch, D.H. ChipCytometry for multiplexed detection of protein and mRNA markers on human FFPE tissue samples. STAR Protoc. 2022, 3, 101374. [Google Scholar] [CrossRef] [PubMed]
- Kinkhabwala, A.; Herbel, C.; Pankratz, J.; Yushchenko, D.A.; Ruberg, S.; Praveen, P.; Reiss, S.; Rodriguez, F.C.; Schafer, D.; Kollet, J.; et al. MACSima imaging cyclic staining (MICS) technology reveals combinatorial target pairs for CAR T cell treatment of solid tumors. Sci. Rep. 2022, 12, 1911. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.R.; Fallahi-Sichani, M.; Chen, J.Y.; Sorger, P.K. Cyclic Immunofluorescence (CycIF), A Highly Multiplexed Method for Single-cell Imaging. Curr. Protoc. Chem. Biol. 2016, 8, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.H.; Boettiger, A.N.; Moffitt, J.R.; Wang, S.; Zhuang, X. RNA imaging. Spatially resolved, highly multiplexed RNA profiling in single cells. Science 2015, 348, aaa6090. [Google Scholar] [CrossRef] [PubMed]
- Eng, C.L.; Lawson, M.; Zhu, Q.; Dries, R.; Koulena, N.; Takei, Y.; Yun, J.; Cronin, C.; Karp, C.; Yuan, G.C.; et al. Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH. Nature 2019, 568, 235–239. [Google Scholar] [CrossRef]
- Stickels, R.R.; Murray, E.; Kumar, P.; Li, J.; Marshall, J.L.; Di Bella, D.J.; Arlotta, P.; Macosko, E.Z.; Chen, F. Highly sensitive spatial transcriptomics at near-cellular resolution with Slide-seqV2. Nat. Biotechnol. 2021, 39, 313–319. [Google Scholar] [CrossRef]
- Goltsev, Y.; Samusik, N.; Kennedy-Darling, J.; Bhate, S.; Hale, M.; Vazquez, G.; Black, S.; Nolan, G.P. Deep Profiling of Mouse Splenic Architecture with CODEX Multiplexed Imaging. Cell 2018, 174, 968–981.e915. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Bhatt, R.; Brown, C.; Brown, E.A.; Buhr, D.L.; Chantranuvatana, K.; Danaher, P.; Dunaway, D.; Garrison, R.G.; Geiss, G.; et al. High-plex imaging of RNA and proteins at subcellular resolution in fixed tissue by spatial molecular imaging. Nat. Biotechnol. 2022, 40, 1794–1806. [Google Scholar] [CrossRef] [PubMed]
- Janesick, A.; Shelansky, R.; Gottscho, A.D.; Wagner, F.; Williams, S.R.; Rouault, M.; Beliakoff, G.; Morrison, C.A.; Oliveira, M.F.; Sicherman, J.T.; et al. High resolution mapping of the tumor microenvironment using integrated single-cell, spatial and in situ analysis. Nat. Commun. 2023, 14, 8353. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yang, M.; Deng, Y.; Su, G.; Enninful, A.; Guo, C.C.; Tebaldi, T.; Zhang, D.; Kim, D.; Bai, Z.; et al. High-Spatial-Resolution Multi-Omics Sequencing via Deterministic Barcoding in Tissue. Cell 2020, 183, 1665–1681.e1618. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Stuart, T.; Kowalski, M.H.; Choudhary, S.; Hoffman, P.; Hartman, A.; Srivastava, A.; Molla, G.; Madad, S.; Fernandez-Granda, C.; et al. Dictionary learning for integrative, multimodal and scalable single-cell analysis. Nat. Biotechnol. 2023, 42, 293–304. [Google Scholar] [CrossRef] [PubMed]
- Wolf, F.A.; Angerer, P.; Theis, F.J. SCANPY: Large-scale single-cell gene expression data analysis. Genome Biol. 2018, 19, 15. [Google Scholar] [CrossRef] [PubMed]
- Amezquita, R.A.; Lun, A.T.L.; Becht, E.; Carey, V.J.; Carpp, L.N.; Geistlinger, L.; Marini, F.; Rue-Albrecht, K.; Risso, D.; Soneson, C.; et al. Orchestrating single-cell analysis with Bioconductor. Nat. Methods 2020, 17, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Werba, G.; Weissinger, D.; Kawaler, E.A.; Zhao, E.; Kalfakakou, D.; Dhara, S.; Wang, L.; Lim, H.B.; Oh, G.; Jing, X.; et al. Single-cell RNA sequencing reveals the effects of chemotherapy on human pancreatic adenocarcinoma and its tumor microenvironment. Nat. Commun. 2023, 14, 797. [Google Scholar] [CrossRef] [PubMed]
- Glasner, A.; Rose, S.A.; Sharma, R.; Gudjonson, H.; Chu, T.; Green, J.A.; Rampersaud, S.; Valdez, I.K.; Andretta, E.S.; Dhillon, B.S.; et al. Conserved transcriptional connectivity of regulatory T cells in the tumor microenvironment informs new combination cancer therapy strategies. Nat. Immunol. 2023, 24, 1020–1035. [Google Scholar] [CrossRef]
- Young, M.D.; Behjati, S. SoupX removes ambient RNA contamination from droplet-based single-cell RNA sequencing data. Gigascience 2020, 9, giaa151. [Google Scholar] [CrossRef]
- Fleming, S.J.; Chaffin, M.D.; Arduini, A.; Akkad, A.D.; Banks, E.; Marioni, J.C.; Philippakis, A.A.; Ellinor, P.T.; Babadi, M. Unsupervised removal of systematic background noise from droplet-based single-cell experiments using CellBender. Nat. Methods 2023, 20, 1323–1335. [Google Scholar] [CrossRef]
- Carpen, L.; Falvo, P.; Orecchioni, S.; Mitola, G.; Hillje, R.; Mazzara, S.; Mancuso, P.; Pileri, S.; Raveane, A.; Bertolini, F. A single-cell transcriptomic landscape of innate and adaptive intratumoral immunity in triple negative breast cancer during chemo- and immunotherapies. Cell Death Discov. 2022, 8, 106. [Google Scholar] [CrossRef]
- Germain, P.L.; Lun, A.; Garcia Meixide, C.; Macnair, W.; Robinson, M.D. Doublet identification in single-cell sequencing data using scDblFinder. F1000Res 2021, 10, 979. [Google Scholar] [CrossRef]
- Liang, L.; Yu, J.; Li, J.; Li, N.; Liu, J.; Xiu, L.; Zeng, J.; Wang, T.; Wu, L. Integration of scRNA-Seq and Bulk RNA-Seq to Analyse the Heterogeneity of Ovarian Cancer Immune Cells and Establish a Molecular Risk Model. Front. Oncol. 2021, 11, 711020. [Google Scholar] [CrossRef]
- Hafemeister, C.; Satija, R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 2019, 20, 296. [Google Scholar] [CrossRef]
- Ma, K.Y.; Schonnesen, A.A.; Brock, A.; Van Den Berg, C.; Eckhardt, S.G.; Liu, Z.; Jiang, N. Single-cell RNA sequencing of lung adenocarcinoma reveals heterogeneity of immune response-related genes. JCI Insight 2019, 4, e121387. [Google Scholar] [CrossRef]
- Bacher, R.; Chu, L.F.; Leng, N.; Gasch, A.P.; Thomson, J.A.; Stewart, R.M.; Newton, M.; Kendziorski, C. SCnorm: Robust normalization of single-cell RNA-seq data. Nat. Methods 2017, 14, 584–586. [Google Scholar] [CrossRef]
- Wang, X.; Frederick, J.; Wang, H.; Hui, S.; Backman, V.; Ji, Z. Spike-in normalization for single-cell RNA-seq reveals dynamic global transcriptional activity mediating anticancer drug response. NAR Genom. Bioinform. 2021, 3, lqab054. [Google Scholar] [CrossRef]
- Vallejos, C.A.; Marioni, J.C.; Richardson, S. BASiCS: Bayesian Analysis of Single-Cell Sequencing Data. PLoS Comput. Biol. 2015, 11, e1004333. [Google Scholar] [CrossRef]
- Wang, Y.; Fan, J.L.; Melms, J.C.; Amin, A.D.; Georgis, Y.; Barrera, I.; Ho, P.; Tagore, S.; Abril-Rodriguez, G.; He, S.; et al. Multimodal single-cell and whole-genome sequencing of small, frozen clinical specimens. Nat. Genet. 2023, 55, 19–25. [Google Scholar] [CrossRef]
- Prazanowska, K.H.; Lim, S.B. An integrated single-cell transcriptomic dataset for non-small cell lung cancer. Sci. Data 2023, 10, 167. [Google Scholar] [CrossRef]
- Lopez, R.; Regier, J.; Cole, M.B.; Jordan, M.I.; Yosef, N. Deep generative modeling for single-cell transcriptomics. Nat. Methods 2018, 15, 1053–1058. [Google Scholar] [CrossRef]
- Korsunsky, I.; Millard, N.; Fan, J.; Slowikowski, K.; Zhang, F.; Wei, K.; Baglaenko, Y.; Brenner, M.; Loh, P.R.; Raychaudhuri, S. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 2019, 16, 1289–1296. [Google Scholar] [CrossRef]
- Zhao, S.; Ye, B.; Chi, H.; Cheng, C.; Liu, J. Identification of peripheral blood immune infiltration signatures and construction of monocyte-associated signatures in ovarian cancer and Alzheimer’s disease using single-cell sequencing. Heliyon 2023, 9, e17454. [Google Scholar] [CrossRef]
- Satija, R.; Farrell, J.A.; Gennert, D.; Schier, A.F.; Regev, A. Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol. 2015, 33, 495–502. [Google Scholar] [CrossRef]
- Zheng, S.C.; Stein-O’Brien, G.; Augustin, J.J.; Slosberg, J.; Carosso, G.A.; Winer, B.; Shin, G.; Bjornsson, H.T.; Goff, L.A.; Hansen, K.D. Universal prediction of cell-cycle position using transfer learning. Genome Biol. 2022, 23, 41. [Google Scholar] [CrossRef]
- Dong, B.; Miao, J.; Wang, Y.; Luo, W.; Ji, Z.; Lai, H.; Zhang, M.; Cheng, X.; Wang, J.; Fang, Y.; et al. Single-cell analysis supports a luminal-neuroendocrine transdifferentiation in human prostate cancer. Commun. Biol. 2020, 3, 778. [Google Scholar] [CrossRef]
- Prazanowska, K.H.; Hong, J.; Lim, S.B. Single-cell insights into the dynamic tumor microenvironment changes during immunotherapy of non-small cell lung cancer. Transl. Lung Cancer Res. 2023, 12, 1816–1821. [Google Scholar] [CrossRef]
- Sha, Y.; Wang, S.; Zhou, P.; Nie, Q. Inference and multiscale model of epithelial-to-mesenchymal transition via single-cell transcriptomic data. Nucleic Acids Res. 2020, 48, 9505–9520. [Google Scholar] [CrossRef]
- Cao, J.; Spielmann, M.; Qiu, X.; Huang, X.; Ibrahim, D.M.; Hill, A.J.; Zhang, F.; Mundlos, S.; Christiansen, L.; Steemers, F.J.; et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature 2019, 566, 496–502. [Google Scholar] [CrossRef]
- Zhang, L.; Li, Z.; Skrzypczynska, K.M.; Fang, Q.; Zhang, W.; O’Brien, S.A.; He, Y.; Wang, L.; Zhang, Q.; Kim, A.; et al. Single-Cell Analyses Inform Mechanisms of Myeloid-Targeted Therapies in Colon Cancer. Cell 2020, 181, 442–459.e429. [Google Scholar] [CrossRef]
- Thelen, M.; Wennhold, K.; Lehmann, J.; Garcia-Marquez, M.; Klein, S.; Kochen, E.; Lohneis, P.; Lechner, A.; Wagener-Ryczek, S.; Plum, P.S.; et al. Cancer-specific immune evasion and substantial heterogeneity within cancer types provide evidence for personalized immunotherapy. NPJ Precis Oncol. 2021, 5, 52. [Google Scholar] [CrossRef]
- Crinier, A.; Dumas, P.Y.; Escaliere, B.; Piperoglou, C.; Gil, L.; Villacreces, A.; Vely, F.; Ivanovic, Z.; Milpied, P.; Narni-Mancinelli, E.; et al. Single-cell profiling reveals the trajectories of natural killer cell differentiation in bone marrow and a stress signature induced by acute myeloid leukemia. Cell. Mol. Immunol. 2021, 18, 1290–1304. [Google Scholar] [CrossRef]
- Murgas, K.A.; Elkin, R.; Riaz, N.; Saucan, E.; Deasy, J.O.; Tannenbaum, A.R. Multi-scale geometric network analysis identifies melanoma immunotherapy response gene modules. Sci. Rep. 2024, 14, 6082. [Google Scholar] [CrossRef]
- Chen, J.H.; Nieman, L.T.; Spurrell, M.; Jorgji, V.; Elmelech, L.; Richieri, P.; Xu, K.H.; Madhu, R.; Parikh, M.; Zamora, I.; et al. Human lung cancer harbors spatially organized stem-immunity hubs associated with response to immunotherapy. Nat. Immunol. 2024, 25, 644–658. [Google Scholar] [CrossRef]
- Shen, C.; Jiang, X.; Li, M.; Luo, Y. Hepatitis Virus and Hepatocellular Carcinoma: Recent Advances. Cancers 2023, 15, 533. [Google Scholar] [CrossRef]
- Mahmood, M.; Liu, E.M.; Shergold, A.L.; Tolla, E.; Tait-Mulder, J.; Huerta-Uribe, A.; Shokry, E.; Young, A.L.; Lilla, S.; Kim, M.; et al. Mitochondrial DNA mutations drive aerobic glycolysis to enhance checkpoint blockade response in melanoma. Nat. Cancer 2024, 5, 1–14. [Google Scholar] [CrossRef]
- Dominguez Conde, C.; Xu, C.; Jarvis, L.B.; Rainbow, D.B.; Wells, S.B.; Gomes, T.; Howlett, S.K.; Suchanek, O.; Polanski, K.; King, H.W.; et al. Cross-tissue immune cell analysis reveals tissue-specific features in humans. Science 2022, 376, eabl5197. [Google Scholar] [CrossRef]
- Fu, R.; Gillen, A.E.; Sheridan, R.M.; Tian, C.; Daya, M.; Hao, Y.; Hesselberth, J.R.; Riemondy, K.A. clustifyr: An R package for automated single-cell RNA sequencing cluster classification. F1000Research 2020, 9, 223. [Google Scholar] [CrossRef]
- Sekaran, K.; Varghese, R.P.; Zayed, H.; El Allali, A.; George Priya Doss, C. Single-cell transcriptomic analysis reveals crucial oncogenic signatures and its associative cell types involved in gastric cancer. Med. Oncol. 2023, 40, 305. [Google Scholar] [CrossRef]
- Hao, Y.; Hao, S.; Andersen-Nissen, E.; Mauck, W.M., 3rd; Zheng, S.; Butler, A.; Lee, M.J.; Wilk, A.J.; Darby, C.; Zager, M.; et al. Integrated analysis of multimodal single-cell data. Cell 2021, 184, 3573–3587.e3529. [Google Scholar] [CrossRef]
- Aran, D.; Looney, A.P.; Liu, L.; Wu, E.; Fong, V.; Hsu, A.; Chak, S.; Naikawadi, R.P.; Wolters, P.J.; Abate, A.R.; et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat. Immunol. 2019, 20, 163–172. [Google Scholar] [CrossRef]
- Xi, N.M.; Li, J.J. Protocol for executing and benchmarking eight computational doublet-detection methods in single-cell RNA sequencing data analysis. STAR Protoc. 2021, 2, 100699. [Google Scholar] [CrossRef]
- Vallejos, C.A.; Risso, D.; Scialdone, A.; Dudoit, S.; Marioni, J.C. Normalizing single-cell RNA sequencing data: Challenges and opportunities. Nat. Methods 2017, 14, 565–571. [Google Scholar] [CrossRef]
- Buettner, F.; Natarajan, K.N.; Casale, F.P.; Proserpio, V.; Scialdone, A.; Theis, F.J.; Teichmann, S.A.; Marioni, J.C.; Stegle, O. Computational analysis of cell-to-cell heterogeneity in single-cell RNA-sequencing data reveals hidden subpopulations of cells. Nat. Biotechnol. 2015, 33, 155–160. [Google Scholar] [CrossRef]
- Ding, B.; Zheng, L.; Zhu, Y.; Li, N.; Jia, H.; Ai, R.; Wildberg, A.; Wang, W. Normalization and noise reduction for single cell RNA-seq experiments. Bioinformatics 2015, 31, 2225–2227. [Google Scholar] [CrossRef]
- Lun, A.T.; Bach, K.; Marioni, J.C. Pooling across cells to normalize single-cell RNA sequencing data with many zero counts. Genome Biol. 2016, 17, 75. [Google Scholar] [CrossRef]
- Lun, A.T.; McCarthy, D.J.; Marioni, J.C. A step-by-step workflow for low-level analysis of single-cell RNA-seq data with Bioconductor. F1000Res 2016, 5, 2122. [Google Scholar] [CrossRef]
- Lotfollahi, M.; Wolf, F.A.; Theis, F.J. scGen predicts single-cell perturbation responses. Nat. Methods 2019, 16, 715–721. [Google Scholar] [CrossRef]
- Chervov, A.; Zinovyev, A. Computational challenges of cell cycle analysis using single cell transcriptomics. arXiv 2022, arXiv:2208.05229. [Google Scholar]
- Townes, F.W.; Hicks, S.C.; Aryee, M.J.; Irizarry, R.A. Feature selection and dimension reduction for single-cell RNA-Seq based on a multinomial model. Genome Biol. 2019, 20, 295. [Google Scholar] [CrossRef]
- Germain, P.L.; Sonrel, A.; Robinson, M.D. pipeComp, a general framework for the evaluation of computational pipelines, reveals performant single cell RNA-seq preprocessing tools. Genome Biol. 2020, 21, 227. [Google Scholar] [CrossRef]
- Luecken, M.D.; Theis, F.J. Current best practices in single-cell RNA-seq analysis: A tutorial. Mol. Syst. Biol. 2019, 15, e8746. [Google Scholar] [CrossRef]
- Xiang, R.; Wang, W.; Yang, L.; Wang, S.; Xu, C.; Chen, X. A Comparison for Dimensionality Reduction Methods of Single-Cell RNA-seq Data. Front. Genet. 2021, 12, 646936. [Google Scholar] [CrossRef]
- Kobak, D.; Berens, P. The art of using t-SNE for single-cell transcriptomics. Nat. Commun. 2019, 10, 5416. [Google Scholar] [CrossRef]
- Freytag, S.; Tian, L.; Lonnstedt, I.; Ng, M.; Bahlo, M. Comparison of clustering tools in R for medium-sized 10x Genomics single-cell RNA-sequencing data. F1000Research 2018, 7, 1297. [Google Scholar] [CrossRef]
- Duo, A.; Robinson, M.D.; Soneson, C. A systematic performance evaluation of clustering methods for single-cell RNA-seq data. F1000Research 2018, 7, 1141. [Google Scholar] [CrossRef]
- Traag, V.A.; Waltman, L.; van Eck, N.J. From Louvain to Leiden: Guaranteeing well-connected communities. Sci. Rep. 2019, 9, 5233. [Google Scholar] [CrossRef]
- Heumos, L.; Schaar, A.C.; Lance, C.; Litinetskaya, A.; Drost, F.; Zappia, L.; Lucken, M.D.; Strobl, D.C.; Henao, J.; Curion, F.; et al. Best practices for single-cell analysis across modalities. Nat. Rev. Genet. 2023, 24, 550–572. [Google Scholar] [CrossRef]
- Liu, Z.; Sun, D.; Wang, C. Evaluation of cell-cell interaction methods by integrating single-cell RNA sequencing data with spatial information. Genome Biol. 2022, 23, 218. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, R.; Zhang, S.; Song, S.; Jiang, C.; Han, G.; Wang, M.; Ajani, J.; Futreal, A.; Wang, L. iTALK: An R package to characterize and illustrate intercellular communication. bioRxiv 2019, 507871. [Google Scholar] [CrossRef]
- Cillo, A.R.; Kurten, C.H.L.; Tabib, T.; Qi, Z.; Onkar, S.; Wang, T.; Liu, A.; Duvvuri, U.; Kim, S.; Soose, R.J.; et al. Immune Landscape of Viral- and Carcinogen-Driven Head and Neck Cancer. Immunity 2020, 52, 183–199.e9. [Google Scholar] [CrossRef]
- Song, X.; Dong, M.; Liu, M. PyMiner: A method for metabolic pathway design based on the uniform similarity of substrate-product pairs and conditional search. PLoS ONE 2022, 17, e0266783. [Google Scholar] [CrossRef]
- Efremova, M.; Vento-Tormo, M.; Teichmann, S.A.; Vento-Tormo, R. CellPhoneDB: Inferring cell-cell communication from combined expression of multi-subunit ligand-receptor complexes. Nat. Protoc. 2020, 15, 1484–1506. [Google Scholar] [CrossRef]
- Jin, S.; Guerrero-Juarez, C.F.; Zhang, L.; Chang, I.; Ramos, R.; Kuan, C.H.; Myung, P.; Plikus, M.V.; Nie, Q. Inference and analysis of cell-cell communication using CellChat. Nat. Commun. 2021, 12, 1088. [Google Scholar] [CrossRef]
- Noel, F.; Massenet-Regad, L.; Carmi-Levy, I.; Cappuccio, A.; Grandclaudon, M.; Trichot, C.; Kieffer, Y.; Mechta-Grigoriou, F.; Soumelis, V. Dissection of intercellular communication using the transcriptome-based framework ICELLNET. Nat. Commun. 2021, 12, 1089. [Google Scholar] [CrossRef]
- Cabello-Aguilar, S.; Alame, M.; Kon-Sun-Tack, F.; Fau, C.; Lacroix, M.; Colinge, J. SingleCellSignalR: Inference of intercellular networks from single-cell transcriptomics. Nucleic Acids Res. 2020, 48, e55. [Google Scholar] [CrossRef]
- Chan, I.S.; Knutsdottir, H.; Ramakrishnan, G.; Padmanaban, V.; Warrier, M.; Ramirez, J.C.; Dunworth, M.; Zhang, H.; Jaffee, E.M.; Bader, J.S.; et al. Cancer cells educate natural killer cells to a metastasis-promoting cell state. J. Cell Biol. 2020, 219, e202001134. [Google Scholar] [CrossRef]
- Wang, S.; Zheng, H.; Choi, J.S.; Lee, J.K.; Li, X.; Hu, H. A systematic evaluation of the computational tools for ligand-receptor-based cell-cell interaction inference. Brief. Funct. Genom. 2022, 21, 339–356. [Google Scholar] [CrossRef]
- Pong, A.; Mah, C.K.; Yeo, G.W.; Lewis, N.E. Computational cell-cell interaction technologies drive mechanistic and biomarker discovery in the tumor microenvironment. Curr. Opin. Biotechnol. 2024, 85, 103048. [Google Scholar] [CrossRef]
- Xie, J.; Deng, W.; Deng, X.; Liang, J.Y.; Tang, Y.; Huang, J.; Tang, H.; Zou, Y.; Zhou, H.; Xie, X. Single-cell histone chaperones patterns guide intercellular communication of tumor microenvironment that contribute to breast cancer metastases. Cancer Cell Int. 2023, 23, 311. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Smalley, I.; Chen, Z.; Wu, J.Y.; Phadke, M.S.; Teer, J.K.; Nguyen, T.; Karreth, F.A.; Koomen, J.M.; Sarnaik, A.A.; et al. Single-cell Characterization of the Cellular Landscape of Acral Melanoma Identifies Novel Targets for Immunotherapy. Clin. Cancer Res. 2022, 28, 2131–2146. [Google Scholar] [CrossRef] [PubMed]
- Dimitrov, D.; Turei, D.; Garrido-Rodriguez, M.; Burmedi, P.L.; Nagai, J.S.; Boys, C.; Ramirez Flores, R.O.; Kim, H.; Szalai, B.; Costa, I.G.; et al. Comparison of methods and resources for cell-cell communication inference from single-cell RNA-Seq data. Nat. Commun. 2022, 13, 3224. [Google Scholar] [CrossRef] [PubMed]
- Browaeys, R.; Saelens, W.; Saeys, Y. NicheNet: Modeling intercellular communication by linking ligands to target genes. Nat. Methods 2020, 17, 159–162. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Zhang, J.; Wu, Z.; Sun, X. Inferring microenvironmental regulation of gene expression from single-cell RNA sequencing data using scMLnet with an application to COVID-19. Brief. Bioinform. 2021, 22, 988–1005. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.; Sheng, J.; Gao, D.; Li, F.; Durrans, A.; Ryu, S.; Lee, S.B.; Narula, N.; Rafii, S.; Elemento, O.; et al. Transcriptome analysis of individual stromal cell populations identifies stroma-tumor crosstalk in mouse lung cancer model. Cell Rep. 2015, 10, 1187–1201. [Google Scholar] [CrossRef] [PubMed]
- Baruzzo, G.; Cesaro, G.; Di Camillo, B. Identify, quantify and characterize cellular communication from single-cell RNA sequencing data with scSeqComm. Bioinformatics 2022, 38, 1920–1929. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, T.; Hu, X.; Wang, M.; Wang, J.; Zou, B.; Tan, P.; Cui, T.; Dou, Y.; Ning, L.; et al. CellCall: Integrating paired ligand-receptor and transcription factor activities for cell-cell communication. Nucleic Acids Res. 2021, 49, 8520–8534. [Google Scholar] [CrossRef]
- Hu, Y.; Peng, T.; Gao, L.; Tan, K. CytoTalk: De novo construction of signal transduction networks using single-cell transcriptomic data. Sci. Adv. 2021, 7, eabf1356. [Google Scholar] [CrossRef]
- Dries, R.; Zhu, Q.; Dong, R.; Eng, C.L.; Li, H.; Liu, K.; Fu, Y.; Zhao, T.; Sarkar, A.; Bao, F.; et al. Giotto: A toolbox for integrative analysis and visualization of spatial expression data. Genome Biol. 2021, 22, 78. [Google Scholar] [CrossRef]
- Cang, Z.; Nie, Q. Inferring spatial and signaling relationships between cells from single cell transcriptomic data. Nat. Commun. 2020, 11, 2084. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Li, X.; Coleman, K.; Schroeder, A.; Ma, N.; Irwin, D.J.; Lee, E.B.; Shinohara, R.T.; Li, M. SpaGCN: Integrating gene expression, spatial location and histology to identify spatial domains and spatially variable genes by graph convolutional network. Nat. Methods 2021, 18, 1342–1351. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Yang, X. De novo reconstruction of cell interaction landscapes from single-cell spatial transcriptome data with DeepLinc. Genome Biol. 2022, 23, 124. [Google Scholar] [CrossRef] [PubMed]
- Arnol, D.; Schapiro, D.; Bodenmiller, B.; Saez-Rodriguez, J.; Stegle, O. Modeling Cell-Cell Interactions from Spatial Molecular Data with Spatial Variance Component Analysis. Cell Rep. 2019, 29, 202–211.e206. [Google Scholar] [CrossRef] [PubMed]
- Armingol, E.; Officer, A.; Harismendy, O.; Lewis, N.E. Deciphering cell-cell interactions and communication from gene expression. Nat. Rev. Genet. 2021, 22, 71–88. [Google Scholar] [CrossRef]
- Wang, X.; Almet, A.A.; Nie, Q. The promising application of cell-cell interaction analysis in cancer from single-cell and spatial transcriptomics. Semin. Cancer Biol. 2023, 95, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Noel, G.; Fontsa, M.L.; Garaud, S.; De Silva, P.; de Wind, A.; Van den Eynden, G.G.; Salgado, R.; Boisson, A.; Locy, H.; Thomas, N.; et al. Functional Th1-oriented T follicular helper cells that infiltrate human breast cancer promote effective adaptive immunity. J. Clin. Invest. 2021, 131, e139905. [Google Scholar] [CrossRef] [PubMed]
- Azizi, E.; Carr, A.J.; Plitas, G.; Cornish, A.E.; Konopacki, C.; Prabhakaran, S.; Nainys, J.; Wu, K.; Kiseliovas, V.; Setty, M.; et al. Single-Cell Map of Diverse Immune Phenotypes in the Breast Tumor Microenvironment. Cell 2018, 174, 1293–1308.e1236. [Google Scholar] [CrossRef] [PubMed]
- Bernard, V.; Semaan, A.; Huang, J.; San Lucas, F.A.; Mulu, F.C.; Stephens, B.M.; Guerrero, P.A.; Huang, Y.; Zhao, J.; Kamyabi, N.; et al. Single-Cell Transcriptomics of Pancreatic Cancer Precursors Demonstrates Epithelial and Microenvironmental Heterogeneity as an Early Event in Neoplastic Progression. Clin. Cancer Res. 2019, 25, 2194–2205. [Google Scholar] [CrossRef]
- Davidson, S.; Efremova, M.; Riedel, A.; Mahata, B.; Pramanik, J.; Huuhtanen, J.; Kar, G.; Vento-Tormo, R.; Hagai, T.; Chen, X.; et al. Single-Cell RNA Sequencing Reveals a Dynamic Stromal Niche That Supports Tumor Growth. Cell Rep. 2020, 31, 107628. [Google Scholar] [CrossRef]
- Guo, X.; Zhang, Y.; Zheng, L.; Zheng, C.; Song, J.; Zhang, Q.; Kang, B.; Liu, Z.; Jin, L.; Xing, R.; et al. Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nat. Med. 2018, 24, 978–985. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Hernandez, M.O.; Zhao, Y.; Mehta, M.; Tran, B.; Kelly, M.; Rae, Z.; Hernandez, J.M.; Davis, J.L.; Martin, S.P.; et al. Tumor Cell Biodiversity Drives Microenvironmental Reprogramming in Liver Cancer. Cancer Cell 2019, 36, 418–430.e416. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Sun, B.F.; Chen, C.Y.; Zhou, J.Y.; Chen, Y.S.; Chen, H.; Liu, L.; Huang, D.; Jiang, J.; Cui, G.S.; et al. Single-cell RNA-seq highlights intra-tumoral heterogeneity and malignant progression in pancreatic ductal adenocarcinoma. Cell Res. 2019, 29, 725–738. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Zhang, S.; Liu, Y.; He, X.; Qu, M.; Xu, G.; Wang, H.; Huang, M.; Pan, J.; Liu, Z.; et al. Single-cell RNA sequencing reveals the heterogeneity of liver-resident immune cells in human. Cell Discov. 2020, 6, 22. [Google Scholar] [CrossRef] [PubMed]
- Subedi, N.; Verhagen, L.P.; Bosman, E.M.; van Roessel, I.; Tel, J. Understanding natural killer cell biology from a single cell perspective. Cell Immunol. 2022, 373, 104497. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Siebert, J.R.; Burns, R.; Gerbec, Z.J.; Bonacci, B.; Rymaszewski, A.; Rau, M.; Riese, M.J.; Rao, S.; Carlson, K.S.; et al. Heterogeneity of human bone marrow and blood natural killer cells defined by single-cell transcriptome. Nat. Commun. 2019, 10, 3931. [Google Scholar] [CrossRef]
- Wang, Y.; Liang, Y.; Xu, H.; Zhang, X.; Mao, T.; Cui, J.; Yao, J.; Wang, Y.; Jiao, F.; Xiao, X.; et al. Single-cell analysis of pancreatic ductal adenocarcinoma identifies a novel fibroblast subtype associated with poor prognosis but better immunotherapy response. Cell Discov. 2021, 7, 36. [Google Scholar] [CrossRef]
- Krishna, C.; DiNatale, R.G.; Kuo, F.; Srivastava, R.M.; Vuong, L.; Chowell, D.; Gupta, S.; Vanderbilt, C.; Purohit, T.A.; Liu, M.; et al. Single-cell sequencing links multiregional immune landscapes and tissue-resident T cells in ccRCC to tumor topology and therapy efficacy. Cancer Cell 2021, 39, 662–677.e666. [Google Scholar] [CrossRef] [PubMed]
- Leader, A.M.; Grout, J.A.; Maier, B.B.; Nabet, B.Y.; Park, M.D.; Tabachnikova, A.; Chang, C.; Walker, L.; Lansky, A.; Le Berichel, J.; et al. Single-cell analysis of human non-small cell lung cancer lesions refines tumor classification and patient stratification. Cancer Cell 2021, 39, 1594–1609.e1512. [Google Scholar] [CrossRef]
- Jiang, R.; Sun, T.; Song, D.; Li, J.J. Statistics or biology: The zero-inflation controversy about scRNA-seq data. Genome Biol. 2022, 23, 31. [Google Scholar] [CrossRef]
- Huang, M.; Wang, J.; Torre, E.; Dueck, H.; Shaffer, S.; Bonasio, R.; Murray, J.I.; Raj, A.; Li, M.; Zhang, N.R. SAVER: Gene expression recovery for single-cell RNA sequencing. Nat. Methods 2018, 15, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Hou, W.; Ji, Z.; Ji, H.; Hicks, S.C. A systematic evaluation of single-cell RNA-sequencing imputation methods. Genome Biol. 2020, 21, 218. [Google Scholar] [CrossRef] [PubMed]
- An, U.; Pazokitoroudi, A.; Alvarez, M.; Huang, L.; Bacanu, S.; Schork, A.J.; Kendler, K.; Pajukanta, P.; Flint, J.; Zaitlen, N.; et al. Deep learning-based phenotype imputation on population-scale biobank data increases genetic discoveries. Nat. Genet. 2023, 55, 2269–2276. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Daugharthy, E.R.; Scheiman, J.; Kalhor, R.; Ferrante, T.C.; Terry, R.; Turczyk, B.M.; Yang, J.L.; Lee, H.S.; Aach, J.; et al. Fluorescent in situ sequencing (FISSEQ) of RNA for gene expression profiling in intact cells and tissues. Nat. Protoc. 2015, 10, 442–458. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Allen, W.E.; Wright, M.A.; Sylwestrak, E.L.; Samusik, N.; Vesuna, S.; Evans, K.; Liu, C.; Ramakrishnan, C.; Liu, J.; et al. Three-dimensional intact-tissue sequencing of single-cell transcriptional states. Science 2018, 361, eaat5691. [Google Scholar] [CrossRef] [PubMed]
- Xia, C.; Babcock, H.P.; Moffitt, J.R.; Zhuang, X. Multiplexed detection of RNA using MERFISH and branched DNA amplification. Sci. Rep. 2019, 9, 7721. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, C.; Grau, S.; Jager, C.; Sondermann, P.; Brunker, P.; Waldhauer, I.; Hennig, M.; Ruf, A.; Rufer, A.C.; Stihle, M.; et al. Unique carbohydrate-carbohydrate interactions are required for high affinity binding between FcgammaRIII and antibodies lacking core fucose. Proc. Natl. Acad. Sci. USA 2011, 108, 12669–12674. [Google Scholar] [CrossRef] [PubMed]
- Majewska, N.I.; Tejada, M.L.; Betenbaugh, M.J.; Agarwal, N. N-Glycosylation of IgG and IgG-Like Recombinant Therapeutic Proteins: Why Is It Important and How Can We Control It? Annu. Rev. Chem. Biomol. Eng. 2020, 11, 311–338. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, M.; Kogadeeva, M.; Gengenbacher, M.; McEwen, G.; Mollenkopf, H.J.; Zamboni, N.; Kaufmann, S.H.E.; Sauer, U. Integration of Metabolomics and Transcriptomics Reveals a Complex Diet of Mycobacterium tuberculosis during Early Macrophage Infection. mSystems 2017, 2, e00057-17. [Google Scholar] [CrossRef]
- Keren, L.; Bosse, M.; Marquez, D.; Angoshtari, R.; Jain, S.; Varma, S.; Yang, S.R.; Kurian, A.; Van Valen, D.; West, R.; et al. A Structured Tumor-Immune Microenvironment in Triple Negative Breast Cancer Revealed by Multiplexed Ion Beam Imaging. Cell 2018, 174, 1373–1387.e1319. [Google Scholar] [CrossRef]
- Medaglia, C.; Giladi, A.; Stoler-Barak, L.; De Giovanni, M.; Salame, T.M.; Biram, A.; David, E.; Li, H.; Iannacone, M.; Shulman, Z.; et al. Spatial reconstruction of immune niches by combining photoactivatable reporters and scRNA-seq. Science 2017, 358, 1622–1626. [Google Scholar] [CrossRef] [PubMed]
- Vento-Tormo, R.; Efremova, M.; Botting, R.A.; Turco, M.Y.; Vento-Tormo, M.; Meyer, K.B.; Park, J.E.; Stephenson, E.; Polanski, K.; Goncalves, A.; et al. Single-cell reconstruction of the early maternal-fetal interface in humans. Nature 2018, 563, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Giladi, A.; Cohen, M.; Medaglia, C.; Baran, Y.; Li, B.; Zada, M.; Bost, P.; Blecher-Gonen, R.; Salame, T.M.; Mayer, J.U.; et al. Dissecting cellular crosstalk by sequencing physically interacting cells. Nat. Biotechnol. 2020, 38, 629–637. [Google Scholar] [CrossRef] [PubMed]
- Regev, A.; Teichmann, S.A.; Lander, E.S.; Amit, I.; Benoist, C.; Birney, E.; Bodenmiller, B.; Campbell, P.; Carninci, P.; Clatworthy, M.; et al. The Human Cell Atlas. eLife 2017, 6, e27041. [Google Scholar] [CrossRef] [PubMed]
- Li, W.V.; Li, J.J. An accurate and robust imputation method scImpute for single-cell RNA-seq data. Nat. Commun. 2018, 9, 997. [Google Scholar] [CrossRef] [PubMed]
- Franzosa, E.A.; Sirota-Madi, A.; Avila-Pacheco, J.; Fornelos, N.; Haiser, H.J.; Reinker, S.; Vatanen, T.; Hall, A.B.; Mallick, H.; McIver, L.J.; et al. Gut microbiome structure and metabolic activity in inflammatory bowel disease. Nat. Microbiol. 2019, 4, 293–305. [Google Scholar] [CrossRef]
- Rozenblatt-Rosen, O.; Regev, A.; Oberdoerffer, P.; Nawy, T.; Hupalowska, A.; Rood, J.E.; Ashenberg, O.; Cerami, E.; Coffey, R.J.; Demir, E.; et al. The Human Tumor Atlas Network: Charting Tumor Transitions across Space and Time at Single-Cell Resolution. Cell 2020, 181, 236–249. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
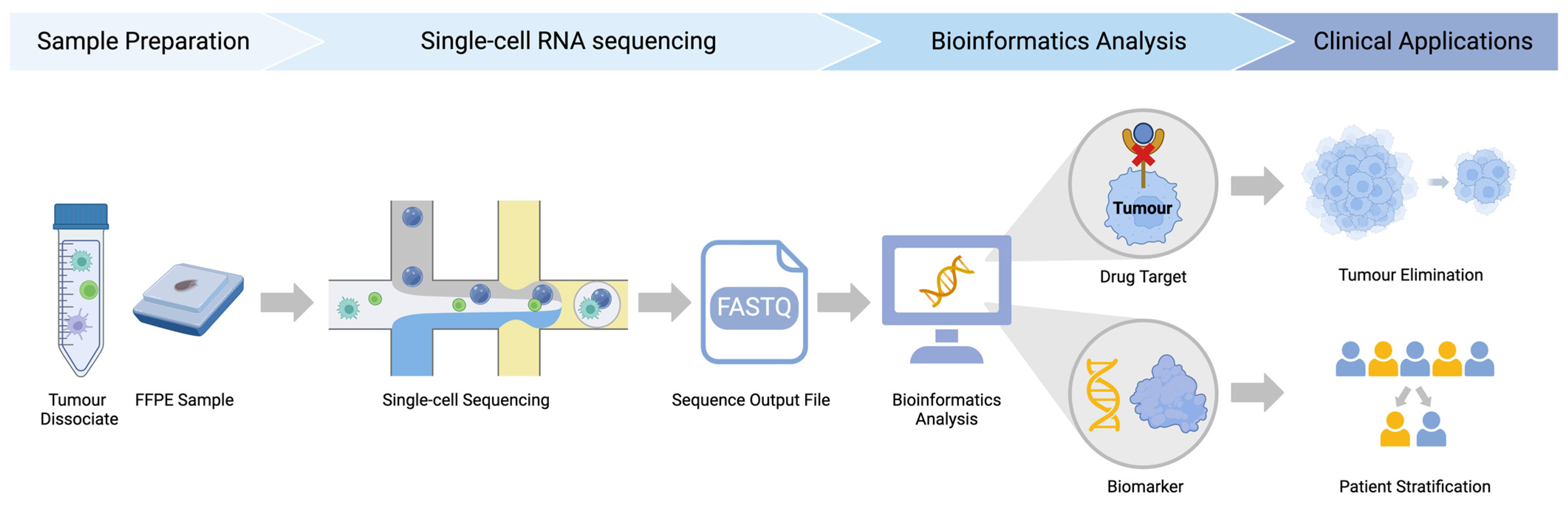{kind=link}
| Cancer Type | Single-Cell Technique(s) | Platform(s) | Analysis Tool(s) | Key Finding(s) | Reference(s) |
|---|---|---|---|---|---|
| Breast | scRNA-seq | Smart-Seq2 | Seurat | Micrometastases display an upregulated OXPHOS pathway with elevated levels of promoting metabolites compared to primary tumors. OXPHOS inhibition greatly attenuates metastasis in lung cancer models, potentiating its value in breast cancer. | [25] |
| Melanoma | scRNA-seq | 10x Genomics Chromium | Seurat | CCR7-SELL-CD4+ T cell subtype strongly correlates to developing severe immune-related adverse events. | [26] |
| Prostate | Spatial transcriptomic | Slide-seq | Conos | Stromal cells, including endothelial cells and pericytes, are more chaotically aligned in cancerous prostate; interactions between IGF1+ fibroblasts and IGF1R+ tumor cells are suggested by their colocalization. | [27] |
| Head and neck | Spatial transcriptomic and proteomic | 10x Genomics Visium | Seurat | Two distinct tumor phenotypes were identified: one situated at the leading edge characterized by an accumulation of highly proliferative tumor cells, and the other located at the core with a high infiltration of immune cells. | [28] |
| Colon | scRNA-seq; Spatial proteomic | 10x Genomics Chromium; PenoCycler | Seurat; CACTI | Single-cell analysis discovered the interactions occurring between SPP1+ macrophages and cancer-associated fibroblasts (CAFs) which are high in integrin receptors; spatial analysis demonstrates the spatial proximity between CAFs and SPP1+ macrophages, further suggesting their interactions. | [29] |
| Lung | scRNA-seq; Spatial transcriptomic | 10x Genomics Chromium; 10x Genomics Visium | Seurat | Four cancer subpopulations were identified. The UBE2C+ cell type is strongly correlated with the invasion of lung adenocarcinoma, reflected by its constant increase during the invasion process. The UBE2C+ subpopulation is predominantly located within the peritumoral region and is associated with highly active tumor activities. | [30] |
| scRNA-seq Data Analysis Step | Method | Package(s) | Advantage(s) | Limitation(s) | Example Study | Platform | Ref. |
|---|---|---|---|---|---|---|---|
| Quality control | Ambient RNA removal | SoupX, CellBender |
|
| [48,49] | R (SoupX) and Terra (CellRender) | [50,51] |
| Doublet removal | scDblFinder |
|
| [52] | R | [53] | |
| Normalization | Scaling-based | sctransform |
|
| [54] | R | [55] |
| Regression-based | SCnorm |
|
| [56] | R | [57] | |
| Spike-in RNA-based | BASiCS |
|
| [58] | R | [59] | |
| Confounder correction | Technical-based | scVI, Harmony |
|
| [60,61] | R (Harmony) and Python (scVI and scGen) | [62,63] |
| Biological-based | Seurat or Scanpy build-ins, Tricycle |
|
| [64] | R (Seurat) and Python (Scanpy and Tricycle) | [46,65,66] | |
| Feature selection | Deviance-based | sctransform |
|
| [67] | R | [55] |
| Highly variable gene-based | Seurat |
|
| [68] | R | [65] | |
| Highly expressed gene-based | Monocle |
|
| [69] | R | [70] | |
| Dimensionality reduction | PCA | Seurat, Scanpy |
|
| [71] | R, Python | [46,65] |
| t-SNE | Seurat, Scanpy |
|
| [72] | R, Python | [46,65] | |
| UMAP | Seurat, Scanpy |
|
| [73] | R, Python | [46,65] | |
| Clustering | Louvain | Seurat, Scanpy |
|
| [74] | R, Python | [46,65] |
| Leiden | Seurat, Scanpy |
|
| [75] | R, Python | [46,65] | |
| Annotation | Classifier-based | CellTypist, Clustifyr |
|
| [76,77] | Python, R | [78,79] |
| Reference-based | Azimuth, SingleR |
|
| [26,80] | R, Python | [81,82] |
| CCC Analysis Tool | Approach | Advantage | Limitation | Platform | Example Study | Ref. |
|---|---|---|---|---|---|---|
| iTALK | Identifies general interaction patterns between different cell types based on gene expression profiles. | Enables the prediction of interactions from multiple samples. | The scoring scheme accounts for only abundant genes and thus likely overlooks interactions between less abundant genes. | R | [108] | [101] |
| PyMINEr | Integrates multi-omics data to identify activating and inhibitory interactions; primarily focuses on identifying metabolic pathways. | Offers the full pipeline from clustering to visualization; there is no requirement for a reference database due to automatic generation; provides additional information on activator and inhibitor. | Signaling pathway-based CCC inference may lead to false prediction due to poor understanding of pathway components, increasing false positive rates. | Python | [109] | [103] |
| CellPhoneDB | Calculates the permutation-based LR scores to identify significantly up- and downregulated interactions. | Gives strengthened inflammation- and proliferation-related gene sets; reduces false positive rates by using heteromeric complex-based inference. | Shortened in epithelial–mesenchymal transition gene sets; may increase false negative rates. | Python; Python web interface | [110] | [104] |
| CellChat | Computes the communication score of each LR pair’s interaction strength that are permuted to identify significant interactions. | Reduces false positive rates by using heteromeric complex-based inference; integrates information of mediators and influencers; enables CCC inference in continuous cell states. | Lacks predictions within cell groups; pairwise comparisons limit analysis under different conditions. | R | [111] | [105] |
| ICELLNET | Calculates both individual and global communication scores to assess cellular communications among single cells or cell types of interests. | Particularly useful in predicting cytokine interactions; enables the incorporation of gene expressions from different datasets; implements experimental validation of the predicted interactions. | Comprises less interactions in the database; lacks information of signaling pathways and gene regulatory networks. | R | [106] | [106] |
| SingleCellSignalR | Uses regularized product scores to stabilize the noise and variability present in the dataset. | The regularized product score generates a stable threshold to reduce false positive rates. | Unable to integrate information from multiple samples. | R | [112] | [107] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tian, J.; Bai, X.; Quek, C. Single-Cell Informatics for Tumor Microenvironment and Immunotherapy. Int. J. Mol. Sci. 2024, 25, 4485. https://doi.org/10.3390/ijms25084485
Tian J, Bai X, Quek C. Single-Cell Informatics for Tumor Microenvironment and Immunotherapy. International Journal of Molecular Sciences. 2024; 25(8):4485. https://doi.org/10.3390/ijms25084485
Chicago/Turabian StyleTian, Jiabao, Xinyu Bai, and Camelia Quek. 2024. "Single-Cell Informatics for Tumor Microenvironment and Immunotherapy" International Journal of Molecular Sciences 25, no. 8: 4485. https://doi.org/10.3390/ijms25084485