
A Rare De Novo Mutation in the TRIM8 Gene in a 17-Year-Old Boy with Steroid-Resistant Nephrotic Syndrome: Case Report
 , , , , ,
, , , , ,
Abstract
:1. Introduction
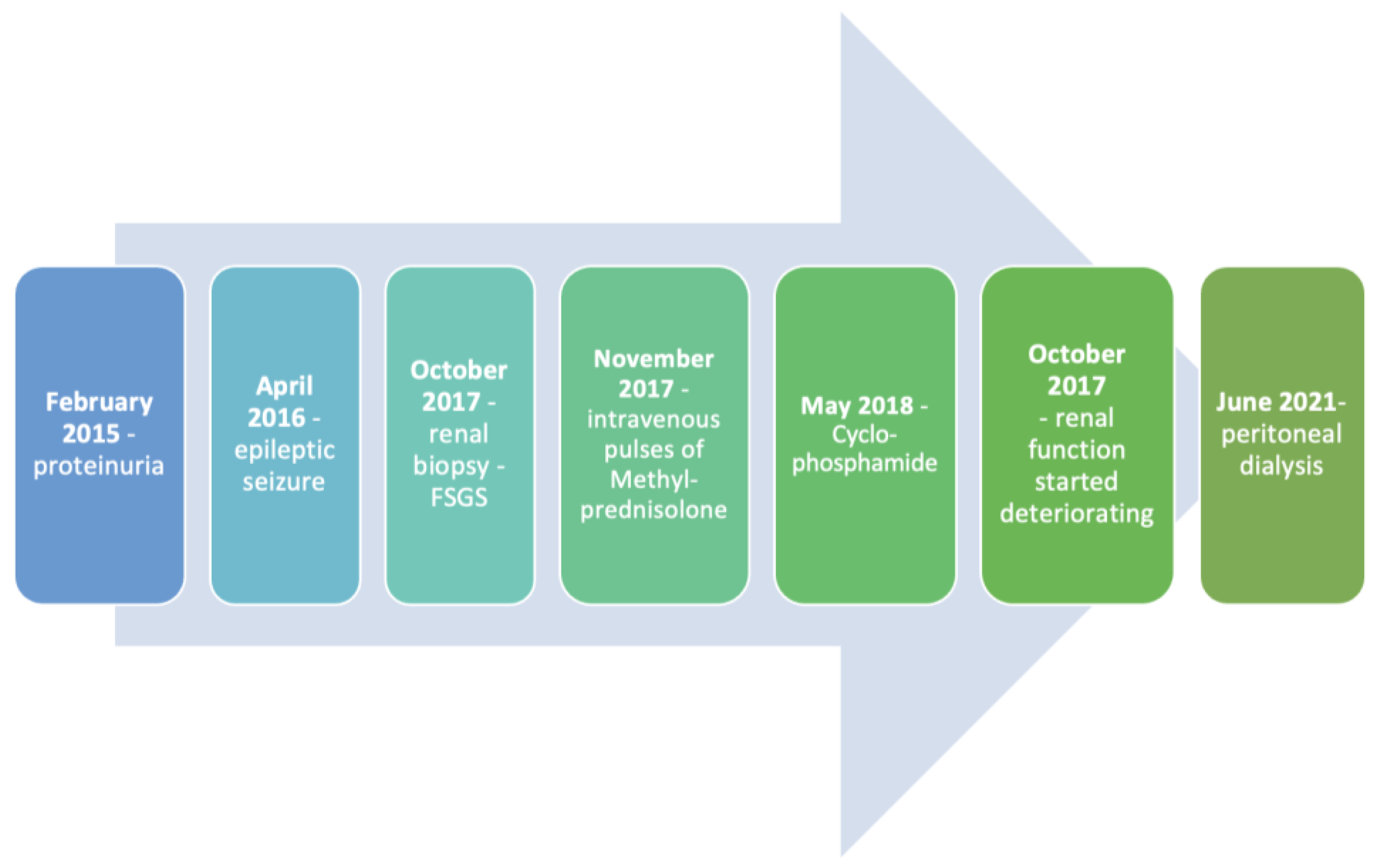
2. Case Report
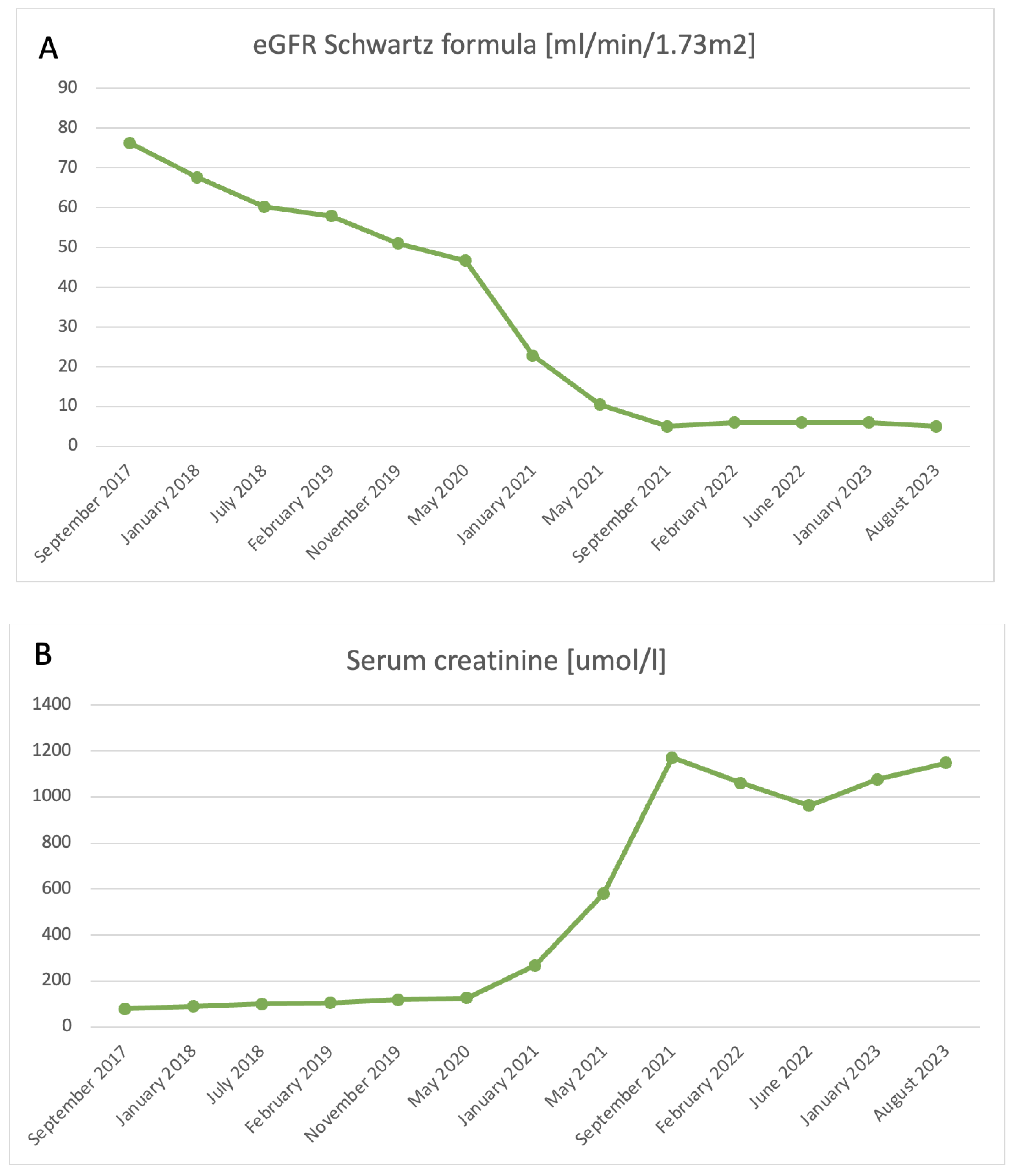
2.1. Renal Manifestations
2.2. Neurological Manifestations
2.3. Psychological Evaluation
2.4. Immunological Evaluation
2.5. Anamnesis Vitae
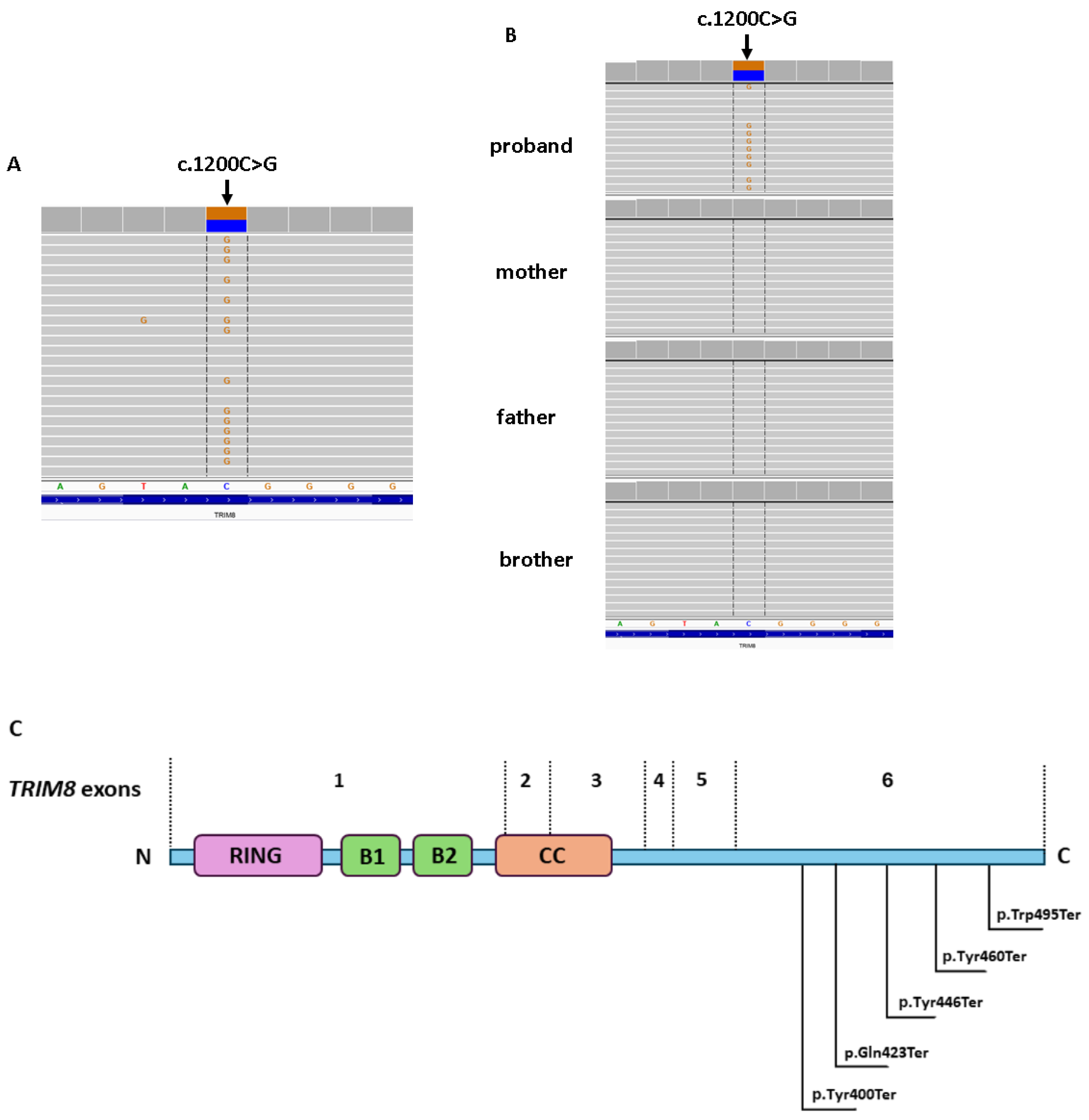
3. Whole-Exome Sequencing
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Eddy, A.A.; Symons, J.M. Nephrotic syndrome in childhood. Lancet 2003, 362, 629–639. [Google Scholar] [CrossRef]
- Tullus, K.; Webb, H.; Bagga, A. Management of steroid-resistant nephrotic syndrome in children and adolescents. Lancet Child Adolesc. Health 2018, 2, 880–890. [Google Scholar] [CrossRef] [PubMed]
- Noone, D.G.; Iijima, K.; Parekh, R. Idiopathic nephrotic syndrome in children. Lancet 2018, 392, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Trautmann, A.; Lipska-Zietkiewicz, B.S.; Schaefer, F. Exploring the clinical and genetic spectrum of steroid resistant nephrotic syndrome: The PodoNet registry. Front. Pediatr. 2018, 6, 200. [Google Scholar] [CrossRef] [PubMed]
- Warejko, J.K.; Tan, W.; Daga, A.; Schapiro, D.; Lawson, J.A.; Shril, S.; Lovric, S.; Ashraf, S.; Rao, J.; Hermle, T.; et al. Whole Exome Sequencing of Patients with Steroid-Resistant Nephrotic Syndrome. Clin. J. Am. Soc. Nephrol. 2018, 13, 53–62. [Google Scholar] [CrossRef]
- Trautmann, A.; Schnaidt, S.; Lipska-Zietkiewicz, B.S.; Bodria, M.; Ozaltin, F.; Emma, F.; Anarat, A.; Melk, A.; Azocar, M.; Oh, J.; et al. Long-term outcome of steroid-resistant nephrotic syndrome in children. J. Am. Soc. Nephrol. 2017, 28, 3055–3065. [Google Scholar] [CrossRef] [PubMed]
- Lovric, S.; Ashraf, S.; Tan, W.; Hildebrandt, F. Genetic testing in steroid-resistant nephrotic syndrome: When and how? Nephrol. Dial. Transplant. 2016, 31, 1802–1813. [Google Scholar] [CrossRef] [PubMed]
- Wiggins, R.C. The spectrum of podocytopathies: A unifying view of glomerular diseases. Kidney Int. 2007, 71, 1205–1214. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Hildebrandt, F. Whole exome sequencing identifies monogenic forms of nephritis in a previously unsolved cohort of children with steroid-resistant nephrotic syndrome and hematuria. Pediatr. Nephrol. 2022, 37, 1567–1574. [Google Scholar] [CrossRef] [PubMed]
- Schapiro, D.; Daga, A.; Lawson, J.A.; Majmundar, A.J.; Lovric, S.; Tan, W.; Warejko, J.K.; Fessi, I.; Rao, J.; Airik, M.; et al. Panel sequencing distinguishes monogenic forms of nephritis from nephrosis in children. Nephrol. Dial. Transplant. 2019, 34, 474–485. [Google Scholar] [CrossRef] [PubMed]
- Trautmann, A.; Bodria, M.; Ozaltin, F.; Gheisari, A.; Melk, A.; Azocar, M.; Anarat, A.; Caliskan, S.; Emma, F.; Gellermann, J.; et al. Spectrum of steroid-resistant and congenital nephrotic syndrome in children: The PodoNet registry cohort. Clin. J. Am. Soc. Nephrol. 2015, 10, 592–600. [Google Scholar] [CrossRef] [PubMed]
- Knoers, N.; Antignac, C.; Bergmann, C.; Dahan, K.; Giglio, S.; Heidet, L.; Lipska-Ziętkiewicz, B.S.; Noris, M.; Remuzzi, G.; Vargas-Poussou, R.; et al. Genetic testing in the diagnosis of chronic kidney disease: Recommendations for clinical practice. Nephrol. Dial. Transplant. 2021, 37, 239–254. [Google Scholar] [CrossRef] [PubMed]
- Reymond, A.; Meroni, G.; Fantozzi, A.; Merla, G.; Cairo, S.; Luzi, L.; Riganelli, D.; Zanaria, E.; Messali, S.; Cainarca, S.; et al. The tripartite motif family identifies cell compartments. EMBO J. 2001, 20, 2140–2151. [Google Scholar] [CrossRef]
- Sakai, Y.; Fukai, R.; Matsushita, Y.; Miyake, N.; Saitsu, H.; Akamine, S.; Sasazuki, M.; Ishizaki, Y.; Sanefuji, M.; Torisu, H.; et al. De novo truncating mutation of TRIM8 causes early-onset epileptic encephalopathy. Ann. Hum. Genet. 2016, 80, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Assoum, M.; Lines, M.A.; Elpeleg, O.; Darmency, V.; Whiting, S.; Edvardson, S.; Devinsky, O.; Heinzen, E.; Hernan, R.R.; Antignac, C.; et al. Further delineation of the clinical spectrum of de novo TRIM8 truncating mutations. Am. J. Med. Genet. Part A 2018, 176, 2470–2478. [Google Scholar] [CrossRef] [PubMed]
- Warren, M.; Takeda, M.; Partikian, A.; Opas, L.; Fine, R.; Yano, S. Association of a de novo nonsense mutation of the TRIM8 gene with childhood-onset focal segmental glomerulosclerosis. Pediatr. Nephrol. 2020, 35, 1129–1132. [Google Scholar] [CrossRef] [PubMed]
- Shirai, Y.; Miura, K.; Kaneko, N.; Ishizuka, K.; Endo, A.; Hashimoto, T.; Kanda, S.; Harita, Y.; Hattori, M. A novel de novo truncating TRIM8 variant associated with childhood-onset focal segmental glomerulosclerosis without epileptic encephalopathy: A case report. BMC Nephrol. 2021, 22, 417. [Google Scholar] [CrossRef] [PubMed]
- Badura-Stronka, M.; Kuszel, Ł.; Wencel-Warot, A.; Cudnoch, K.; Wołyńska, K.; Rutkowska, K.; Steinborn, B.; Płoski, R. Broadening the phenotypic spectrum of the presumably epilepsy-related SV2A gene variants. Epilepsy Res. 2023, 190, 107101. [Google Scholar] [CrossRef]
- Li, X.; Wei, Y.; Wang, M.; Jia, L.; Shi, Z.; Yang, X.; Ju, T.; Kuang, Q.; Xia, Z.; Gao, C. Two Children with Steroid-Resistant Significant Proteinuria Due to Nonsense Mutations of the TRIM8 Gene: A Case Report and Literature Review. Front. Pediatr. 2022, 10, 918373. [Google Scholar] [CrossRef]
- Sukenik-Halevy, R.; Perlman, S.; Ruhrman-Shahar, N.; Engel, O.; Orenstein, N.; Gonzaga-Jauregui, C.; Shuldiner, A.R.; Magal, N.; Hagari, O.; Azulay, N.; et al. The prevalence of prenatal sonographic findings in postnatal diagnostic exome sequencing performed for neurocognitive phenotypes: A cohort study. Prenat. Diagn. 2022, 42, 717–724. [Google Scholar] [CrossRef] [PubMed]
- Toniato, E.; Chen, X.P.; Losman, J.; Flati, V.; Donahue, L.; Rothman, P. TRIM8/GERP RING finger protein interacts with SOCS-1. J. Biol. Chem. 2002, 277, 37315–37322. [Google Scholar] [CrossRef] [PubMed]
- Schneider, W.M.; Chevillotte, M.D.; Rice, C.M. Interferon- stimulated genes: A complex web of host defenses. Annu. Rev. Immunol. 2014, 32, 513–545. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Guo, H. De novo truncating variants of TRIM8 and atypical neuro-renal syndrome: A case report and literature review. Ital. J. Pediatr. 2023, 15, 46. [Google Scholar] [CrossRef] [PubMed]
- Stratakis, C.A.; Berthon, A. Molecular mechanisms of ARMC5 mutations in adrenal pathophysiology. Curr. Opin. Endocr. Metab. Res. 2019, 8, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Zilbermint, M.; Xekouki, P.; Faucz, F.R.; Berthon, A.; Gkourogianni, A.; Schernthaner-Reiter, M.H.; Sinaii, M.B.N.; Quezado, M.M.; Merino, M.; Hodes, A.; et al. Primary Aldosteronism and ARMC5 Variants. J. Clin. Endocrinol. Metab. 2015, 100, 900–909. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Lao, L.; Mao, J.; Jin, W.; Luo, H.; Charpentier, T.; Qi, S.; Peng, J.; Hu, B.; Marcinkiewicz, M.M.; et al. Armc5 deletion causes developmental defects and compromises T-cell immune responses. Nat. Commun. 2017, 8, 13834. [Google Scholar] [CrossRef] [PubMed]
- Chuang, H.C.; Wang, X.; Tan, T.H. MAP4K Family Kinases in Immunity and Inflammation. Adv. Immunol. 2016, 129, 277–314. [Google Scholar] [PubMed]
- Gawande, D.Y.; Narasimhan, K.K.S.; Bhatt, J.M.; Pavuluri, R.; Kesherwani, V.; Suryavanshi, P.S.; Shelkar, G.P.; Dravid, S.M. Glutamate delta 1 receptor regulates autophagy mechanisms and affects excitatory synapse maturation in the somatosensory cortex. Pharmacol. Res. 2022, 178, 106144. [Google Scholar] [CrossRef] [PubMed]
- Niitsuma, S.; Kudo, H.; Kikuchi, A.; Hayashi, T.; Kumakura, S.; Kobayashi, S.; Okuyama, Y.; Kumagai, N.; Niihori, T.; Aoki, Y.; et al. Biallelic variants/mutations of IL1RAP in patients with steroid-sensitive nephrotic syndrome. Int. Immunol. 2019, 32, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Weng, P.L.; Majmundar, A.J.; Khan, K.; Lim, T.Y.; Shril, S.; Jin, G.; Musgrove, J.; Wang, M.; Ahram, D.F.; Aggarwal, V.S.; et al. De novo TRIM8 variants impair its protein localization to nuclear bodies and cause developmental delay, epilepsy, and focal segmental glomerulosclerosis. Am. J. Hum. Genet. 2021, 108, 357–367. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Parameter | Percentage 1st Assessment | Percentage 2nd Assessment | Absolute Number (Cells/μL) 1st Assessment | Absolute Number (Cells/μL) 2nd Assessment | Normal Percentage Value | Normal Absolute Number (Cells/μL) | |
|---|---|---|---|---|---|---|---|
| Lymphocytes | 44 | 51.1 | 27.4–56.0 | ||||
| Monocytes | 7.8 | 7.3 | 6.1–10.5 | ||||
| Granulocytes | 48.3 | 41.6 | 36.2–64.9 | ||||
| Lymphocytes | CD45+/SSC low | 2748 | 3050 | 1500–3900 | |||
| T Lymphocytes | CD3+/CD45+ | 81.5 | 78.1 | 2239 | 2381 | 52.9–79.1 | 1000–2700 |
| Cytotoxic T cells | CD3+CD8+/CD45+ | 32.6 | 31.1 | 895 | 949 | 18.2–33.2 | 300–1100 |
| Helper T cells | CD3+CD4+/CD45+ | 39.4 | 35.2 | 1083 | 1072 | 27.4–54.3 | 500–1600 |
| Helper/cytotoxic T cells | CD3+CD4+CD8+/CD45+ | 1.4 | 0.4 | 37 | 13 | 0.5–1.8 | 10–50 |
| NK cells | CD16+56+CD3-/CD45+ | 6.3 | 6.1 | 173 | 187 | 5.2–28.6 | 100–830 |
| T cells with NK phenotype | CD3+CD16+CD56+/CD45+ | 4.9 | 4.3 | 134 | 132 | 1.2–6.9 | 27–164 |
| B Lymphocytes | CD19+/CD45+ | 10.67 | 14.54 | 293 | 443 | 9.4–22.8 | 200–600 |
| Helper/cytotoxic T-cell ratio | CD4+/CD8+ | 1.2 | 1.1 | 1.1–2.7 | |||
| Helper memory + effector memory lymphocytes | CD4+CD45RO+/CD3+CD4+ | 50.1 | Not done | 543 | Not done | 27.2–62.0 | 200–570 |
| Helper naive + activated lymphocytes | CD4+CD45RA+/CD3+CD4+ | 50.5 | Not done | 547 | Not done | 31.1–66.3 | 180–880 |
| Cytotoxic memory + effector memory lymphocytes | CD8+CD45RO+/CD3+CD8+ | 22.4 | Not done | 200 | Not done | 15.9–46.4 | 80–300 |
| Cytotoxic naive + activated lymphocytes | CD8+CD45RA+/CD3+CD8+ | 81.1 | Not done | 726 | Not done | 44.1–77.1 | 170–730 |
| Helper T cells | |||||||
| Recent thymic emigrants | CD31+CD45RA+/CD3+CD4+ | 42.6 | Not done | 461 | Not done | >30 | |
| Central naive helper T cells | CD31-CD45RA+/CD3+CD4+ | 5.2 | Not done | 57 | Not done | ||
| Naive helper T cells | CD27+CD45RO-/CD3+CD4+ | 49.5 | Not done | 536 | Not done | 49.3–72.0 | |
| Memory helper T cells | CD27+CD45RO+/CD3+CD4+ | 40.6 | Not done | 440 | Not done | 24.5–44.4 | |
| Effector memory helper T cells | CD27-CD45RO+/CD3+CD4+ | 9.5 | Not done | 103 | Not done | 2.1–5.5 | |
| Effector helper T cells | CD27-CD45RO-/CD3+CD4+ | 0.3 | Not done | 4 | Not done | 0.1–8.7 | |
| Regulatory helper T cells | CD127-CD25+/CD3+CD4+ | 6.5 | Not done | 70 | Not done | 2.3–7.1 | |
| Cytotoxic T cells | |||||||
| Naive cytotoxic T cells | CD27+CD45RO-/CD3+CD8+ | 69 | Not done | 617 | Not done | 62.3–86.3 | |
| Memory cytotoxic T cells | CD27+CD45RO+/CD3+CD8+ | 16.8 | Not done | 150 | Not done | 12.2–27.2 | |
| Effector memory cytotoxic T cells | CD27-CD45RO+/CD3+CD8+ | 5.6 | Not done | 50 | Not done | 0.8–6.2 | |
| Effector cytotoxic T cells | CD27-CD45RO-/CD3+CD8+ | 8.62 | Not done | 77 | Not done | 0.8–13.2 | |
| Lymphocytes B | |||||||
| Transitional B lymphocytes | CD38+IgM++/CD19+ | 10.49 | 6.31 | 31 | 28 | 1.4–13.0 | 10–60 |
| Immature B lymphocytes | CD19+CD21low/limfocyty | 1.11 | 0.74 | 0.6–1.9 | |||
| Immature B lymphocytes | CD21low/CD19+ | 10.4 | 5.09 | 2.9–13.2 | 10–50 | ||
| Mature B lymphocytes | CD27-/CD19+ | 74 | 78.91 | 217 | 350 | 52.1–86.7 | 110–450 |
| Naive mature B lymphocytes | IgD+cd27-/CD19+ | 65.19 | 76.15 | 191 | 338 | 51.3–82.5 | 120–430 |
| Memory B lymphocytes | CD27+/CD19+ | 26 | 21.09 | 76 | 94 | 13.3–47.9 | 50–200 |
| Memory “non-switched” B lymphocytes (marginal zone) | CD27+IgD+/CD19+ | 18.04 | 16.04 | 53 | 71 | 4.6–18.2 | 20–70 |
| Memory “switched” B lymphocytes | CD27+IgD-/CD19+ | 7.85 | 5.09 | 23 | 23 | 8.7–25.6 | 30–110 |
| Memory “switched” B lymphocytes | CD27+IgD-/PBL | 0.84 | 0.74 | 1.5–4.0 | |||
| IgM-only memory B lymphocytes | IgD-IgM+/CD19+IgD-CD27+ | 39.27 | 15.31 | 3.9–18.8 | |||
| IgM-only memory B lymphocytes | IgD-IgM+CD27+/CD19+ | 3.08 | 0.78 | 0.4–3.1 | |||
| Activated B lymphocytes | CD38lowCD21low/CD19+ | 7.94 | 2.1 | 23 | 9 | 2.7–8.7 | 10–40 |
| Plasmablasts | CD38+++IgM-/CD19+ | 0.4 | 0.21 | 1 | 1 | 0.6–6.5 | 0–20 |
| CD40 Ligand | |||||||
| Stimulated | CD154+/CD3+CD8- | 51.5 | |||||
| Stimulated | CD69+/CD3+CD4+ | 99.5 | |||||
| Gene | Variant | Proband | Mother | Father | Brother |
|---|---|---|---|---|---|
| GRID1 | c.2771del | Heterozygous | Excluded | Heterozygous | Heterozygous |
| MAP4K5 | c.2293G>C | Heterozygous | Heterozygous | Excluded | Heterozygous |
| TRIM8 | c.1200C>G | Heterozygous | Excluded | Excluded | Excluded |
| IL1RAP | c.911A>G | Heterozygous | Heterozygous | Excluded | Excluded |
| ARMC5 | c.49G>C | Heterozygous | Heterozygous | Excluded | Heterozygous |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Badeńska, M.; Pac, M.; Badeński, A.; Rutkowska, K.; Czubilińska-Łada, J.; Płoski, R.; Bohynikova, N.; Szczepańska, M. A Rare De Novo Mutation in the TRIM8 Gene in a 17-Year-Old Boy with Steroid-Resistant Nephrotic Syndrome: Case Report. Int. J. Mol. Sci. 2024, 25, 4486. https://doi.org/10.3390/ijms25084486
Badeńska M, Pac M, Badeński A, Rutkowska K, Czubilińska-Łada J, Płoski R, Bohynikova N, Szczepańska M. A Rare De Novo Mutation in the TRIM8 Gene in a 17-Year-Old Boy with Steroid-Resistant Nephrotic Syndrome: Case Report. International Journal of Molecular Sciences. 2024; 25(8):4486. https://doi.org/10.3390/ijms25084486
Chicago/Turabian StyleBadeńska, Marta, Małgorzata Pac, Andrzej Badeński, Karolina Rutkowska, Justyna Czubilińska-Łada, Rafał Płoski, Nadezda Bohynikova, and Maria Szczepańska. 2024. "A Rare De Novo Mutation in the TRIM8 Gene in a 17-Year-Old Boy with Steroid-Resistant Nephrotic Syndrome: Case Report" International Journal of Molecular Sciences 25, no. 8: 4486. https://doi.org/10.3390/ijms25084486